


Abstract
We have devised an efficient method for replicating and stably maintaining entire mitochondrial genomes in Escherichia coli and have shown that we can engineer these mitochondrial DNA (mtDNA) genome clones using standard molecular biological techniques. In general, we accomplish this by inserting an E.coli replication origin and selectable marker into isolated, circular mtDNA at random locations using an in vitro transposition reaction and then transforming the modified genomes into E.coli. We tested this approach by cloning the 16.3 kb mouse mitochondrial genome and found that the resulting clones could be engineered and faithfully maintained when we used E.coli hosts that replicated them at moderately low copy numbers. When these recombinant mtDNAs were replicated at high copy numbers, however, mtDNA sequences were partially or fully deleted from the original clone. We successfully electroporated recombinant mouse mitochondrial genomes into isolated mouse mitochondria devoid of their own DNA and detected robust in organello RNA synthesis by RT–PCR. This approach for modifying mtDNA and subsequent in organello analysis of the recombinant genomes offers an attractive experimental system for studying many aspects of vertebrate mitochondrial gene expression and is a first step towards true in vivo engineering of mammalian mitochondrial genomes.
INTRODUCTION
The typical animal cell contains hundreds of mitochondria that produce the majority of the cell’s ATP through oxidative phosphorylation (1) and that regulate multiple cellular processes, including apoptosis (2). Mitochondria are generally thought to have arisen from an intracellular bacterial symbiont of the first ancestral eukaryotic cells that presumably provided most of the energy metabolism for this symbiotic pairing (3). As these early protomitochondria co-evolved with the emerging eukaryotic host, most of the genetic information from the circular protomitochondrial genome was either lost or was transferred to the nuclear genome. Because this evolution of mitochondrial genomes occurred in conjunction with the evolution of eukaryotic cells, the mitochondrial genomes of different eukaryotic lineages differ in size, gene content and even in the genetic code that they use. The mammalian mitochondrial genome has been reduced to ∼16.5 kb in size and encodes genes for only 13 protein products, all of which are critical components of the electron transport chain, as well as two rRNAs and 22 tRNAs that are required for the mitochondrial translation system. All of the other genes needed for the biogenesis, maintenance and regulation of this organelle are now encoded in the nucleus. Nevertheless, the mitochondrial genome remains critical for normal mitochondrial function, and mutations in this genome are known to cause a wide range of human diseases (4).
Although the level of interest in mitochondrial genomes is understandably high, no practical means have yet been found to directly modify mitochondrial sequences in animals for either basic or applied research purposes. Mitochondria in the yeast Saccharomyces cerevisiae have been successfully transformed with both foreign and mitochondrial genes using a biolistic delivery system (5,6), but similar techniques have only proven suitable for a limited number of organisms and have not yet been adapted to animals. As an alternative to modifying mitochondrial genomes in vivo, attempts have been made to clone and then modify mitochondrial genomes in host organisms, but these experiments have met with mixed results. Recent efforts to clone and engineer the mouse mitochondrial genome in Escherichia coli ultimately proved to be unsuccessful due to the high instability of the mitochondrial clones (7,8), and a strategy in which the genome was cloned and modified in yeast and then shuttled back into E.coli was adopted.
In this report we describe an efficient method for replicating and stably maintaining entire mitochondrial genomes in E.coli, and demonstrate the effectiveness of this procedure by cloning mouse mitochondrial genomes isolated from mouse liver. We found that we could readily engineer these mitochondrial DNA (mtDNA) genome clones using standard molecular biological techniques, and report for the first time that full mitochondrial genomes can be transferred back into purified, transcriptionally active mouse mitochondria for subsequent in organello analyses.
MATERIALS AND METHODS
Escherichia coli strains, cell lines, plasmids and culture media
Escherichia coli strains DH5α λatt::pirwt, DH5α λatt::pir200 (9) and DH5α λatt::pir116 (M. Koob, unpublished results) were used for maintaining plasmids containing the R6K γ origin of replication (γ-ori). p2CγCmR (9), which contains the γ-ori and a chloramphenicol resistance gene (CmR), was used as a template for making a transposon DNA fragment (see below). The mouse cell line LL/2 (ATCC CRL-1642) (10) was grown in DMEM (Life Technologies, Rockville, MD) in the presence of heat-inactivated 10% fetal bovine serum at 37°C in a humidified 10% CO2 incubator. The mtDNA-less ρ0 LL/2 cell line (this report), a derivative of LL/2 cells, was grown in DMEM supplemented with 10% fetal bovine serum, 50 µg/ml uridine and 0.1 mg/ml pyruvate.
Isolation of mtDNA-less (ρ0) LL/2 cell line
The mtDNA-less ρ0 LL/2 cell line was isolated by a modification of a method described earlier (11,12), which involves treatment of LL/2 cells with high concentrations of ethidium bromide. LL/2 cells were exposed to 5 µg/ml ethidium bromide for 4 weeks in medium supplemented with 50 µg/ml uridine and 0.1 mg/ml pyruvate. After 4 weeks, clonal ρ0 LL/2 cell lines were obtained by diluting these ethidium bromide-treated cells to single cells per well on 96-well plates. The cloned cells were then cultured in normal medium supplemented with 50 µg/ml uridine and 0.1 mg/ml pyruvate. The ρ0 state of the cloned cells was verified by a PCR assay using L-strand (5′-ACC CAA CGC GGC AAA CTA ACC-3′) and H-strand (5′-TCT TGT TCG TCT GCC AGG CT-3′) primers and by assaying for the unique growth requirements of the ρ0 cells (13).
DNA manipulation
Mini-scale preparations of plasmid DNA were prepared by the alkaline lysis method (14) and large quantities of plasmid DNA were prepared by the PEG precipitation method and other recombinant DNA techniques were performed essentially as previously described (14). Restriction enzymes and T4 DNA ligase were purchased from New England Biolabs (Beverly, MA) and used as recommended by the manufacturer. AmpliTaq DNA polymerase was purchased from Applied Biosystems (Branchburg, NJ) and deoxyoligonucleotides (oligos) were synthesized by Life Technologies (Rockville, MD) or IDT (Coralville, IA). Ampicillin (Amp), tetracycline and chloramphenicol (Cm) were used at concentrations of 50, 12.5 and 12.5 µg/ml, respectively.
