


Abstract
Despite the great potential of single nucleotide polymorphism (SNP) markers in evolutionary studies, in particular for inferring population genetic parameters, SNP analysis has almost exclusively been limited to humans and ‘genomic model’ organisms, due to the lack of available sequence data in non-model organisms. Here, we describe a rapid and cost effective method to isolate candidate SNPs in non-model organisms. This SNP isolation strategy consists basically in the direct sequencing of amplified fragment length polymorphism bands. In a first application of this method, 10 unique DNA fragments that contained 24 SNPs were discovered in 11.11 kb of sequenced genomic DNA of a non-model species, the brown trout (Salmo trutta).
INTRODUCTION
Single nucleotide polymorphisms (SNPs) are the most abundant resources of genetic variation among individuals of a species. They are currently in the center of interest concerning genome-wide studies, especially in human disease gene mapping (1) and human evolution (2). However, despite the great potential of SNP markers in evolutionary studies, in particular for inferring population genetic parameters (3,4), SNP analysis has almost exclusively been limited to humans and ‘genomic model’ organisms (5). Because of the lack of available sequence data in non-model organisms, the major hurdle consists in the development of efficient and flexible SNP isolation strategies (5).
SNPs are usually discovered by the sequencing of homologous fragments of genomic DNA (6). Several screening strategies for SNP isolation have been described so far: locus-specific amplification (LSA) and comparative sequencing from multiple individuals (7), expressed sequence tags (ESTs) sequencing (8), whole-genome shotgun sequencing (9) and, more recently, reduced representation shotgun sequencing (RRS; 10). Finally, high-density oligonucleotide arrays have been shown to be useful for efficient SNP discovery and screening in humans (11). Unfortunately, all these procedures are currently too expensive and too time consuming, particularly for small- to medium-scale DNA sequencing laboratories (12,13). The development of a more efficient method for the isolation of SNPs, which is especially suited to develop a couple of hundred unlinked SNP markers for population genetic studies in any species including domesticated species, is therefore highly desirable (e.g. bovine SNP-based markers for individual identification and paternity testing; 8).
In order to define an efficient SNP isolation strategy, we first need to identify the most laborious and expensive steps of ‘traditional’ SNP isolation procedures. As a matter of fact, the aforementioned methods require either the design of a specific pair of primers for each DNA fragment in order to amplify homologous copies of loci (e.g. LSA), or an additional cloning step (e.g. RRS). Consequently, the time and expenses required for these procedures are proportional to the number of fragments screened. Thus, the primary aim should be the reduction of primers needed for the LSA and/or to avoid extensive cloning.
The amplified fragment length polymorphism (AFLP) method is a PCR-based DNA fingerprinting technique (14) that has been successfully applied to a wide range of organisms with a broad application in systematics, pathotyping, population genetics and quantitative trait loci mapping (15). Recently, two studies reported the isolation of SNP markers from AFLP bands involving both a band-specific PCR amplification and a cloning step (13,16).
In this study, we present a highly efficient and flexible AFLP-based SNP isolation strategy that does not require any cloning steps and that allows a reduction of the number of primers needed for the LSA to a handful, virtually independent of the number of DNA fragments screened. We tested the method by applying it to a non-model species, the brown trout (Salmo trutta), which is an abundant freshwater fish in Europe. Finally, we discuss the potential of this method for controlling the degree of the ascertainment bias due to sampling strategies (compare with 2).
MATERIALS AND METHODS
Biological material and DNA extraction
Whole genomic DNA was extracted from 16 brown trout fin samples that had been stored in absolute ethanol using a standard phenol–chloroform extraction protocol (http://www.inapg.inra.fr/dsa/microsat/microsat.htm). Samples had been collected from six populations in Switzerland. Detailed information on the sampling locations of the analyzed trout individuals is available from the authors upon request.
AFLP analysis
We used the ‘AFLP analysis system 1’ kit (Gibco BRL, Rockville, USA; cat. no. 10544-013) following the instructions of the manufacturer to generate AFLP fingerprints. The protocol of the kit has originally been described in detail by Vos et al. (14), who summarized the method as follows. The AFLP technique is based on the selective PCR of restriction fragments from a total digest of genomic DNA. The technique involves three steps: (i) restriction of the DNA and ligation of oligonucleotide adapters; (ii) selective amplification of sets of restriction fragments and (iii) gel analysis of amplified fragments. PCR amplification of restriction fragments is achieved by using the adapter and restriction site sequence as target sites for primer annealing. The selective amplification is achieved by the use of primers that extend into the restriction fragments, amplifying only those fragments in which the primer extensions match the nucleotides flanking the restriction site.
