

Abstract
Various strategies of interrupting highly active antiretroviral therapy (HAART) are being investigated for the treatment of human immunodeficiency virus (HIV) infection. Interruptions of greater than 2 weeks frequently result in rebound of plasma HIV RNA. In order to discern changes in the viral population that might occur during cycles of treatment interruption, we evaluated the homology of HIV-1 envelope gene sequences over time in 12 patients who received four to seven cycles of 4 weeks off HAART followed by 8 weeks on HAART by using the heteroduplex tracking assay and novel statistical tools. HIV populations in 9 of 12 patients diverged from those found in the first cycle in at least one subsequent cycle. The substantial genetic changes noted in HIV env did not correlate with increased or decreased log changes in levels of plasma HIV RNA (P > 0.5). Thus, genetic changes in HIV env itself did not contribute in a systematic way to changes in levels of plasma viremia from cycle to cycle of treatment interruption. In addition, the data suggest that there may be multiple compartments contributing to the rebound of plasma viremia and to viral diversity from cycle to cycle of intermittent therapy.
While highly active antiretroviral therapy (HAART) has significantly reduced human immunodeficiency virus (HIV) mortality (4), it is clear that HIV replication persists despite prolonged treatment that maintains plasma viremia below the limit of detection (18, 31, 43). Thus, lifelong treatment may be required for many HIV-infected individuals. Unfortunately, the long-term use of HAART is associated with an increasingly broad array of toxicities (2, 16, 22, 26, 29, 30, 35, 39), adherence to regimens is difficult (23), and the monetary cost of therapy and monitoring is prohibitive for many individuals and countries (3). In an attempt to reduce the requirement for lifelong therapy, several approaches to treatment interruption have been studied with individuals who have maintained suppression of plasma viremia and relatively high CD4+ T-cell counts while receiving HAART. One strategy, structured treatment interruptions (STI), allows bursts of plasma viremia during interruptions of HAART with the goal of enhancing HIV-specific immune responses (i.e., autoimmunization) (19, 32, 36, 37; B. Hirschel, C. Fagard, A. Oxenius, H. F. Gunthard, and F. Garcia, 9th Conf. Retrovir. Opportunistic Infect., poster 528-M, 2001).
In patients who began HAART during acute HIV infection, significant reductions in plasma viremia have been noted from cycle to cycle of STI, suggesting that successful autoimmunization had occurred (36). There was also a correlation in those individuals between an enhanced HIV-specific immune response and reductions in levels of HIV RNA from cycle to cycle of treatment interruptions. In individuals who began therapy during the chronic stage of infection, the results have been inconsistent, and a significant reduction in HIV plasma viremia from the first to subsequent cycles off HAART occurred in a much smaller proportion of patients (19, 32, 37; Hirschel et al., 9th Conf. Retrovir. Opportunistic Infect.). Furthermore, while there was a correlation between reductions in plasma HIV RNA levels from cycle to cycle and enhanced HIV-specific immune responses in certain reports (34, 37), other studies have failed to observe an immunologic benefit of treatment interruptions (32; Hirschel et al., 9th Conf. Retrovir. Opportunistic Infect.). Virologic factors may also contribute to changes in plasma viremia from cycle to cycle of treatment interruptions; for example, fluctuations in levels of plasma viremia may be correlated with genetic and biologic changes in HIV that result in differing replication kinetics.
A pool of HIV-infected resting CD4+ T cells that harbor replication-competent HIV is established early in infection (7), and this reservoir persists in the presence of long-term HAART despite the fact that plasma HIV RNA levels remain below the limit of detection (8, 15, 41). Persistent HIV replication during HAART also suggests the existence of reservoirs of infection that are impervious to antiretroviral drugs (7, 8, 15, 41). Several investigators have found that, in the plasma of certain individuals, HIV RNA that rebounded following a single interruption of HAART was genetically homologous to HIV isolated from resting CD4+ T cells prior to treatment interruptions (6, 24, 42). However, in other individuals, rebounding plasma viremia seemed to originate from different potential reservoirs (6, 42), such as lymphoid tissue, the reproductive tract, and other tissues (5, 8, 15, 27, 33). Regardless of the source of heterogeneous HIV from cycle to cycle, the appearance of divergent populations during multiple treatment interruptions may provide insights into the inability of the immune system to control viremia: i.e., diverse HIV populations may require a diverse immune response in order to control replication (1, 36).
Utilizing a heteroduplex tracking assay (HTA) to measure genetic diversity, we evaluated the homology of HIV-1 envelope from plasma HIV RNA obtained during multiple cycles of STI, implemented as 4 weeks off HAART followed by 8 weeks on HAART. We found that 9 of 12 individuals had divergent HIV env from the first to at least one subsequent cycle of treatment interruptions. There was no correlation between genetic changes in HIV env and changes in levels of plasma HIV RNA. These data provide important insights into the complexity of HIV rebound during multiple long-cycle antiretroviral treatment interruptions that may account, in part, for the limited success of such strategies for the purpose of autoimmunization in patients with chronic HIV infection.
MATERIALS AND METHODS
Patients.
Individuals participating in a clinical protocol approved by the National Institute of Allergy and Infectious Diseases Internal Review Board to evaluate the effects of repeated cycles of 4 weeks off HAART followed by 8 weeks on HAART were included in this analysis. Patient characteristics are provided in Table 1. The first 15 patients to reach a minimum of four cycles of intermittent therapy were evaluated; 3 patients were excluded from the final analysis due to an inability to consistently amplify viral gene products. Plasma HIV levels were determined in all patients every 4 weeks; patients had the option to undergo additional evaluations during any of the weeks during the periods of 4 weeks off HAART. Plasma HIV-1 levels were determined by branched-chain DNA (limit of detection of 50 copies per ml; Bayer Corporation, Tarrytown, N.Y.)