Purification of mtDNA from mouse liver
The liver of a freshly killed mouse was removed and placed in cold isolation medium (0.32 M sucrose, 1 mM potassium EDTA, 10 mM Tris–HCl pH 7.4). All subsequent steps were performed at 4°C or on ice. The tissue was minced with scissors and rinsed several times with the same medium. The chopped tissue was suspended in isolation medium (4 ml/g liver) and homogenized in a Potter–Elvehjem tissue grinder (Kontes Galss Co., Vineland, NJ). The homogenate was transferred to 50 ml centrifuge tubes and centrifuged at low speed (1000 g, 5 min). The supernatant was then centrifuged at high speed (13 000 g, 20 min). The resultant supernatant was discarded, the pellet was resuspended in 5 ml of DNase I buffer (210 mM mannitol, 70 mM sucrose, 5 mM Tris–HCl pH 7.4, 10 mM MgCl2) containing 0.5 mM phenylmethylsulfonyl fluoride. The suspension was incubated with 4000 Kunitz units of DNase I (Sigma) at 37°C for 1 h. After incubation, the suspension was washed three times in 10 vol of washing buffer (210 mM mannitol, 70 mM sucrose, 5 mM Tris–HCl pH 7.4, 10 mM EDTA) by centrifugation at 13 000 g for 20 min. The washed pellet was resuspended in 5 ml of sucrose–TE buffer (20% sucrose, 50 mM Tris–HCl pH 7.4, 10 mM EDTA) and loaded on top of sucrose layers consisting of 15 ml of 1.5 M sucrose (lower layer) and 15 ml of 1.0 M sucrose (upper layer), both containing 10 mM Tris–HCl pH 7.4, 5 mM EDTA. After centrifugation at 25 000 r.p.m. for 30 min in a Beckman SW28 rotor, the interface fraction (red-brown color) between the 1.0 and 1.5 M sucrose gradients was collected and washed twice with 4 vol of washing buffer by centrifugation at 18 000 g for 20 min. The mitochondrial pellet was suspended in 3 ml of STE buffer (100 mM NaCl, 10 mM EDTA, 50 mM Tris–HCl pH 7.4) and incubated with 330 µl of 10% SDS and 400 µl of proteinase K (10 mg/ml) at 50°C for 3 h. After incubation, the digest was extracted with 3 ml of phenol/chloroform saturated with TE (10 mM Tris–HCl pH 7.4, 1 mM EDTA) by shaking gently for 10 min and the extraction was repeated with 3 ml of chloroform. The DNA in the aqueous phase was precipitated by the addition of 370 µl of 3 M sodium acetate and 6 ml of absolute ethanol (30 min at –20°C). The DNA was collected by centrifugation, washed with 3 ml of 70% ethanol, dried and resuspended in TE.
In vitro transposon insertion reaction
A synthetic transposon in which the γ-ori and a chloramphenicol resistance gene are flanked by Tn5 mosaic ends (ME) was generated by a PCR using BamHI-linearized p2CγCmR as template and the primers MESalCmR (5′-CTG TCT CTT ATA CAC ATC TGT CGA CAG AAG CCA CTG GAG CA-3′; ME sequence italicized, SalI restriction site underlined) and MESmaγori (5′-CTG TCT CTT ATA CAC ATC TCC CGG GCT AAT TCT GTC AGC CGT T-3; SmaI restriction site underlined). A total of 30 amplification cycles were performed: 1 min at 95°C, 1 min at 55°C, followed by 90 s at 72°C. PCR products were purified from low melting point agarose gels (FMC, Rockland, ME) using AgarACE™ (Promega, Madison, WI). For the in vitro transposon insertion reaction, a purified hyperactive Tn5 transposase (EZ::TN; Epicentre, Madison, WI) was used in a reaction consisting of 1 U transposase, 200 ng mouse mitochondrial DNA and 10 ng PCR-amplified transposon in the buffer provided by the supplier. After incubating the reaction mixture for 2 h at 37°C, the reaction was terminated by adding 1 µl of the supplied 10× stop solution and by heating at 70°C for 10 min. An aliquot of 1 µl of the in vitro transposon insertion reaction mixture was used for electrotransformation of DH5α λatt::pirwt.
Cloning of mtDNA fragments with promoter activity in E.coli and the chloramphenicol acetyltransferase (CAT) assay
Mouse mtDNA digested with AluI, HaeIII and Sau3AI was ligated into the HincII and BglII sites 5′ of the promoter-less chloramphenicol acetyltransferase gene (CAT, CmR) of the ‘promoter trapping’ vector pANTSγ-Cm (Fig. 3A). The ligation mixture was transformed into E.coli strain DH5α λatt::pir200 and recombinant clones were selected on Luria broth (LB)-Cm plates (12.5 µg/ml Cm). DNA isolated from these colonies was sequenced and retransformed into DH5α λatt::pirwt (LB-Amp selection). The CAT activity from the same recombinant clones was assayed in both the pir200 and pirwt strains grown in LB-Amp medium to an optical density of ∼0.6 at 600 nm. CAT activity was assayed using the FAST CAT® Green (deoxy) Chloramphenicol Acetyltransferase Assay Kit (Molecular Probes Inc., Eugene, OR) according to the manufacturer’s instructions. A PhosphorImager Storm 840 (Molecular Dynamics, Sunnyvale, CA) system was used to measure band intensity of the acetylated products after fractionating on a thin layer chromatography plate (Merck KGaA, Darmstadt, Germany). Values of the intensities were calculated relative to the CAT activity of the vector-only low copy control sample.
Figure 3.

Identification of mtDNA fragments that serve as transcriptional promoters in E.coli. (A) Schematic outline of the approach used to clone fragments from the mouse mitochondrial genome with promoter activity. AluI-, HaeIII- and Sau3AI-digested mouse mtDNA fragments were cloned 5′ of the promoter-less CAT (CmR) gene in the pANTSγ-Cm vector and recombinant clones expressing Cm resistance were selected on LB-Cm plates. (B) Comparisons of CAT activities generated from mtDNA promoter fragment clones at low and high copy numbers. The recombinant plasmid clones obtained from the promoter screen were grown in each of two E.coli strains that replicated them at either low copy numbers (pirwt) or at high copy numbers (pir200), and the CAT activities of both cultures were assayed. The black and gray bars shown for each clone indicate the relative CAT activities from low copy and high copy strains, respectively, with the value of the low copy vector-only control arbitrarily set to 1.