Electrophoretic separation and visualization of the AFLP bands was performed according to Griffiths and Orr (17). From the 64 primer combinations provided by the kit (EcoRI/MseI), we used the following eight randomly chosen primer combinations: E-ACC/M-CTG, E-ACT/M-CTG, E-ACT/ M-CTA, E-ACA/M-CTA, E-ACA/M-CTC, E-ACC/M-CTC, E-ACC/M-CAG and E-AAG/M-CAG.
Isolation of bands of interest
The following band selection criteria, which are illustrated in Figure 1, were applied as an enrichment strategy for the isolation of several homologous copies of a particular DNA fragment. (i) Only clearly visible and distinct bands that were present at least in two individuals (i.e. that displayed the same electrophoretic mobility) were selected. This criterion is based on the assumption that equal mobility is a good predictor of homology and aims at ensuring that at least two homologous copies of any selected DNA fragment can potentially be analyzed. (ii) Bands that were present in all screened individuals were not chosen, because we argued that bands that are polymorphic in terms of presence and absence are less prone to represent repeated DNA elements.
Figure 1.
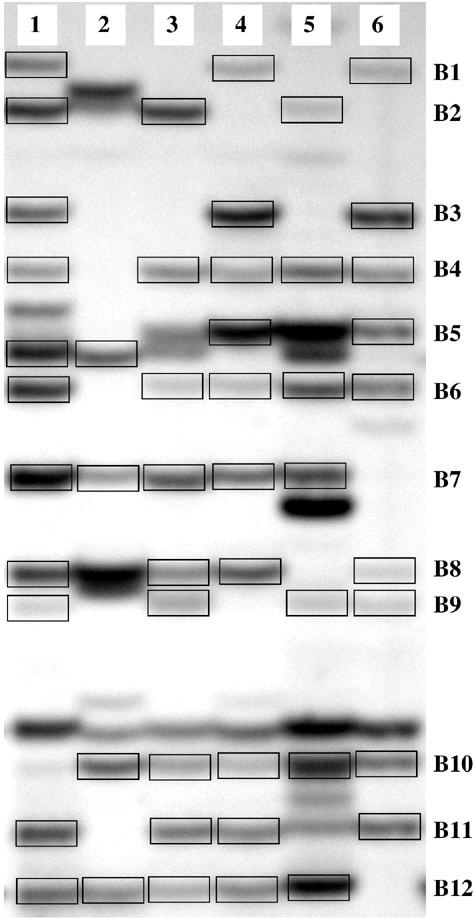
AFLP profile in six brown trout individuals (1–6) generated with the primer combination E-AAG/M-CAG. The approximate size range of the bands displayed in the figure is 250–350 bp. Boxes indicate copies of the 12 AFLP bands (B1–B12) that comply with the selection criteria for direct sequencing described in the Materials and Methods section.
Bands were selected by eye, excised from the dried gel with a clean scalpel and placed individually into PCR tubes containing 50 µl of 1× PCR buffer [Qiagen, Basel, Switzerland; buffer contains Tris–HCl, KCl, (NH4)2SO4, concentrations kept confidential by company]. The gel pieces were then incubated for 2 h at 68°C in order to elute the DNA fragments. Then incubation buffer was stored at 4°C up to several weeks before using it as a template for PCR re-amplification of the bands.
Band re-amplification and sequencing
PCR amplifications were carried out in volumes of 20 µl using a PTC100™-machine (MJ Research, Waltham, USA). Two microliters of eluted DNA mix was added to the PCR mix composed of 10 µl deionized water, 2 µl of PCR 10× buffer (Qiagen), 2 µl 2.5 mM dNTP, 2 pmol of each EcoRI-core and MseI-core primers and 1 U of Taq polymerase. Reaction conditions were as follows: an initial 2 min denaturation step at 94°C, 30 cycles consisting of 30 s at 94°C, 30 s at 50°C, 2 min at 72°C extension, and a final extension step at 72°C for 5 min. PCR products were then checked with agarose gel (1.5%) electrophoresis and purified with QIAquick® PCR purification kit (Qiagen). Single strand sequencing was done either with EcoRI-core primer or MseI-core primer by using the BigDye Terminator v3.0™ cycle sequencing kit (Applied BioSystems, Foster City, USA) according to the manufacturer’s instructions on an automated capillary electrophoresis sequencer, model ABI 3100 (Applied BioSystems).