TABLE 1.
Characteristics of the patients in this study
| Patient | Baseline no. of CD4 cells/mm3 before interruptions | Nadir in no. of CD4 cells/mm3a | No. of pre-HAART plasma HIV RNA copies/mlb |
|---|---|---|---|
| 101 | 415 | 257 | 217,006 |
| 102 | 1,419 | 453 | 117,136 |
| 103 | 581 | 246 | 158,099 |
| 104 | 645 | 371 | 8,806 |
| 105 | 772 | 390 | 5,333 |
| 106 | 709 | 508 | 4,790 |
| 107 | 730 | 382 | 20,641 |
| 108 | 537 | 337 | 18,950 |
| 111 | 466 | 205 | 451,375 |
| 114 | 1,090 | 426 | 80,060 |
| 115 | 387 | 209 | 75,317 |
| 121 | 617 | 258 | 153,333 |
| Mean | 697 | 337 | 109,237 |
Lowest reported CD4+ T-cell count.
Highest reported level of HIV plasma viremia. Patients may have been receiving dual or single antiretroviral drugs.
RNA isolation and RT-PCR.
Patient plasma samples stored at −80°C (at least 1 ml) were thawed and centrifuged at 17,530 × g for 1 h at 4°C. The plasma was removed, and 750 μl of Trizol-LS (Invitrogen, Rockville, Md.) was added to the pellet. Tubes were incubated for 5 min at room temperature, and RNA was extracted with 200 μl of chloroform. RNA was precipitated by addition of an equal volume of isopropanol and washed twice with 70% ethanol. The air-dried pellets were dissolved in 25 μl of diethyl pyrocarbonate (DEPC) · H2O, and 7 μl was reverse transcribed in 20 μl of reverse transcription mix containing 1× reverse transcriptase (RT) buffer (Life Sciences, St. Petersburg, Fla.), 1 mM deoxynucleoside triphosphates (dNTPs) (Applied Biosystems, Inc., Foster City, Calif.), 30 U of avian myeloblastosis virus RT (Life Sciences), 20 U of RNase inhibitor (Roche Diagnostic, Indianapolis, Ind.), and 30 pmol of antisense primer ED12 (9). Reactions were carried out at 42°C for 45 min followed by heat inactivation at 70°C for 10 min.
Two microliters of HIV cDNA was used as the template for amplification of the V3-V5 coding region of the env gene by a nested PCR amplification strategy with primer pairs ED12 and ED5 (10) in the first round and ED7 and ED8 (38) in the second round. In both rounds of PCR, the reaction mixture contained 200 pmol of forward primer (ED5 or ED7), 200 pmol of reverse primer (ED12 or ED8), 1× PCR buffer, 2.5 mM MgCl2, 400 μM dNTPs, and 2.5 U of Taq polymerase (Invitrogen, Inc., Frederick, Md.) in a volume of 50 μl. The first-round amplification conditions were 4 cycles at 94°C for 45 s, 55°C for 45 s, and 72°C for 1 min followed by 32 cycles at 94°C for 30 s, 57°C for 30 s, and 72°C for 45 s with a 10-min extension at 72°C. One microliter of the first-round PCR product was amplified in a second round of PCR under the following conditions: 4 cycles at 94°C for 45 s, 53°C for 45 s, and 72°C for 1 min followed by 32 cycles at 94°C for 30 s, 55°C for 30 s, and 72°C for 45 s with a final extension at 72°C for 10 min.
HTA.
We wanted to maximize our ability to observe and differentiate between two classes of viral variants. The first class includes those variants that were prevalent prior to therapy and their direct descendants. The second class includes those existing at very low to undetectable frequency in the blood prior to therapy, which have greater levels of sequence difference from prevailing variants than expected in direct descendants (>1.5% different from prevailing virus), and which are thus likely to represent different viral lineages (9). For this analysis, we used patient-specific, radiolabeled single-stranded HTA probes (12) generated from each patient from a time point within the earliest off-drug cycle. The probe was derived from the second PCR product and labeled in a 50-μl reaction mixture containing 200 pmol of biotin-labeled ED7 primer, 200 pmol of ED8 primer, 1× PCR buffer, 2.5 mM MgCl2, 25 μM dNTPs, 0.2 μCi of [α-32P]dTTP, and 5 U of Taq polymerase (Invitrogen). Labeling was performed by amplification for five cycles at 94°C for 1 min, 55°C for 2 min, and 72°C for 2 min followed by an extension for 10 min at 72°C. The HTA probe was purified by binding the biotin moiety of the PCR product to streptavidin-coated magnetic beads (Dynal Biotech, Inc., Lake Success, N.Y.) and isolating the complementary strand with 0.1 N NaOH solution as previously described (13).
For HTA, the radiolabeled probe was annealed to target DNA derived from the nested RT-PCR of longitudinally collected, reverse-transcribed plasma HIV-1 RNA. Heteroduplexes were separated on a 5% acrylamide gel at 250 V for 2 h 50 min, stained with ethidium bromide, and visualized with the Kodak Imaging System (Eastman Kodak Company, Rochester, N.Y.). Gels were dried, exposed to phosphor screens overnight, and analyzed with ImageQuant (Molecular Dynamics, Inc., Sunnyvale, Calif.).
Gel analysis.
We developed statistics to compare two populations based on the number of bands having the same or different mobilities between two lanes, as well as the relative intensities of those bands. Band mobility was measured as the Rf, or fraction of the total distance traveled by homoduplex DNA. Band intensity was measured as the relative fraction of total lane signal (see below).