Electroporation of mitochondria
Exponentially growing LL/2 cells were harvested by centrifugation, washed twice with 1 mM Tris–HCl pH 7.0, 0.13 M NaCl, 5 mM KCl and 7.5 mM MgCl2. The cell pellet was resuspended in half the cell volume with 1/10× IB (4 mM Tris–HCl pH 7.4, 2.5 mM NaCl, 0.5 mM MgCl2) and the cells were broken using a Pellet Pestle tissue grinder (Kontes Glass Co.). The homogenate was mixed with a one-ninth volume of the packed cell volume of 10× IB (400 mM Tris–HCl pH 7.4, 250 mM NaCl, 50 mM MgCl2) resulting in a buffer concentration of roughly 1× IB (40 mM Tris–HCl pH 7.4, 25 mM NaCl, 5 mM MgCl2). The unbroken cells and nuclei were removed by two consecutive low speed centrifugations (2000 r.p.m. for 5 min). The supernatant was placed into new 1.5 ml Eppendorf tubes and centrifuged at full speed for 10 min to obtain a crude mitochondrial pellet. Mitochondria were rinsed once with 500 µl of 1× IB, centrifuged again at full speed for 10 min and resuspended into 0.33 M sucrose/10% glycerol (at a concentration of 100 mg mitochondrial protein/ml) for electroporation. Electroporations were performed essentially as described (15). An aliquot of 10 µg of cloned mouse mtDNA was added to 50 µl of 100 mg/ml mitochondrial suspension and the mixture was transferred into a cold electroporation cuvette (0.1 cm gap cuvette; Bio-Rad). Electroporation was carried out using a Bio-Rad Gene Pulser at a capacitance of 25 µF, a resistance of 400 Ω and a field strength of 10–16 kV/cm. After electroporation, 1 ml of incubation buffer (40 mM Tris–HCl pH 7.4, 25 mM NaCl, 5 mM MgCl2, 10% glycerol) was added to the electroporation cuvette. The electroporated mitochondria were rapidly mixed by pipetting, transferred to a new Eppendorf tube and washed three times with incubation buffer by centrifugation.
In orgenello RNA synthesis of electroporated mitochondria and RT–PCR analysis
For in organello RNA synthesis (16), the final mitochondrial pellet was resuspended in 50 µl of incubation buffer (10% glycerol, 40 mM Tris–HCl pH 7.4, 25 mM NaCl, 5 mM MgCl2, 1 mM pyruvate, 1 mM ATP and 1 mg/ml BSA) and incubated at 37°C for 2 h. After incubation, the mitochondrial suspension was pelleted at full speed in a microcentrifuge for 10 min and washed twice with incubation buffer. The pellets were suspended in 200 µl of DNase I buffer (10% glycerol, 10 mM Tris–HCl pH 8.0, 1 mM MgCl2) and incubated with 200 Kunitz units of DNase I (Sigma) at 37°C for 30 min. After incubation, the mitochondria were pelleted and washed twice with washing buffer (10% glycerol, 10 mM Tris–HCl pH 7.4, 1 mM EDTA) to inactivate and remove the nuclease. The mitochondrial samples were lysed with 300 µl of lysis buffer (0.5% SDS, 10 mM Tris–HCl pH 7.4, 150 mM NaCl, 1 mM EDTA) and incubated with 100 µg proteinase K for 15 min at 37°C. Phenol extraction was then performed, total mitochondrial nucleic acids were collected by ethanol precipitation and residual DNA contaminants were removed using RNase-free DNase I (Promega) as directed by the manufacturer. RT–PCR analysis was carried out using a SuperScript First-Strand Synthesis system (Life Technologies) and the following primers: CmR-F (5′-GTA CCT ATA ACC AGA CCG TTC AGC-3′), CmR-R (5′-CAG CGG CAT CAG CAC CTT GTC-3′), 16S rRNA-F (5′-GCA GCC ACC AAT AAA GAA AG-3′), 16S rRNA-R (5′-GGA CCC TCG TTT AGC CGT TC-3′), MusMt-MluIA (5′-AGG TGG ATT ATT TAT AGT GTG ATT ATT GCC-3′), ND6-F (5′-GGA GAT CTT GAT GTA TGA GGT TGA TGA TGT TGG A-3′) and ND6-R (5′-CCC GCA AAC AAA GAT CAC CC-3′). The following PCR cycle parameters were used for each of the RT–PCR reactions: 95°C for 5 min, followed by 30 cycles of 95°C for 30 s, 60°C for 45 s and 72°C for 30 s, and finally 72°C for 10 min.
RESULTS
Cloning the mouse mitochondrial genome in E.coli
We devised a scheme for cloning complete mitochondrial genomes in E.coli that uses an in vitro transposition reaction to insert an E.coli origin of DNA replication (ori) and selectable marker at random locations into circular mtDNA. To make a synthetic transposon containing the γ-ori from the R6K plasmid (17,18) and the CmR gene, we performed a PCR amplification using linearized plasmid p2CγCmR as template and the PCR primers MESalCmR and MESmaγori (Materials and Methods). These primers contain the 19 bp Tn5 transposase recognition sequences (mosaic end or ME) and the unique restriction sites for SalI and SmaI, respectively, at their 5′ ends and plasmid-specific sequences at their 3′ ends. The 1.5 kb PCR-amplified transposon, therefore, consisted of the CmR marker and the γ-ori flanked by inverted Tn5 ME sequences at each of its ends. To perform the in vitro transposition reaction, this linear synthetic transposon was incubated with hyperactive Tn5 transposase and purified, circular mouse mtDNA. The products from this transposition reaction were electroporated into an E.coli strain containing a chromosomal copy of the R6K pir gene, which encodes the replication initiator protein π needed for γ-ori replication (17,18), and transformants were selected on Cm plates.
We characterized three transposon-inserted mouse mtDNA clones obtained using this cloning strategy by restriction enzyme mapping and sequencing. Figure 1 shows the schematic representation (Fig. 1A) and restriction enzyme analysis (Fig. 1B–D) of the mouse mtDNA clones. All of the transposons were inserted into the mouse mtDNA in the same orientation. When we sequenced the transposon junctions of these clones using vector-specific sequencing primers, we found that the transposons were inserted into the ND1, COXII and ND5 genes on the mouse mtDNA at nucleotides 3347, 7688 and 13371, respectively. Tn5 transposition reactions typically generate a 9 bp target duplication immediately flanking the transposon insertion site (19) and we found these sequence duplications in each of the mouse mtDNA clones. Extensive sequencing of these clones identified several sequence polymorphisms present in the mtDNA of the mouse strain used as the source of the mtDNA but further confirmed the overall sequence integrity of the constructs.