Candidate SNP identification
Sequences were first base-called and aligned using the Sequence Analysis program (Applied BioSystems) and the BioEdit v5.0.9 sequence alignment editor (18). Then nucleotide BLAST searches were carried out for quickly revealing similarities between the obtained sequences and database sequences using the NCBI server (http://www.ncbi.nlm.nih.gov/BLAST). BLAST parameters were set to default values; nr for database (includes all GenBank, EMBL, DDBJ, PDB sequences but no EST, STS, GSS, or phase 0, 1 or 2 HTGS sequences) and Expect threshold = 10. Each sequence was processed with filtering programs for masking repeated elements (SINEs) and VNTRs (minisatellites and microsatellites). The Repeat Masker program (A.F.D.Smit and P.Green, unpublished; http://repeatmasker.genome.washington.edu/cgi-bin/RM2_req.pl) was used to search and detect sequences that were homologous to interspersed repetitive elements of vertebrates. The Tandem repeats finder program (19; http://c3.biomath.mssm.edu/trf.html) was used to locate and display tandem repeats in DNA sequences.
SNP first step validation
Homologous sites from each locus alignment that appeared to exhibit sequence variation, either in heterozygous or homozygous forms were first evaluated by manual inspection (7). SNPs were first checked by eye looking at the alignments and chromatograms. Heterozygous positions were detected as multiple peaks at the same position in DNA sequence chromatograms as shown in Figure 2 (compare with 20). Then, each ABI sequence trace was re-processed with the LifeTrace base-calling program (21), to obtain base quality values and to discern true allelic variations from base-calling errors and sequencing artifacts. Only SNPs that were surrounded by bases showing good qualities (LifeTrace score q ≥ 20) were taken in account and considered as valuable ‘candidate’ SNPs.
Figure 2.

(A) Electropherogram data of a candidate SNP discovered in brown trout by direct sequencing of a re-amplified AFLP band. (B) Electropherogram data of the homologous region in two clones derived from the re-amplified band shown in (A). (C) Electropherogram data of the candidate SNP region obtained by LSA and direct sequencing of the same individual as in (A). The position of the SNP is indicated by asterisks.
SNP second step validation by cloning
In order to confirm first step validation of some candidate SNPs and to assess the causes of low quality sequence data observed for some bands, we cloned several re-amplified bands using the AdvanTAge™ PCR cloning kit according to the manufacturer’s instructions (Clontech, Basel, Switzerland). In total four individual fragments corresponding to two different loci either showing homozygote or heterozygote SNP allelic states, and three individual fragments that showed low quality signal after direct sequencing (i.e. high background data), were cloned using freshly amplified and unpurified PCR products. Cloned products were then purified using the QIAprep Spin Miniprep Kit™ (Qiagen). Eight to 10 independent clones for each fragment were sequenced with M13 universal forward primer and compared on the basis of respective sequences alignment.
An alternative and independent method was applied for a second step SNP validation. Specific primer pairs for the bands containing candidate SNPs were designed. With these primers, the candidate SNP containing regions were PCR amplified from genomic DNA of the same trout individuals that had been analyzed in the AFLP screen. The PCR products were then purified and directly sequenced with the specific primers following the same protocols as were used for the re-amplification and sequencing of AFLP bands given above.
RESULTS
AFLP data, re-amplification and sequencing success
Regarding the band selection criteria (detailed in the Materials and Methods section), the eight AFLP primer combinations yielded on average 40 selectable bands or ∼50% of all visible bands in the size range of 100–700 bp. Thus, we observed a total of 320 bands that were shared at least between two individuals. Re-amplification success of the extracted individual band copies was nearly 90% (155 out of 176 PCRs, respectively). Fifty-five bands were sequenced on average for 2.56 individuals (range 1–6 individuals), which correspond to 141 individual band copies in total. Forty-eight percent of these 141 individual band copies yielded reliable sequence data (LifeTrace score q ≥ 20) (19) representative for 29 of the 55 analyzed bands. For 24 or 80% of these 29 bands, good quality and identical sequence data were obtained for at least one additional individual. All sequences were submitted to EMBL under the accession nos AJ535561–AJ535589. The BLAST searches indicated that four bands contained DNA sequences similar to known repetitive elements. In all cases, it concerned the Hpa I SINE family of salmonid fishes.