Kodak 1D gel analysis software (Eastman Kodak Co.) was used to identify and quantify bands manually in a computer-assisted way. Band intensity was calculated by interactively modeling bands as Gaussian functions and integrating over those functions. Background signal, estimated by the software along the length of each lane, was subtracted from band intensity. Integration and background subtraction were performed automatically by the software. In some cases, Gaussian modeling also separated bands whose signals overlapped; however, smears without visually distinct positions of signal concentration that could identify centers of bands were treated as part of the background. All bands that were visible in the software's display of the TIFF image of each gel were used; however, bands of very high apparent molecular weight (MW) (Rf of <0.10) were excluded, and faint (<1% of total signal) bands of intermediate MW (Rf of ∼0.3 to 0.7) present in all lanes were excluded, with the assumption that they were nonspecific products or single-stranded DNA (ssDNA). Not all bands included in the analysis were visible in the print reproductions of the figures. (The gels that were analyzed are available upon request.)
An a priori intensity cutoff for the inclusion of bands was not used; rather, visual ascertainment of bands was performed, erring on the side of inclusion. Of 533 bands identified, 28 (5%) had relative intensities of 0.01 or lower, and 15 (3%) had relative intensities of 0.001 or lower; two bands were identified with intensities of <0.0001. There was no correlation (P > 0.65) between the relative intensity of bands and the absolute lane signal for bands that had a relative intensity of <1% (data not shown).
Lane entropy.
Shannon's entropy (12) was applied to band data as a basic measure of viral population diversity within an individual lane. Suppose a lane has n bands. Let the proportion of total signal intensity of band i be denoted pi. Then the lane entropy is given by
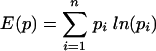 |
pi in this case is always non-zero, since these values always represent observed bands.
ED.
The entropy of the difference (ED) quantifies the difference of mobility and intensity in banding patterns between a pair of lanes. Suppose that between the two sets of bands there are m unique mobilities. Then for any mobility j, 1 < j < m, let pj be the intensity of the band at mobility j in the first lane. If there is no band at mobility j in the first lane, let pj = 0. Define qj similarly for bands in the second lane. Then the ED is defined as
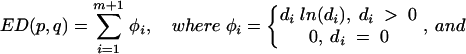 |
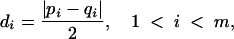 |
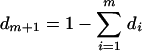 |
ED can be visualized in the following way. Overlay two lanes to produce a composite “lane.” Where band positions coincide, suppose they cancel out each other's signal. Then the entropy of the resulting lane is essentially the ED.
The ED quantifies both changes in relative abundance of variants common to both lanes and changes in the diversity between the lanes. Two lanes with variants of identical mobility and intensity will give an ED of zero, while lane pairs with completely nonoverlapping sets of variants will give the highest ED, with the ED growing with the total number of distinct variants.
Since variations in gel conditions can cause identical heteroduplexes to migrate at slightly different rates, we assumed that bands migrating within 5% of the average of their relative mobilities had the same mobility for the purpose of calculating ED.
This method assumes that bands having the same mobility represent identical or very similar variants. Two targets can have two different sets of changes with respect to a probe, which nevertheless lead to similar heteroduplex mobilities. However, since for each patient we use a probe derived from that patient, all target sequence is evolutionarily closely related to that probe. Since very few new nucleotide differences are expected to arise during the time intervals studied, shared differences with the probe are most likely to account for identical mobility shifts between two targets.
OC.
The overlap coefficient (OC) is a normalization of the ED, which takes the value zero when the banding patterns are identical (i.e., when ED = 0) and which takes a maximum of 1 when there is no overlap at all between lane variants. For banding patterns p and q, if we write
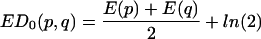 |
where E is again the Shannon entropy of the individual banding pattern, then the OC is given by
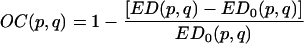 |
ED0 is the value of ED if the patterns were completely nonoverlapping; i.e., if no pair of bands belonged to the same mobility class.
For any patient, the mean OC is the average of OCs calculated for every pair of 4-week HTA lanes. Thus, the mean OC characterized patients based on viral population differences occurring among all STI cycles. This quantified the propensity of a patient to generate unique viral variants at any cycle; the higher the mean OC, the more likely a patient is to have had distinct viral populations between any two cycles.
Scripts (written in PERL) for calculation of these statistics are available upon request.
Statistics.
The Spearman test for correlation was used for each patient, comparing the association between log decreases and log increases in plasma HIV RNA and the corresponding OC for pairs of consecutive cycles. The Student's one-sample t test was used to determine if the mean of these correlations was significantly different from zero. Adjustment of P values for multiple testing was done by the Bonferroni method.
For each patient, the OCs were taken on pairs of adjacent time points and then grouped according to whether the change in plasma viral levels between the pair of time points did or did not achieve at least a 0.5-log viral decrease. For each patient, the average OC over cycles with at least a 0.5-log decrease was calculated, and the average OC for each patient over cycles at which a 0.5-log decrease was not achieved was calculated. The means for each of these groups were calculated and reported with the corresponding 95% confidence interval (CI).
RESULTS
Analysis of week 4 rebound plasma HIV by HTA.
Twelve patients underwent a minimum of four cycles of 4 weeks off HAART followed by 8 weeks on HAART. For each patient, the HTA was performed on plasma HIV env (V3-V5) sequences obtained from the 4th week of each sequential treatment interruption. Although the levels varied from cycle to cycle, each patient had sufficient HIV plasma RNA to perform the necessary amplification during each treatment interruption period analyzed. Double-stranded DNA (dsDNA) heteroduplexes with mismatched nucleotides show reduced mobilities when the degree of divergence exceeds 1 to 2%, and further reduction in mobility occurs when gaps are present due to a deletion or insertion in one of the annealing strands (13). Nine of 12 patients had HIV env populations that diverged significantly (i.e., exhibited several persistent or recurrent bands not found in pretreatment samples, with an Rf of <0.95 relative to the homoduplex band) from the first interruption period to at least one subsequent cycle (Fig. 1A). By this criterion, HIV env remained relatively homogeneous from the first to each subsequent cycle in 3 of the 12 patients (Fig. 1B).