Figure 1.

Cloning of mouse mtDNA by an in vitro transposition reaction. (A) Schematic representation of three mouse mtDNA clones and sequences of the transposon insertion sites. A synthetic transposon (1.5 kb) was constructed containing the γ-ori from plasmid R6K, CmR and two hyperactive 19 bp mosaic end sequences adjacent to SalI and SmaI restriction enzyme sites. The transposons were inserted into the ND1, COXII and ND5 genes on mouse mtDNA, respectively, and all insertions generated 9 bp duplications of the target DNA (upper case). (B–D) Restriction enzyme analyses of transposon-inserted mouse mtDNA clones. Plasmids pMusMtTN-ND1 (B), pMusMtTN-COXII (C) and pMusMtTN-ND5 (D) were digested with one or two unique restriction enzymes at 37°C for 2 h and were electrophoresed on 1% agarose gels. Lane M, λ DNA HindIII digest; lanes 1, 6 and 11, DNA digested with SalI; lanes 2, 7 and 12, DNA digested with SalI + MluI; lanes 3, 8 and 13, DNA digested with SalI + BspEI; lanes 4, 9 and 14, DNA digested with SalI + SphI; lanes 5, 10 and 15, DNA digested with SalI + BglII.
Comparison of the stability of mouse mtDNA clones in E.coli at low and high copy number
To examine the stability of the cloned mouse mtDNA in E.coli, the three mapped mouse mtDNA clones were transformed into one of two E.coli strains. One of these strains contained the wild-type pir gene (pirwt) in the chromosome and replicated the mtDNA clones at a moderately low number of copies/cell (10–15 copies), and the other strain contained a mutant pir gene (pir116) (20) that replicated the clones at a relatively high number of copies/cell (∼200 copies). After transforming the mtDNA plasmids into the pirwt strain, cultures were grown in LB medium at 30 or 37°C, plasmid DNA was isolated at regular time intervals and the restriction patterns of these DNA preparations were compared. As shown in Figure 2A, no deletions or rearrangements were observed in the mouse mtDNA fragments even after several days of culture at either slow growth (30°C) or rapid growth (37°C) temperatures. When the mouse mtDNA clones were transformed into the pir116 strain, however, the transformation efficiency using a standard chemical transformation method was dramatically lower than that seen with the pirwt strain transformed with the same method (Table 1). We electroporated the mtDNA clones into the pir116 strain in order to obtain more transformants and performed restriction analyses of plasmid DNA isolated from the few colonies that were formed. We found that almost all of these colonies contained plasmids in which all or most of the mouse mtDNA sequence was deleted (Fig. 2B).
Figure 2.
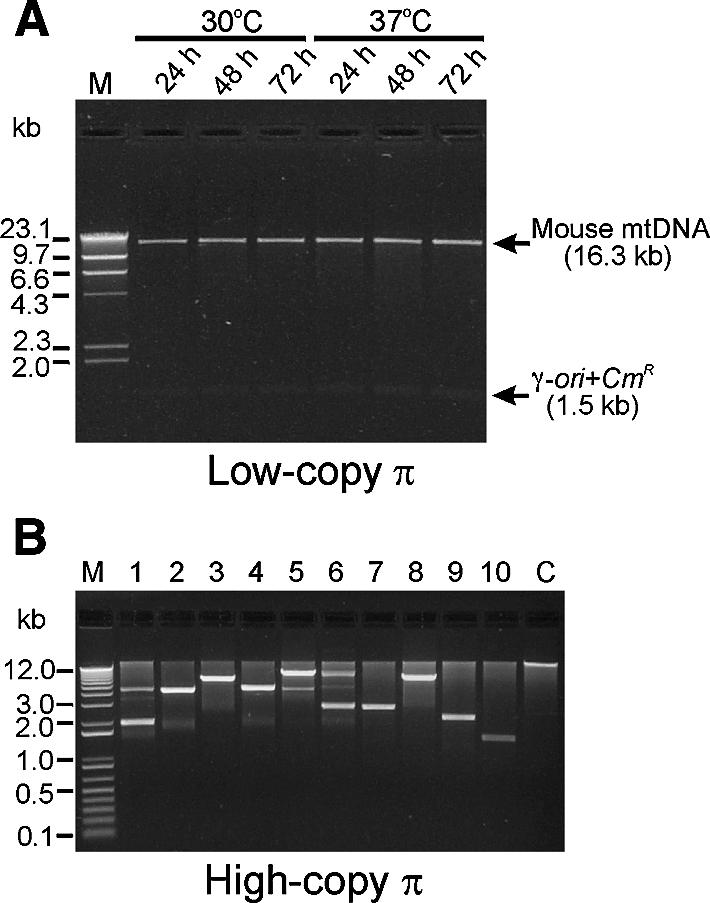
Effect of plasmid copy number on the stability of cloned mouse mtDNA. (A) Stability of the mouse mtDNA in a low copy number E.coli strain. Plasmid pMusMtTN-ND5 was transformed into DH5α λatt::pirwt and was re-isolated after culturing in LB medium containing Cm (12.5 µg/ml) at 30 or 37°C for regular time intervals (24–72 h). Restriction patterns after SalI and SmaI digestion were compared on a 1% agarose gel. The top arrow points to the 16.3 kb mouse mtDNA and the lower to the band of the 1.5 kb γ-ori+CmR transposon. Lane M, λ DNA HindIII digest. (B) Stability of the mouse mtDNA in a high copy number E.coli strain. Mouse mtDNA clones were transformed into DH5α λatt::pir116 and were re-isolated from the recombinant E.coli after culturing in LB medium containing Cm at 37°C for 20 h. Restriction patterns after SalI digestion were compared on a 1% agarose gel. Lane M, 1 kb plus DNA ladder (Life Technologies); lanes 1–10, DNAs from DH5α λatt::pir116 strain; lane C, DNA from DH5α λatt::pirwt strain.