The size range of the sequenced bands was 140–680 bp. A non-parametric Mann–Whitney U-test (compare with 22) comparing the band sizes of successfully sequenced bands with the sizes of the remaining bands provided no indication that sequencing success was related to band size (P-value = 0.375). The sequence analysis of three cloned PCR products revealed the presence of multiple non-homologous DNA fragments in bands that yielded low quality sequence data (i.e. high background signal) when directly sequenced.
SNP discovery, first and second step validation of candidate SNPs
In total, 24 candidate SNPs were discovered among 10 of the 29 successfully sequenced bands. Up to seven candidate SNPs were found within a single band. The 29 bands sum up to ∼11.11 kb DNA sequence, thus, on average one SNP out of every 463 base positions. Nineteen candidate SNPs were detected within individuals, i.e. they occurred in heterozygous state; the remaining five candidates were discovered by comparing band copies from different individuals of the same population (Table 1). In these cases, individual band copies represent either homozygous states (two copies of the same allele) or a single allele copy. No candidate SNPs were detected by comparisons between individuals from different populations. The ratio between transitions and transversions was 1.4.
Table 1. Candidate SNPs discovered in directly sequenced brown trout AFLP bands.
| Within individuals as heterozygous state | Between individuals of the same population as homozygous state | Between individuals of different populations as homozygous state | |
|---|---|---|---|
| No. of candidate SNPs | 19 | 5 | 0 |
| No. of AFLP bands concerned | 8 | 3 | 0 |
| No. of sequences or pairs of sequences analyzed | 68 | 44 | 20 |
For six candidate SNPs, the allelic states were confirmed by the sequencing of several clones derived from PCR products of band re-amplifications. A total of 12 candidate SNPs were confirmed by the independent PCR amplification and sequencing of AFLP band segments with specific primers. Furthermore, no inconsistencies were detected with this second validation method, i.e. all candidate SNPs were confirmed in all AFLP band segments that could successfully be amplified. An example for both second step validation procedures is given in Figure 2.
DISCUSSION
Our analysis of brown trout samples suggests that direct sequencing of AFLP bands represents a highly efficient method to isolate candidate SNPs in non-model species. Using the method described here, we were able to discover rapidly 24 SNPs analyzing 55 AFLP bands. The successful validation of candidate SNPs by cloning and by LSA shows that SNP discovery through direct sequencing of AFLP bands is reliable and does not lead to extensive false inclusion of base-calling errors and sequencing artifacts if candidate SNP identification is based on stringent base-calling quality criteria (i.e. LifeTrace score q ≥ 20). These results are encouraging, indicating that SNPs can be isolated from AFLP fingerprints in numbers that are suitable for population genetics studies. A rough extrapolation of the 24 SNPs identified obtained from analyzing 55 AFLP bands indicates that an isolation of a couple of hundred candidate SNPs involves about the same expenses and time effort as is usually invested to isolate microsatellite markers for population genetic studies in non-model species (23). Thus, further tests of the applicability of this SNP isolation strategy in other species would be very promising.
The major advantage of the method resides in the fact that several copies of the same DNA fragment can be sequenced without the need of cloning step and specific primer design. In most cases, the sequencing of one particular AFLP band yields already information of two homologous copies. Indeed our data shows that most candidate SNPs are discovered within individuals, i.e. in heterozygous state. Furthermore, our assumption for selecting bands of interest, regarding equal electrophoretic mobility, is a good indicator for sequence homology. This was at least confirmed for 80% of the bands that could successfully be sequenced. Thus, the number of screened homologous copies can easily be increased by sequencing bands of equal electrophoretic mobility of several individuals, which obviously increases the probability of detecting existing SNPs in a particular DNA fragment.
Theoretically AFLP fingerprints can be generated for any organisms (14,15). However, the efficiency of the AFLP-based SNP isolation strategy depends critically on the proportion of AFLP bands that represent unique DNA fragments and that are not difficult to sequence due to structural problems. Repetitive elements probably do not represent a major problem in this context, as it was shown that AFLP fingerprints of higher plant species, which contain high numbers of repeated sequences, consisted predominantly of unique AFLP fragments, but are characterized by the presence of a small number of more intense fragments (14). We hypothesize that such bands can effectively be avoided by not selecting bands that are present in all screened individuals [compare with band selection criterion (ii) in the Materials and Methods section].