FIG. 1.


Week 4 HTA and plasma HIV RNA levels from multiple HAART interruptions. Plasma HIV RNA was determined by branched-chain DNA assay and is reported as the number of copies per milliliter (•). Nine of 12 patients (patients 101, 103, 104, 106, 107, 108, 114, 115, and 121) had heterogeneous HIV env during four to seven cycles of treatment interruptions (A), while 3 patients (patients 102, 105, and 111) maintained similar levels of HIV env as determined by HTA (B). The sample from which the probe was made is indicated by an asterisk.
In order to quantify the sequence divergence between populations with a greater degree of sensitivity, we utilized the ED, a measure of the divergence in band mobility and intensity of HIV env between each pair of time points. Among the three patients without evidence for significant divergence in HIV env from cycle to cycle, the mean entropy was 0.57 (range, 0.41 to 0.75) versus 1.26 (range, 0.85 to 1.83) among the patients with apparent divergence (data not shown). In order to normalize the ED, the OC was evaluated by comparing adjacent lanes as a measure of the likelihood of divergent HIV env at any given interruption. Similar to visual inspection (Fig. 1) and the ED results, the mean OC of the three patients who did not appear to have significant alterations in mobility or intensity in adjacent lanes was 0.39 (range, 0.38 to 0.40) versus 0.75 (range, 0.53 to 0.85) among the nine patients with apparent diversity in intensity and mobility (P < 0.001). Therefore, these statistical analyses validated the subjective interpretation of the gels. (The ED and OC for all lane pairs are available upon request.)
In order to evaluate the possibility that divergence in HIV env was the result of random errors in PCR, the reproducibility of variant population sampling (9, 11) was analyzed. A minimum of 1,000 copies of HIV env RNA were reverse transcribed and amplified in two independent PCRs from various time points for three patients (Fig. 2A). As determined by visual inspection, the results were highly reproducible. In addition, the OC was determined for each pair of samples. The OC ranged from 0.00 to 0.30 (Fig. 2). In order to evaluate the possibility that various levels of input RNA above 1,000 copies could result in the appearance of variant populations, HTA was performed on a single time point for patient 101 by utilizing a fivefold serial dilution of cDNA-derived HIV RNA. Two independent PCRs were done per dilution (Fig. 2B). As determined by visual inspection, the results were highly reproducible. The OC ranged from 0.15 to 0.29 for each pair of samples, and the average OC across all samples was 0.19.
FIG. 2.

Reproducibility of HTA results. Independent PCRs for paired samples from week 4 of multiple cycles of HAART interruption were evaluated in three patients to analyze the reproducibility of variant population sampling (A). The OC for each pair is provided. In addition, the effects of different numbers of input HIV RNA copies on HTA were evaluated by serial dilutions of a single sample from patient 101 (B). Two independent PCRs were performed for each sample. The OC for each sample pair is provided. The overall OC was 0.19.
Comparison of genetic changes in HIV env and plasma HIV RNA levels.
Seven patients experienced a >0.5-log decrease in maximal plasma HIV RNA rebound compared to the previous period off HAART during a total of 10 interruption intervals (one to two events per patient) (patients 101, 102, 105, 107, 114, 115, and 121). The overall average of the OCs during these episodes of decreased plasma HIV RNA (0.62; 95% CI: 0.44, 0.80) was similar to that of the 22 episodes in the same individuals with a <0.5-log decrease in plasma HIV RNA compared to adjacent cycles off HAART (0.51; 95% CI: 0.36, 0.67). In addition, five patients (patients 103, 104, 106, 108, and 111) did not experience a single episode of >0.5-log decrease in plasma HIV RNA compared to the previous cycle off HAART during 23 treatment interruptions and had an average OC of 0.62 (95% CI: 0.45, 0.80), yet the average OC was similar to that of the seven patients with significant decreases in plasma HIV RNA. Therefore, it was not surprising that there was no correlation between the log change in plasma viremia and a change in the overall average OC (mean correlation for log decreases, 0.23; range, −1 to 1; and mean correlation for log increases, 0.02; range, −1 to 1; P > 0.5).
Although there was no correlation between the average OC and log changes in plasma HIV RNA from adjacent cycles off HAART, it was possible that a log decrease of >0.5 from the first to the last cycle off HAART was associated with divergence in HIV env compared to that in patients with no log change or a log increase in plasma HIV RNA. The average OC of the six patients (patients 102, 106, 108, 115, 114, and 121) with a >0.5-log decrease in plasma HIV RNA over all cycles off HAART was 0.67 (95% CI: 0.53, 0.81). This was not significantly different from the average OC of the six patients (patients 101, 103, 104, 105, 107, and 111) with no change or a >0.5-log increase in plasma HIV RNA from the first to the last cycle off HAART of 0.65 (95% CI: 0.48, 0.82) (P > 0.5).
Analysis of multiple time points during treatment interruptions.
Four of the nine patients with divergent HIV env during multiple treatment interruptions had plasma viral isolates available from multiple time points during each interruption. For each of these individuals, the first week shown was the first level of plasma HIV RNA of >50 copies per ml during each cycle (Fig. 3). Given the complexity of the gel patterns and the difficulty of interpreting them visually for changes in mobility and intensity, we plotted the OC for each patient for all pairs of lanes as colors on a grid in order to more easily interpret genetic shifts in plasma HIV populations within and between interruptions (Fig. 4). Warm (more red) colors indicate greater divergence of the banding patterns between two lanes, while cool (more blue) colors indicate greater homogeneity of banding patterns. (The OC for all lane pairs is available upon request.)