Table 1. Transformation efficiency of the mouse mtDNA clones in two different E.coli strains.
| Plasmid | Transformation efficiency (colonies/µg DNA) | |
|---|---|---|
| DH5α λatt::pirwta | DH5α λatt::pir116a | |
| p2CγCmR | 1.2 × 105 | 1.3 × 105 |
| pMusMtTN-ND1 | 7.5 × 103 | 1 |
| pMusMtTN-COXII | 9.0 × 103 | 2 |
| pMusMtTN-ND5 | 8.5 × 103 | 1 |
aEscherichia coli strain.
Identification of mtDNA fragments with transcriptional promoter activity in E.coli
We hypothesized that transcription of mtDNA in E.coli was a likely underlying cause of the apparent toxicity of the mtDNA sequences at high copy number. To test this hypothesis, we cloned mtDNA fragments into a ‘promoter-trap’ vector designed to directly select recombinant clones in which a promoter-less CmR (CAT) gene is transcribed only if an inserted DNA fragment has promoter activity (Fig. 3A). We used AluI, HaeIII and Sau3AI to digest mouse mtDNA into relatively small fragments and cloned them into the HincII or BglII sites upstream of the CAT gene in pANTSγ-Cm. We then selected recombinant clones with inserts that had promoter activity by transforming them into an E.coli strain that replicated the plasmids at high copy number (pir200) and plating on LB-Cm plates. The mouse mtDNA fragments identified in this screen are listed in Table 2, with the direction of the promoter activity relative to the sequence numbering indicated by an arrow. The plasmids containing these inserts were transformed into the low copy E.coli strain (pirwt) and the CAT activity generated from each clone was measured at both low and high copy numbers (Fig. 3B). For each of the mtDNA fragment clones, the CAT activity generated in the high copy strain was significantly higher than that generated in the low copy strain, with many of the fragments only producing background levels of CAT activity at low copy numbers.
Table 2. Functional E.coli promoter-like sequences identified in mouse mtDNA.
| Clone no. | Restriction fragment | Position in mouse mtDNAa | Promoter direction |
|---|---|---|---|
| 1 | HaeIII | 2216–2857 | (←) |
| 2 | HaeIII | 2857–3336 | (←) |
| 3 | Sau3AI | 4276–5887 | (→) |
| 4 | HaeIII | 4671–6622 | (→) |
| 5 | Sau3AI | 5884–5991 | (←) |
| 6 | AluI | 6336–6717 | (←) |
| 7 | AluI | 7709–8254 | (←) |
| 8 | Sau3AI | 8165–8940 | (→) |
| 9 | AluI | 8491–9045 | (→) |
| 10 | HaeIII | 8812–9757 | (←) |
| 11 | AluI | 9045–9138 | (←) |
| 12 | AluI | 10978–11083 | (→) |
| 13 | HaeIII | 11119–11193 | (→) |
| 14 | Sau3AI | 12028–13403 | (←) |
| 15 | AluI | 13844–14913 | (→) |
| 16 | Sau3AI | 14749–15333 | (←) |
| 17 | HaeIII | 15190–15740 | (←) |
aNucleotide numbering as in accession no. NC_001569.
Modification of pMusMtTN plasmids in E.coli
We have made numerous modifications to the pMusMtTN plasmids using standard cloning techniques and have found these plasmids to be amenable to most of the alterations we have attempted as long as the recombinant clones were propagated in pirwt E.coli strains. To determine if any segments of the mouse mtDNA genome contain sequences that might make modifying or subcloning these segments difficult, we isolated and subcloned each of four segments spanning the entire mtDNA sequence. We digested the appropriate clones with the restriction enzyme pairs BglII/MluI, MluI/BspEI, BspEI/SphI and SphI/BglII (see Fig. 1A), isolated the smaller DNA segments between these restriction sites and cloned them into a plasmid with a replication ori from pBR322. We obtained the expected subclones from each of these segments. We then recloned these subcloned fragments back into the full pMusMtTN constructs, and did not experience a problem with any of these segments except the SphI–BglII fragment, from which only a small number of recombinant clones were obtained. To further localize the source of this cloning difficulty, we subdivided this mtDNA segment into SphI–XhoI and XhoI–BglII fragments. We found that the SphI–XhoI segment could be cloned with a normal level of efficiency whereas the number of clones obtained from the XhoI–BglII segment was once again noticeably reduced. We sequenced the XhoI–BglII segment in the clones obtained and found that they still contained the full, unaltered mtDNA sequence.
In organello RNA synthesis from electroporated mitochondria using cloned mouse mtDNA
We constructed p2CγSSB (Fig. 4A) as a control template to develop a RT–PCR assay for transcription of foreign DNA introduced into purified mitochondria. This plasmid was designed so that the CAT gene of the plasmid would be transcribed from the H-strand mitochondrial promoter (HSP) once the construct was introduced into physiologically active mitochondria. We used electroporation to introduce p2CγSSB into wild-type and mtDNA-less (ρ0) mitochondria purified from mouse LL/2 and ρ0 LL/2 cell lines, respectively. In order to optimize the electroporation reaction, we performed multiple electroporation reactions with field strengths ranging from 10 to 16 kV/cm (25 µF capacitance, 400 Ω resistance). We then incubated the electroporated mitochondria in a buffer suitable for in organello RNA synthesis, purified total RNA from these mitochondria and performed RT–PCR analysis to identify the RNA transcribed from the electroporated DNA. For these experiments, the RNA transcribed across the CAT gene by the HSP mitochondrial promoter was analyzed using the CAT-specific primers CmR-R and CmR-F (Materials and Methods). As shown in Figure 4B and C, mitochondrial RNA transcripts were clearly detected from DNA electroporated into both wild-type and ρ0 mitochondria. Lanes 4 and 6 of Figure 4B and C show positive signals for both 12 and 14 kV/cm electroporation conditions, respectively. No signal was detected in the control reactions without reverse transcriptase (–RT) (Fig. 4B and C, lanes 3 and 5) or in the no electroporation controls, in which plasmid p2CγSSB was mixed with isolated mitochondria without electroporation (Fig. 4B and C, lanes 1 and 2).
Figure 4.