The analysis of cloned AFLP bands, which did not yield interpretable sequence data when directly sequenced, suggests that the extent of size homoplasy among AFLP markers may be the most influential factor determining the proportion of unique DNA fragments that can be analyzed for a given species. Moreover, simulation studies indicated very strong expected size homoplasy for two plant models and that the chance of size homoplasy decreases with increasing fragment size (24). Such a correlation should result in a higher rate of sequencing failures for small AFLP bands than for large AFLP bands; however, probably due to the limited number of bands analyzed in this study, we were not able to detect a significant difference in the mean size between bands that could be successfully sequenced and bands that could not. In any case, it seems logical to assume that the higher the density of visible bands in a particular AFLP fingerprint, the higher the probability of size homoplasy. Based on our study, we are not able to give accurate recommendations on the band density of AFLP fingerprints that would be suitable for our SNP isolation strategy regarding the problem of size homoplasy. For practical reasons, the number of restriction fragments that are amplified and detected on a denaturing polyacrylamide gel should not exceed 100. Above this number many bands cannot be clearly separated from each other, and thus cannot unambiguously be excised from the gel. Strategies for ‘tuning’ the band densities in AFLP fingerprints are for example discussed in detail in Vos et al. (14).
The efficiency of the described method also depends on the SNP density in the organism under study. Although it was not in the scope of this study to obtain statistical sound estimates on SNP density in brown trout, it is noteworthy in this context that the SNP density estimated from the brown trout AFLP bands (one SNP out of every 463 base positions) is similar to recent estimates for the human genome (one SNP out of every 300–500 base positions; 3).
Concerning the inference of population genetic parameters, ‘traditional’ SNP isolation strategies are prone to introduce a problem known as the ascertainment bias (compare with 2). This is especially true for methods that take advantage of DNA pooling procedures of several individuals (e.g. RRS; 10) in order to increase the probability of SNP detection. Ascertainment bias refers to a systematic distortion in measuring the true frequency of a phenomenon due to the way in which the data are obtained. Theoretical treatment of this problem clearly indicates that the information on how SNPs were isolated (e.g. in which sample, in which population etc.) is fundamental for an appropriate analysis (4). Moreover, it was also empirically shown that SNP-based variability estimates are highly dependent on the population used to isolate the SNP markers (5). Although not illustrated in detail in this paper, our method provides some means to control for ascertainment bias by knowing the exact source of a candidate SNP and by providing in some cases information on the allelic state of that SNP. Moreover, using the AFLP fingerprinting procedure enables one to screen a large set of individuals, and thus various populations. Therefore, with the here described SNP isolation strategy, anyone considering the development of an SNP panel in any given species can easily adapt the isolation strategy (e.g. choice of individuals and populations) according to the questions one intends to address.
Acknowledgments
ACKNOWLEDGEMENTS
We thank Laurent Excoffier, Grant Hamilton and other members of the CMPG for fruitful discussions and technical assistance. We thank two anonymous reviewers for helpful suggestions and Eric Desmarais for sharing his knowledge on fingerprinting and polyacrylamide band gel cutting and soaking. This work was supported by the BUWAL research grant F-01-9825.
DDBJ/EMBL/GenBank accession nos+ AJ535561–AJ535589
REFERENCES
- 1.Wang D.G, Fan,J.-B., Siao,C.-J., Berno,A., Young,P., Sapolsky,R., Ghandour,G., Perkins,N., Winchester,E. and Spencer,J. (1998) Large-scale identification, mapping and genotyping of single-nucleotide polymorphisms in the human genome. Science, 280, 1077–1082. [DOI] [PubMed] [Google Scholar]
- 2.Stoneking M. (2001) Single nucleotide polymorphisms: from the evolutionary past. Nature, 409, 821–822. [DOI] [PubMed] [Google Scholar]