FIG. 3.

HTA and plasma HIV RNA levels for patients with multiple HIV env isolates within cycles off HART. Plasma HIV RNA was determined by branched-chain DNA assay and is reported as the number of copies per milliliter (•). Four patients (patients 101, 104, 105, and 115) had weekly evaluations during each of 4 weeks off HAART during a cycle of 4 weeks off HAART followed by 8 weeks on HAART. The first sample shown for each cycle is the first sample with >50 copies of HIV RNA per ml. The sample from which the probe was made is indicated by an asterisk.
FIG. 4.

OC plots for patients with divergent HIV env variants within cycles off HAART. The plot reduces the complexity of the gel data to a single comparative measure to assist in interpretation of the gels. The OC, a measure of divergence in mobility and intensity of bands between pairs of lanes, is calculated and displayed for each pair of lanes or time points. Warm (more red) colors indicate greater divergence of the banding patterns between two lanes, while cool (more blue) colors indicate greater homogeneity of banding patterns. cyc, cycle; Pt, patient.
The OC plots demonstrate that patients 101 and 104 had substantially divergent HIV env levels at all cycles subsequent to the initial rebound. This is indicated by the orange-to-red boxes along the first row and column (Fig. 4). Patient 115 replicated virus similar to the initial isolate during cycle 1 of week 2 (cycle 1/week 2) to cycle 2/week 2, cycle 3/week 3, and cycle 5/week 3, as indicated by the greenish boxes, but relatively divergent virus in cycles 3/week 4, 4/week 3, and 5/week 4 as indicated by the orange-yellow boxes at each intersection (Fig. 4). In contrast, patient 105 had relatively homogenous HIV env from the initial viral isolates from the first to each of the subsequent cycles, with the possible exception of cycles 5 and 6, as indicated by the green-yellow boxes (Fig. 4).
Patient 101 appeared to generate distinct HIV populations in almost every sample. The predominance of orange-to-red colors throughout the plot indicates the divergence of HIV env within as well as between cycles of treatment interruptions. A clear exception is shown by cycle 2/week 2 and cycle 5/week 4, where the blue box at the intersection of these time points on the OC plot indicated homology (Fig. 4). Patient 105 tended to vary around a relatively stable spectrum of variants. However, oscillations between different sets of variants at late time points can be observed as warm-colored columns at cycle 5/week 2 and cycle 6/weeks 1 and 3 (Fig. 4). Patients 104 and 115 exhibited intermediate levels of divergence. Virus in patient 104 from cycle 1/week 1 was divergent compared to that at all other time points, and cycle 4/week 4 was relatively divergent from multiple time points (Fig. 4). Virus from patient 115 was characterized by fluctuations in closely related variants (rapidly migrating bands in Fig. 3), with the occasional outgrowth of rather highly divergent variants (slowly migrating bands). This behavior is reflected in the haphazard color pattern in the OC plot.
DISCUSSION
In the present study, we have examined the heterogeneity of HIV env in patients who underwent cyclic interruptions of HAART. Although HIV env heterogeneity was found in 9 of 12 patients who underwent multiple cycles of 4 weeks off HAART followed by 8 weeks on HAART, there was no correlation between divergence in HIV env and changes in levels of plasma HIV RNA for all of the patients evaluated together (P > 0.5). These data indicate that genetic changes per se in HIV env do not contribute in a systematic way to fluctuations in plasma HIV RNA levels. Furthermore, three of four patients who had specimens available from the initial time point in each cycle when plasma HIV RNA was greater than 50 copies per ml had initially genetically divergent HIV env from cycle to cycle (Fig. 3 and 4). Thus, the HIV env gene associated with most rebounds in plasma viremia was divergent from the initial population during multiple interruptions in antiretroviral therapy. In addition, fluctuations in HIV env within the limited 4 weeks of treatment interruptions were observed during at least 1 cycle in all four patients. The env region of HIV is known to accumulate nucleotide changes within untreated patients at a rate of about 1% per year (38), while the rate of nucleotide evolution in patients on effective HAART is <0.05% per year, if it can be detected at all. Thus, continued evolution of virus in any of our patients over the course of the study should be less than that in an untreated patient. The significant HTA mobility shifts that we observed typically correspond to >1 to 2% difference (13). Therefore, it was possible that preexisting viral variants originating from evolutionary lineages distinct from that of strains prevailing before therapy, rather than newly evolved variants, are frequently responsible for viral outgrowth during antiretroviral STI. It was also possible that ongoing evolution during STI was responsible for the divergence in HIV env that we observed during multiple cycles of treatment interruptions. Sequence analysis and phylogenetic evaluation are necessary to further explore this issue.
The anatomical source of divergent lineages remains unclear. Several investigators have demonstrated that rebound plasma HIV RNA following a single interruption of HAART was genetically homologous to HIV isolated from resting CD4+ T cells prior to treatment interruptions in certain individuals (6, 24, 42), whereas in other individuals, rebounding plasma virus seemed to originate from other potential reservoirs (6, 42). Distinct genetic variants of HIV have been detected in multiple fluid, anatomical, and cellular compartments: e.g., brain (5, 27), lung (25), spleen (14, 20), kidney (28), genital secretions (33), and natural killer cells (40). Thus, during multiple interruptions of HAART, it is possible that plasma HIV can originate from different reservoirs or sites within a reservoir.