DNA transcription assay by in organello RNA synthesis using isolated mouse mitochondria. (A) A schematic representation of p2CγSSB, a control plasmid containing the prokaryotic CAT gene and the D-loop region, L-strand promoter and H-strand promoter (LSP and HSP) sequences from mouse mtDNA. This plasmid was designed so that the CAT gene is transcribed by the HSP promoter when it is introduced into transcriptionally active mitochondria. (B and C) RT–PCR analyses for CAT transcripts in electroporated mouse mitochondria. Plasmid p2CγSSB was electroporated into the isolated mouse wild-type (B) and ρ0 (C) mitochondria and then in organello RNA syntheses of those electroporated mitochondria were performed in incubation buffer for 2 h at 37°C. After isolating total RNA from the mitochondria, RT–PCR was carried out using CAT-specific primers CmR-F and CmR-R (see Materials and Methods). Expected band size of the RT–PCR CAT products was 470 bp. Lane M, 100 bp DNA ladder (Life Technologies); lanes 1 and 2, no electroporation control with plasmid; lanes 3 and 4, 12 kV/cm electroporation; lanes 5 and 6, 14 kV/cm electroporation. Control lanes in which reverse transcriptase (RT) was omitted are indicated below each panel by minus signs.
Once we had established robust electroporation and transcription assay conditions, we electroporated the mouse mtDNA clone pMusMtTN-COXII into isolated mouse ρ0 mitochondria and performed in organello RNA synthesis as before. In this experiment, we used RT–PCR to assay for the expression of the 16S rRNA and ND6 genes, which are transcribed from HSP and LSP, respectively. As shown in Figure 5A and B, lanes 2 and 4, RNA transcripts were clearly detected from each strand of the modified mitochondrial genome electroporated into the ρ0 mitochondria. We performed these experiments using a variety of electroporation conditions and found that using 16 kV/cm for the electroporation reaction most often resulted in the most efficient transfer of the 17.8 kb mouse mtDNA construct into transcriptionally active ρ0 mitochondria (data not shown).
Figure 5.
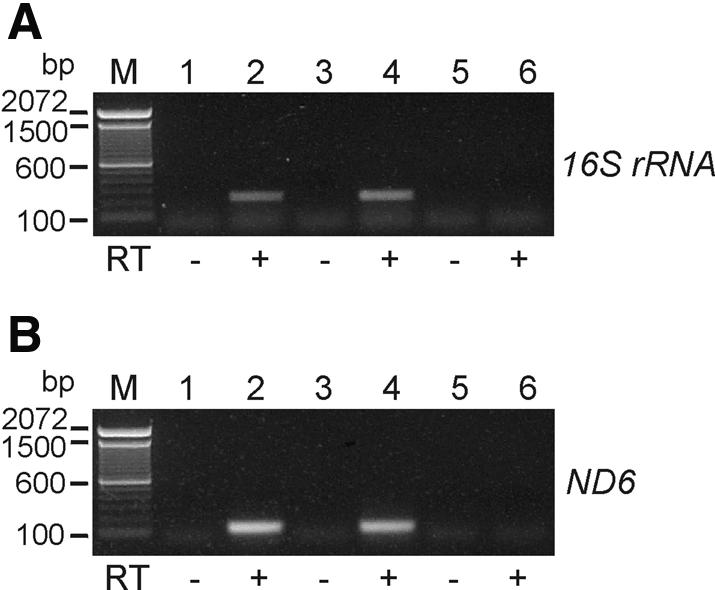
DNA transcription assay of a recombinant mitochondrial genome by in organello RNA synthesis using isolated mouse ρ0 mitochondria. The full mouse mtDNA clone, pMusMtTN-COXII, was electroporated into isolated ρ0 mitochondria and in organello RNA syntheses were performed for 2 h at 37°C. RT–PCR was carried out using 16S rRNA-specific (A) or ND6-specific (B) primers. 16S rRNA-R and ND6-R primers were used as gene-specific primers for the first strand synthesis of 16S rRNA and ND6 transcription, respectively, and the 16S rRNA-F and MusMt-MluIA primer set or ND6-F and ND6-R primer set was used for subsequent PCR amplification, respectively (see Materials and Methods). Expected band sizes of the RT–PCR products were 232 bp for 16S rRNA (see panel A) and 123 bp for ND6 (see panel B), respectively. Lane M, 100 bp DNA ladder; lanes 1 and 2, 12 kV/cm electroporation; lanes 3 and 4, 16 kV/cm electroporation; lanes 5 and 6, no electroporation control with plasmid. Control lanes in which reverse transcriptase (RT) was omitted are indicated below each panel by minus signs.
DISCUSSION
No practical means have yet been found to directly modify the mitochondrial genomes of vertebrate organisms. To overcome this limitation, we are pursuing an experimental approach in which we transfer circular mitochondrial genomes into E.coli, where they can be readily manipulated and modified using standard molecular techniques. Once the mtDNA genome has been engineered, we use electroporation to transfer it back into isolated mitochondria devoid of their own DNA and assay transcriptional activity using in organello RNA synthesis followed by RT–PCR analysis. Ultimately, we hope to transfer engineered mitochondrial genomes back into the mitochondria of intact, living cells in order to make both tissue culture and animal mitochondrial models, but an effective method for delivering DNA into the mitochondria inside cells is not yet available. In the meantime, however, the approach we describe for modifying mtDNA sequences and for subsequent in organello analyses of the recombinant genomes should be applicable to a wide array of research studies.
We use an efficient in vitro transposition reaction to insert an E.coli replication ori and selectable marker into isolated mtDNA (Fig. 1), which we feel offers a number of advantages for the cloning of mitochondrial genomes. This cloning procedure takes advantage of the fact that (i) DNA must be circular in order to replicate in E.coli and (ii) that the mtDNA genome is typically the only circular DNA in most eukaryotic cells. Since the transposition reaction does not circularize linear DNA, the genomic DNA fragments and broken mtDNA fragments that invariably contaminate mtDNA preparations are not cloned by this procedure and so the background is very low. The replication ori and selectable marker are also inserted at random locations and orientations throughout the mitochondrial genome and so many different clones are generated in the same experiment. The most stable of these resulting constructs are readily identified during the initial plasmid analysis as those that have faithfully replicated the mitochondrial genome in E.coli. All of the transposition reactions that generated the mouse mtDNA genome clones described in this work inserted the synthetic transposon in the same orientation with respect to the mtDNA, and this probably reflects an inherent increased stability for this orientation versus the other. Finally, the transposon cloning approach that we describe can be used to clone completely uncharacterized, circular mitochondrial genomes with no prior knowledge of either the sequence content or restriction pattern of that mtDNA.