- 3.Nielsen R. (2000) Estimation of population parameters and recombination rates from single nucleotide polymorphisms. Genetics, 154, 931–942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kuhner M.K., Beerli,P., Yamato,J. and Felsenstein,J. (2000) Usefulness of single nucleotide polymorphism data for estimating population parameters. Genetics, 156, 439–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schlötterer C. and Harr,B. (2002) Single nucleotide polymorphisms derived from ancestral populations show no evidence for biased diversity estimates in Drosophila melanogaster. Mol. Ecol., 11, 947–950. [DOI] [PubMed] [Google Scholar]
- 6.Qingbo L., Zhaowei,L., Monroe,H. and Culiat,C.T. (2002) Integrated platform for detection of DNA sequence variants using capillary array electrophoresis. Electrophoresis, 23, 1499–1511. [DOI] [PubMed] [Google Scholar]
- 7.Primmer C.R., Borge,T., Lindell,J. and Saetre,G.P. (2002) Single-nucleotide polymorphism characterization in species with limited available sequence information: high nucleotide diversity revealed in the avian genome. Mol. Ecol., 11, 603–612. [DOI] [PubMed] [Google Scholar]
- 8.Heaton M.P., Harhay,G.P., Bennett,G.L., Stone,R.T., Grosse,M.W., Casas,E., Keele,J.W., Smith,T.P.L., Chitko-McKown,C.G. and Laegreid,W.W. (2002) Selection and use of SNP markers for animal identification and paternity analysis in U.S. beef cattle. Mamm. Genome, 13, 272–281. [DOI] [PubMed] [Google Scholar]
- 9.Weber J.L. and Myers,E.W. (1997) Human whole-genome shotgun sequencing. Genome Res., 7, 401–409. [DOI] [PubMed] [Google Scholar]
- 10.Altshuler D., Pollara,V.J., Cowles,C.R., Van Etten,W.J., Baldwin,J., Linton,L. and Lander,E.S. (2000) An SNP map of the human genome generated by reduced representation shotgun sequencing. Nature, 407, 513–516. [DOI] [PubMed] [Google Scholar]
- 11.Dong S., Wang,E., Hsie,L., Cao,Y., Chen,X. and Gingeras,T.R. (2001) Flexible use of high-density oligonucleotide arrays for single-nucleotide polymorphism discovery and validation. Genome Res., 11, 1418–1424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ware J.L., Moran,L., Lin,C.-L. and Slatko,B. (2000) Implementation of automation in a small-scale DNA sequencing core facility. J. Biom. Tech., 11, 151–154. [PMC free article] [PubMed] [Google Scholar]
- 13.Meksem K., Ruben,E., Hyten,D., Triwitayakorn,K. and Lightfoot,D.A. (2001) Conversion of AFLP bands into high-throughput DNA markers. Mol. Genet. Genomics, 265, 207–214. [DOI] [PubMed] [Google Scholar]
- 14.Vos P., Hogers,R., Bleeker,M., Reijans,M., van de Lee,T., Hornes,M., Frijters,A., Pot,J., Peleman,J. and Kuiper,M. (1995) AFLP: a new technique for DNA fingerprinting. Nucleic Acids Res., 23, 4407–4414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mueller U.G. and Wolfenbarger,L.L. (1999) AFLP genotyping and fingerprinting. Trends. Ecol. Evol., 14, 389–394. [DOI] [PubMed] [Google Scholar]
- 16.Bensch S., Akesson,S. and Irwin,D.E. (2002) The use of AFLP to find an informative SNP: genetic differences across a migratory divide in willow warblers. Mol. Ecol., 11, 2359–2366. [DOI] [PubMed] [Google Scholar]
- 17.Griffiths R. and Orr,K. (1999) The use of AFLP in the isolation of sex-specific markers. Mol. Ecol., 8, 671–674. [DOI] [PubMed] [Google Scholar]
- 18.Hall T.A. (1999) BIOEDIT: a user-friendly biological sequence alignment, editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp. Ser., 41, 95–98. [Google Scholar]
- 19.Benson G. (1999) Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res., 27, 573–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fahrenkrug S.C., Freking,B.A., Smith,T.P.L., Rohrer,G.A. and Keele,J.W. (2002) Single nucleotide polymorphism (SNP) discovery in porcine expressed genes. Anim. Genet., 33, 186–195. [DOI] [PubMed] [Google Scholar]
- 21.Walther D., Bartha,G. and Morris,M. (2001) Basecalling with LifeTrace. Genome Res., 11, 875–888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sokal R.R. and Rohlf,F.J. (1995) Biometry: The Principles and Practice of Statistics in Biological Research, 3rd Edn. W.H. Freeman and Company, New York, NY.
- 23.Zane L., Bargelloni,L. and Patarnello,T. (2002) Strategies for microsatellite isolation: a review. Mol. Ecol., 11, 1–16. [DOI] [PubMed] [Google Scholar]
- 24.Vekemans X., Beauwens,T., Lemaire,M. and Roldán-Ruiz,I. (2002) Data from amplified fragment length polymorphism (AFLP) markers show indication of size homoplasy and of a relationship between degree of homoplasy and fragment size. Mol. Ecol., 11, 139–151. [DOI] [PubMed] [Google Scholar]