It was possible that the numbers of nucleotide substitutions were similar between the patients with relatively homogeneous and relatively divergent HIV env genes as determined by HTA, but that the patients with relatively divergent HIV env genes also had length variability. Sequence analysis is necessary to evaluate this possibility. However, it has been demonstrated that length variation itself may have biological implications (17, 21).
We have introduced novel descriptive statistics for HTA divergence based on a computer-aided ascertainment of bands. The number of bands in an HTA lane is a direct measurement of the minimum number of different genetic variants in the target population as a whole. Therefore, we have analyzed the absolute number of bands to determine statistically the diversity and divergence of bands by HTA. This approach contrasts with previous reports in which an image analysis of each lane in its entirety was performed (9, 11). This technique may minimize the inclusion of background signal and spurious banding thereby enhancing the statistical power of comparisons.
We have demonstrated that interruptions of therapy that allowed for relatively high levels of plasma HIV RNA resulted in the emergence of divergent HIV env populations in 9 of 12 patients. In addition, we demonstrated that this diversity was not correlated with significant fluctuations in the level of plasma HIV RNA observed from cycle to cycle of intermittent therapy. Thus, the alterations in peak plasma viremia observed during treatment interruptions cannot be explained by diversity in HIV env as determined by HTA. These data suggest that multiple compartments are responsible for rebound plasma HIV during sequential long-cycle antiretroviral treatment interruptions in patients with chronic HIV infection; sequence and phylogenetic analyses will be required to confirm the latter hypothesis.
Acknowledgments
M. Dybul, M. Daucher, and M. A. Jensen contributed equally to this work.
We acknowledge with great appreciation the contribution of the persons living with HIV who participated in this research, as well as clinical staff, particularly the case managers, who cared for them. In addition, we thank Mary Rust for expert editorial assistance and John Weddle for expert graphic design.
This work was supported in part by Public Health Service grants to J.I.M. and the University of Washington Center for AIDS Research. M.A.J. was supported by an NIH Genomics Training grant.
REFERENCES
- 1.Altfeld, M., and B. D. Walker. 2001. Less is more? STI in acute and chronic HIV-1 infection. Nat. Med. 7:881-884. [DOI] [PubMed] [Google Scholar]
- 2.Behrens, G., A. Dejam, H. Schmidt, H. J. Balks, G. Brabant, T. Korner, M. Stoll, and R. E. Schmidt. 1999. Impaired glucose tolerance, beta cell function and lipid metabolism in HIV patients under treatment with protease inhibitors. AIDS 13:F63-F70. [DOI] [PubMed] [Google Scholar]
- 3.Binswanger, H. P. 2001. Public health. HIV/AIDS treatment for millions. Science 292:221-223. [DOI] [PubMed] [Google Scholar]
- 4.Centers for Disease Control and Prevention. 2000. HIV/AIDS Surveillance Report 12. Centers for Disease Control and Prevention, Atlanta, Ga.
- 5.Cheng-Mayer, C., C. Weiss, D. Seto, and J. A. Levy. 1989. Isolates of human immunodeficiency virus type 1 from the brain may constitute a special group of the AIDS virus. Proc. Natl. Acad. Sci. USA 86:8575-8579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chun, T. W., R. T. Davey, Jr., M. Ostrowski, J. S. Justement, D. Engel, J. I. Mullins, and A. S. Fauci. 2000. Relationship between pre-existing viral reservoirs and the re-emergence of plasma viremia after discontinuation of highly active anti-retroviral therapy. Nat. Med. 6:757-761. [DOI] [PubMed] [Google Scholar]
- 7.Chun, T. W., D. Engel, M. M. Berrey, T. Shea, L. Corey, and A. S. Fauci. 1998. Early establishment of a pool of latently infected, resting CD4+ T cells during primary HIV-1 infection. Proc. Natl. Acad. Sci. USA 95:8869-8873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chun, T. W., L. Stuyver, S. B. Mizell, L. A. Ehler, J. A. Mican, M. Baseler, A. L. Lloyd, M. A. Nowak, and A. S. Fauci. 1997. Presence of an inducible HIV-1 latent reservoir during highly active antiretroviral therapy. Proc. Natl. Acad. Sci. USA 94:13193-13197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Delwart, E. L., and C. J. Gordon. 1997. Tracking changes in HIV-1 envelope quasispecies using DNA heteroduplex analysis. Methods 12:348-354. [DOI] [PubMed] [Google Scholar]