The R6K plasmid γ-ori that we have inserted into the mouse mitochondrial genome is strictly dependent on the π protein encoded by the R6K pir gene (17,18) to both initiate DNA replication and to control copy number. By using E.coli strains that produce either the wild type or a mutant form of π from a chromosomal copy of the pir gene, we were able to compare the stability of the same mtDNA/γ-ori constructs when they replicated either at moderately low or at high copy numbers. We found that the entire mouse mitochondrial genome was very stably maintained when its copy number was kept low (Fig. 2A), but was extremely unstable when it was replicated at high copy number (Fig. 2B). The recovery of viable colonies after transformation of the full mitochondrial construct into the high copy strain was orders of magnitude lower than after transformation into the low copy strain, whereas a control plasmid without mitochondrial sequences was transformed into these two strains with equal efficiency (Table 1). Almost all of the plasmids recovered from the high copy number strain had deleted most or all of the mtDNA sequences.
Data from the literature suggest that the clone instability we observed in high copy mitochondrial clones is probably due to the inhibition of E.coli cell metabolism by one or more of the 22 heterologous tRNA genes in the mouse mitochondrial genome. Drouin (21) made the original observation that three of the 23 MboI digestion fragments of human mtDNA were drastically under-represented or absent from a mtDNA library made in pBR322, and reports of undefined human mtDNA sequences that inhibit cloning in E.coli were made by other laboratories (22). All of the fragments that were reported to be difficult or impossible to clone contain tRNA genes. Kearsey et al. (23) made a similar early observation that cloned mtDNA fragments containing the mouse mitochondrial tRNAHis, tRNASer and tRNALeu genes reduce the growth rate of E.coli hosts, and found that this inhibition was removed when the tRNA sequences were deleted. The definitive demonstration that some mitochondrial tRNA genes disrupt normal E.coli metabolism was made by Mita et al. (24), who systematically isolated and sequenced 50 different recombinant clones containing one of the ‘uncloneable’ human mtDNA MboI digestion fragments (21). They found that all 50 of these cloned fragments contained one or more mutations in the tRNAThr gene present on this fragment, with all of these mutations occurring in either the anticodon loop or D-stem region of the tRNA.
Because mtDNA sequences are not likely to be toxic to E.coli unless they are transcribed to RNA, we performed experiments to determine if transcription can be initiated in E.coli from sequences naturally present in the mouse mitochondrial genome. We cloned mtDNA restriction fragments at the 5′ end of a promoter-less CAT (CmR) gene and selected recombinant clones that could grow at high copy numbers on Cm-containing plates. Plasmid clones that were isolated from this screen were sequenced to identify the mtDNA sequences that were driving transcription of the CAT gene and the CAT activity generated from these mtDNA promoter fragments was compared at low and high copy numbers. As seen in Table 2 and in Figure 3, we identified multiple fragments in the mouse mitochondrial genome that serve as transcription promoters in E.coli and found that the level of transcription from these sequences is invariably dramatically higher at high copy numbers than at low copy numbers. These results indicate that mtDNA sequences will be transcribed when present in E.coli regardless of how the cloning vector is designed. However, by keeping the clone copy number low the level of expression of the tRNA genes or other potential mtDNA inhibitory sequences is minimized and the overall mitochondrial clone stability is increased.
Contrary to published results (7,8), we report for the first time that mitochondrial genomes can be readily engineered in E.coli hosts. As was the case with overall construct stability, we believe that we were able to successfully modify our mtDNA clones because we employed a relatively low copy DNA replication system and that previous efforts at engineering a mitochondrial genome in E.coli failed because high copy origins of replication were used. We did, however, identify one segment of the mouse mitochondrial genome that was more difficult to reclone than others, indicating that the toxic effects of the mitochondrial sequences have not been completely eliminated by using the γ-ori. Since spontaneous mutations may occur in mtDNA that reduces its toxicity in E.coli if the copy number is too high, a replication system with even lower copy replication than the π/γ system might be useful in some instances to ensure even more complete sequence stability when engineering these constructs.
Each of the mitochondrial genome clones characterized for this report has a transposon insertion that disrupts one of the mitochondrial genes in the clone. Mitochondrial clones in which the E.coli replication and selection functions are inserted in non-coding regions would be more versatile and may be required for some experiments. Although it would be possible to screen a large mitochondrial genome insertion library to identify this preferred sub-population of clones, the frequency with which these more optimal insertions will occur at random is fairly low because the amount of non-coding sequence is so very small in the mammalian mitochondrial genome. For this reason, we are instead taking the approach of engineering the mitochondrial genome clones we obtained from the transposon cloning in order to restore the disrupted genes and in effect move the insertion site to a non-coding region. Because only two randomly selected clones with insertions in different mitochondrial genes will have between them the entire coding capacity of the mitochondrial genome, a source of mtDNA fragments with uninterrupted genes is readily available for this work.
In order to assess the biological activity of the recombinant mouse mitochondrial genomes, we electroporated them back into isolated mouse mitochondria devoid of their own DNA (ρ0) and detected robust levels of in organello transcription (Fig. 5). We optimized the electroporation conditions for these experiments to those that resulted in the highest levels of mitochondrial transcription by using RT–PCR reactions specific for transcripts from the transferred DNA as the assay for electroporation efficiency (Fig. 4). Collombet et al. (15) first described the transfer of small recombinant plasmids into isolated mouse mitochondria by electroporation and proved the functional integrity of the electroporated mitochondria using enzymatic assays, but did not report subsequent in organello transcription. More recently, Farre and Araya (25) showed that they could study mitochondrial RNA transcription and processing by electroporating small chimeric plasmids containing genes under the control of mitochondrial promoters into purified wheat mitochondria. To our knowledge, the work we describe here is the first report of successful introduction of a full mitochondrial genome into mitochondria and the first reported use of ρ0 mitochondria in DNA transfer experiments. Using ρ0 mitochondria that do not have a DNA genome or any RNA transcripts of their own for these experiments allows us to analyze the transcription of genes that would already be present in ρ+ mitochondria without interference from the native transcripts.
The approach we describe for cloning and engineering complete mitochondrial genomes and for subsequent in organello analysis offers an attractive experimental system for studying many aspects of vertebrate mitochondrial gene expression, and is a first step towards true in vivo engineering of the mitochondrial genomes of vertebrates. By manipulating genes within the complete mitochondrial genome rather than in smaller chimeric constructs, the correct genomic context is maintained and the influences of surrounding sequences are preserved. The in organello analysis system we describe can measure transcription, 5′ and 3′ processing and the relative stability of RNA transcripts from both engineered mutant mitochondrial genes and from foreign genes introduced into the mitochondrial genome in an environment that closely resembles an in vivo system.