- 10.Delwart, E. L., J. I. Mullins, P. Gupta, G. H. Learn, Jr., M. Holodniy, D. Katzenstein, B. D. Walker, and M. K. Singh. 1998. Human immunodeficiency virus type 1 populations in blood and semen. J. Virol. 72:617-623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Delwart, E. L., H. Pan, A. Neumann, and M. Markowitz. 1998. Rapid, transient changes at the env locus of plasma human immunodeficiency virus type 1 populations during the emergence of protease inhibitor resistance. J. Virol. 72:2416-2421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Delwart, E. L., H. Pan, H. W. Sheppard, D. Wolpert, A. U. Neumann, B. Korber, and J. I. Mullins. 1997. Slower evolution of human immunodeficiency virus type 1 quasispecies during progression to AIDS. J. Virol. 71:7498-7508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Delwart, E. L., E. G. Shpaer, J. Louwagie, F. E. McCutchan, M. Grez, H. Rubsamen-Waigmann, and J. I. Mullins. 1993. Genetic relationships determined by a DNA heteroduplex mobility assay: analysis of HIV-1 env genes. Science 262:1257-1261. [DOI] [PubMed] [Google Scholar]
- 14.Epstein, L. G., C. Kuiken, B. M. Blumberg, S. Hartman, L. R. Sharer, M. Clement, and J. Goudsmit. 1991. HIV-1 V3 domain variation in brain and spleen of children with AIDS: tissue-specific evolution within host-determined quasispecies. Virology 180:583-590. [DOI] [PubMed] [Google Scholar]
- 15.Finzi, D., M. Hermankova, T. Pierson, L. M. Carruth, C. Buck, R. E. Chaisson, T. C. Quinn, K. Chadwick, J. Margolick, R. Brookmeyer, J. Gallant, M. Markowitz, D. D. Ho, D. D. Richman, and R. F. Siliciano. 1997. Identification of a reservoir for HIV-1 in patients on highly active antiretroviral therapy. Science 278:1295-1300. [DOI] [PubMed] [Google Scholar]
- 16.Fortgang, I. S., P. C. Belitsos, R. E. Chaisson, and R. D. Moore. 1995. Hepatomegaly and steatosis in HIV-infected patients receiving nucleoside analog antiretroviral therapy. Am. J. Gastroenterol. 90:1433-1436. [PubMed] [Google Scholar]
- 17.Fox, D. G., P. Balfe, C. P. Palmer, J. C. May, C. Arnold, and J. A. McKeating. 1997. Length polymorphism within the second variable region of the human immunodeficiency virus type 1 envelope glycoprotein affects accessibility of the receptor binding site. J. Virol. 71:759-765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Furtado, M. R., D. S. Callaway, J. P. Phair, K. J. Kunstman, J. L. Stanton, C. A. Macken, A. S. Perelson, and S. M. Wolinsky. 1999. Persistence of HIV-1 transcription in peripheral-blood mononuclear cells in patients receiving potent antiretroviral therapy. N. Engl. J. Med. 340:1614-1622. [DOI] [PubMed] [Google Scholar]
- 19.Garcia, F., M. Plana, G. M. Ortiz, S. Bonhoeffer, A. Soriano, C. Vidal, A. Cruceta, M. Arnedo, C. Gil, G. Pantaleo, T. Pumarola, T. Gallart, D. F. Nixon, J. M. Miro, and J. M. Gatell. 2001. The virological and immunological consequences of structured treatment interruptions in chronic HIV-1 infection. AIDS 15:F29-F40. [DOI] [PubMed] [Google Scholar]
- 20.Gratton, S., R. Cheynier, M. J. Dumaurier, E. Oksenhendler, and S. Wain-Hobson. 2000. Highly restricted spread of HIV-1 and multiply infected cells within splenic germinal centers. Proc. Natl. Acad. Sci. USA 97:14566-14571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Groenink, M., R. A. Fouchier, S. Broersen, C. H. Baker, M. Koot, A. B. van't Wout, H. G. Huisman, F. Miedema, M. Tersmette, and H. Schuitemaker. 1993. Relation of phenotype evolution of HIV-1 to envelope V2 configuration. Science 260:1513-1516. [DOI] [PubMed] [Google Scholar]
- 22.Heath, K. V., R. S. Hogg, K. J. Chan, M. Harris, V. Montessori, M. V. O'Shaughnessy, and J. S. Montanera. 2001. Lipodystrophy-associated morphological, cholesterol and triglyceride abnormalities in a population-based HIV/AIDS treatment database. AIDS 15:231-239. [DOI] [PubMed] [Google Scholar]
- 23.Ickovics, J. R., and A. W. Meisler. 1997. Adherence in AIDS clinical trials: a framework for clinical research and clinical care. J. Clin. Epidemiol. 50:385-391. [DOI] [PubMed] [Google Scholar]
- 24.Imamichi, H., K. A. Crandall, V. Natarajan, M. K. Jiang, R. L. Dewar, S. Berg, A. Gaddam, M. Bosche, J. A. Metcalf, R. T. Davey, Jr., and H. C. Lane. 2001. Human immunodeficiency virus type 1 quasi species that rebound after discontinuation of highly active antiretroviral therapy are similar to the viral quasi species present before initiation of therapy. J. Infect. Dis. 183:36-50. [DOI] [PubMed] [Google Scholar]
- 25.Itescu, S., P. F. Simonelli, R. J. Winchester, and H. S. Ginsberg. 1994. Human immunodeficiency virus type 1 strains in the lungs of infected individuals evolve independently from those in peripheral blood and are highly conserved in the C-terminal region of the envelope V3 loop. Proc. Natl. Acad. Sci. USA 91:11378-11382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kopp, J. B., K. D. Miller, J. A. Mican, I. M. Feuerstein, E. Vaughan, C. Baker, L. K. Pannell, and J. Falloon. 1997. Crystalluria and urinary tract abnormalities associated with indinavir. Ann. Intern. Med. 127:119-125. [DOI] [PubMed] [Google Scholar]
- 27.Koyanagi, Y., S. Miles, R. T. Mitsuyasu, J. E. Merrill, H. V. Vinters, and I. S. Chen. 1987. Dual infection of the central nervous system by AIDS viruses with distinct cellular tropisms. Science 236:819-822. [DOI] [PubMed] [Google Scholar]
- 28.Marras, D., L. A. Bruggeman, F. Gao, N. Tanji, M. M. Mansukhani, A. Cara, M. D. Ross, G. L. Gusella, G. Benson, V. D. D'Agati, B. H. Hahn, M. E. Klotman, and P. E. Klotman. 2002. Replication and compartmentalization of HIV-1 in kidney epithelium of patients with HIV-associated nephropathy. Nat. Med. 8:522-526. [DOI] [PubMed] [Google Scholar]
- 29.Miller, K. D., E. Jones, J. A. Yanovski, R. Shankar, I. Feuerstein, and J. Falloon. 1998. Visceral abdominal-fat accumulation associated with use of indinavir. Lancet 351:871-875. [DOI] [PubMed] [Google Scholar]
- 30.Mulligan, K., C. Grunfeld, V. W. Tai, H. Algren, M. Pang, D. N. Chernoff, J. C. Lo, and M. Schambelan. 2000. Hyperlipidemia and insulin resistance are induced by protease inhibitors independent of changes in body composition in patients with HIV infection. J. Acquir. Immune Defic. Syndr. 23:35-43. [DOI] [PubMed] [Google Scholar]
- 31.Natarajan, V., M. Bosche, J. A. Metcalf, D. J. Ward, H. C. Lane, and J. A. Kovacs. 1999. HIV-1 replication in patients with undetectable plasma virus receiving HAART. Highly active antiretroviral therapy. Lancet 353:119-120. [DOI] [PubMed] [Google Scholar]
- 32.Ortiz, G. M., M. Wellons, J. Brancato, H. T. Vo, R. L. Zinn, D. E. Clarkson, K. Van Loon, S. Bonhoeffer, G. D. Miralles, D. Montefiori, J. A. Bartlett, and D. F. Nixon. 2001. Structured antiretroviral treatment interruptions in chronically HIV-1-infected subjects. Proc. Natl. Acad. Sci. USA 98:13288-13293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Overbaugh, J., R. J. Anderson, J. O. Ndinya-Achola, and J. K. Kreiss. 1996. Distinct but related human immunodeficiency virus type 1 variant populations in genital secretions and blood. AIDS Res. Hum. Retrovir. 12:107-115. [DOI] [PubMed] [Google Scholar]
- 34.Papasavvas, E., G. M. Ortiz, R. Gross, J. Sun, E. C. Moore, J. J. Heymann, M. Moonis, J. K. Sandberg, L. A. Drohan, B. Gallagher, J. Shull, D. F. Nixon, J. R. Kostman, and L. J. Montaner. 2000. Enhancement of human immunodeficiency virus type 1-specific CD4 and CD8 T cell responses in chronically infected persons after temporary treatment interruption. J. Infect. Dis. 182:766-775. [DOI] [PubMed] [Google Scholar]
- 35.Periard, D., A. Telenti, P. Sudre, J. J. Cheseaux, P. Halfon, M. J. Reymond, S. M. Marcovina, M. P. Glauser, P. Nicod, R. Darioli, and V. Mooser. 1999. Atherogenic dyslipidemia in HIV-infected individuals treated with protease inhibitors. The Swiss HIV Cohort Study. Circulation 100:700-705. [DOI] [PubMed] [Google Scholar]
- 36.Rosenberg, E. S., M. Altfeld, S. H. Poon, M. N. Phillips, B. M. Wilkes, R. L. Eldridge, G. K. Robbins, R. T. D'Aquila, P. J. Goulder, and B. D. Walker. 2000. Immune control of HIV-1 after early treatment of acute infection. Nature 407:523-526. [DOI] [PubMed] [Google Scholar]
- 37.Ruiz, L., G. Carcelain, J. Martinez-Picado, S. Frost, S. Marfil, R. Paredes, J. Romeu, E. Ferrer, K. Morales-Lopetegi, B. Autran, and B. Clotet. 2001. HIV dynamics and T-cell immunity after three structured treatment interruptions in chronic HIV-1 infection. AIDS 15:F19-F27. [DOI] [PubMed] [Google Scholar]
- 38.Shankarappa, R., J. B. Margolick, S. J. Gange, A. G. Rodrigo, D. Upchurch, H. Farzadegan, P. Gupta, C. R. Rinaldo, G. H. Learn, X. He, X.-L. Huang, and J. I. Mullins. 1999. Consistent viral evolutionary changes associated with the progression of human immunodeficiency virus type 1 infection. J. Virol. 73:10489-10502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Thiebaut, R., V. Daucourt, P. Mercie, D. K. Ekouevi, D. Malvy, P. Morlat, M. Dupon, D. Neau, S. Farbos, C. Marimoutou, and F. Dabis. 2000. Lipodystrophy, metabolic disorders, and human immunodeficiency virus infection: Aquitaine Cohort, France, 1999. Groupe d'Epidemiologie Clinique du Syndrome d'Immunodeficience Acquise en Aquitaine. Clin. Infect. Dis. 31:1482-1487. [DOI] [PubMed] [Google Scholar]
- 40.Valentin, A., M. Rosati, D. J. Patenaude, A. Hatzakis, L. G. Kostrikis, M. Lazanas, K. M. Wyvill, R. Yarchoan, and G. N. Pavlakis. 2002. Persistent HIV-1 infection of natural killer cells in patients receiving highly active antiretroviral therapy. Proc. Natl. Acad. Sci. USA 99:7015-7020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wong, J. K., M. Hezareh, H. F. Gunthard, D. V. Havlir, C. C. Ignacio, C. A. Spina, and D. D. Richman. 1997. Recovery of replication-competent HIV despite prolonged suppression of plasma viremia. Science 278:1291-1295. [DOI] [PubMed] [Google Scholar]
- 42.Zhang, L., C. Chung, B. S. Hu, T. He, Y. Guo, A. J. Kim, E. Skulsky, X. Jin, A. Hurley, B. Ramratnam, M. Markowitz, and D. D. Ho. 2000. Genetic characterization of rebounding HIV-1 after cessation of highly active antiretroviral therapy. J. Clin. Investig. 106:839-845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhang, L., B. Ramratnam, K. Tenner-Racz, Y. He, M. Vesanen, S. Lewin, A. Talal, P. Racz, A. S. Perelson, B. T. Korber, M. Markowitz, and D. D. Ho. 1999. Quantifying residual HIV-1 replication in patients receiving combination antiretroviral therapy. N. Engl. J. Med. 340:1605-1613. [DOI] [PubMed] [Google Scholar]