Acknowledgments
ACKNOWLEDGEMENTS
This work was supported by grants from the University of Minnesota School of Medicine to M.D.K. and from the Korea Research Foundation (KRF-99-D032) to Y.G.Y.
REFERENCES
- 1.Saraste M. (1999) Oxidative phosphorylation at the fin de siecle. Science, 283, 1488–1493. [DOI] [PubMed] [Google Scholar]
- 2.Green D.R. and Reed,J.C. (1998) Mitochondria and apoptosis. Science, 281, 1309–1312. [DOI] [PubMed] [Google Scholar]
- 3.Gray M.W., Burger,G. and Lang,B.F. (1999) Mitochondrial evolution. Science, 283, 1476–1481. [DOI] [PubMed] [Google Scholar]
- 4.Wallace D.C. (1999) Mitochondrial diseases in man and mouse. Science, 283, 1482–1488. [DOI] [PubMed] [Google Scholar]
- 5.Johnston S.A., Anziano,P.Q., Shark,K., Sanford,J.C. and Butow,R.A. (1988) Mitochondrial transformation in yeast by bombardment with microprojectiles. Science, 240, 1538–1541. [DOI] [PubMed] [Google Scholar]
- 6.Fox T.D., Sanford,J.C. and McMullin,T.W. (1988) Plasmids can stably transform yeast mitochondria lacking endogenous mtDNA. Proc. Natl Acad. Sci. USA, 85, 7288–7292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wheeler V.C., Aitken,M. and Coutelle,C. (1997) Modification of the mouse mitochondrial genome by insertion of an exogenous gene. Gene, 198, 203–209. [DOI] [PubMed] [Google Scholar]
- 8.Bigger B., Tolmachov,O., Collombet,J.M. and Coutelle,C. (2000) Introduction of chloramphenicol resistance into the modified mouse mitochondrial genome: cloning of unstable sequences by passage through yeast. Anal. Biochem., 277, 236–242. [DOI] [PubMed] [Google Scholar]
- 9.Koob M., Shaw,A. and Cameron,D.C. (1994) Minimizing the genome of Escherichia coli. Ann. N. Y. Acad. Sci., 745, 1–3. [DOI] [PubMed] [Google Scholar]
- 10.Bertram J.S. and Janik,P. (1980) Establishment of a cloned line of Lewis Lung Carcinoma cells adapted to cell culture. Cancer Lett., 11, 63–73. [DOI] [PubMed] [Google Scholar]
- 11.King M.P. and Attardi,G. (1989) Human cells lacking mtDNA: repopulation with exogenous mitochondria by complementation. Science, 246, 500–503. [DOI] [PubMed] [Google Scholar]
- 12.Bai Y. and Attardi,G. (1998) The mtDNA-encoded ND6 subunit of mitochondrial NADH dehydrogenase is essential for the assembly of the membrane arm and the respiratory function of the enzyme. EMBO J., 17, 4848–4858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.King M.P. and Attardi,G. (1996) Isolation of human cell lines lacking mitochondrial DNA. Methods Enzymol., 264, 304–313. [DOI] [PubMed] [Google Scholar]
- 14.Sambrook J., Fritsch,E.F. and Maniatis,T. (1989) Molecular Cloning: A Laboratory Manual, 2nd Edn. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- 15.Collombet J.M., Wheeler,V.C., Vogel,F. and Coutelle,C. (1997) Introduction of plasmid DNA into isolated mitochondria by electroporation. A novel approach toward gene correction for mitochondrial disorders. J. Biol. Chem., 272, 5342–5347. [DOI] [PubMed] [Google Scholar]
- 16.Gaines G.L. III (1996) In organello RNA synthesis system from HeLa cells. Methods Enzymol., 264, 43–49. [DOI] [PubMed] [Google Scholar]
- 17.Kolter R. and Helinski,D.R. (1982) Plasmid R6K DNA replication. II. Direct nucleotide sequence repeats are required for an active γ-origin. J. Mol. Biol., 161, 45–56. [DOI] [PubMed] [Google Scholar]
- 18.Shafferman A., Kolter,R., Stalker,D. and Helinski,D.R. (1982) Plasmid R6K DNA replication. III. Regulatory properties of the π initiation protein. J. Mol. Biol., 161, 57–76. [DOI] [PubMed] [Google Scholar]
- 19.Goryshin I.Y., Jendrisak,J., Hoffman,L.M., Meis,R. and Reznikoff,W.S. (2000) Insertional transposon mutagenesis by electroporation of released Tn5 transposition complexes. Nat. Biotechnol., 18, 97–100. [DOI] [PubMed] [Google Scholar]
- 20.Greener A., Filutowicz,M.S., McEachern,M.J. and Helinski,D.R. (1990) N-terminal truncated forms of the bifunctional π initiation protein express negative activity on plasmid R6K replication. Mol. Gen. Genet., 224, 24–32. [DOI] [PubMed] [Google Scholar]
- 21.Drouin J. (1980) Cloning of human mitochondrial DNA in Escherichia coli. J. Mol. Biol., 140, 15–34. [DOI] [PubMed] [Google Scholar]
- 22.Tapper D.P., Van Etten,R.A. and Clayton,D.A. (1983) Isolation of mammalian mitochondrial DNA and RNA and cloning of the mitochondrial genome. Methods Enzymol., 97, 426–434. [DOI] [PubMed] [Google Scholar]
- 23.Kearsey S.E., Flanagan,J.G. and Craig,I.W. (1980) Cloning of mouse mitochondrial DNA in E. coli affects bacterial viability. Gene, 12, 249–255. [DOI] [PubMed] [Google Scholar]
- 24.Mita S., Monnat,R.J.,Jr and Loeb,L.A. (1988) Direct selection of mutations in the human mitochondrial tRNAThr gene: reversion of an ‘uncloneable’ phenotype. Mutat. Res., 199, 183–190. [DOI] [PubMed] [Google Scholar]
- 25.Farre J.C. and Araya,A. (2001) Gene expression in isolated plant mitochondria: high fidelity of transcription, splicing and editing of a transgene product in electroporated organelles. Nucleic Acids Res., 29, 2484–2491. [DOI] [PMC free article] [PubMed] [Google Scholar]