

Cells sense cues that guide their growth and development by probing their microenvironment with surface membrane receptors that elicit intracellular signals when they bind their ligands. Cell interactions with extracellular matrix (ECM) molecules differ from those with soluble regulatory factors, however, because in addition to ligation-induced signaling, cells also apply traction to their integrin receptors that mediate ECM adhesion (1). Tensional forces generated in the actin cytoskeleton and resisted by the ECM feed back to alter cell shape and function, and different cellular responses are produced depending on the ECM's ability to physically balance this stress. Rigid substrates that support high levels of isometric tension in the cell promote cell spreading and growth in the presence of soluble mitogens, whereas flexible ECM scaffolds that cannot resist cytoskeletal forces promote cell retraction, turn off growth, and switch on differentiation in the same medium (reviewed in ref. 1). Cells also sense local changes in ECM compliance that can influence their rate of migration (2). Tension-dependent changes in cell shape seem to be critical to this process: planar substrates that contain microfabricated ECM islands on the size of single cells can similarly control whether a cell will grow, differentiate, undergo directional motility, or die, depending on their ability to support or restrict cell spreading (3–5). The integrin receptors that mediate this form of mechanical signaling cluster together within spot weld-like anchoring complexes, known as focal adhesions. Molecules that form the structural backbone of the focal adhesion both mechanically couple integrins to the actin cytoskeleton (6, 7) and orient much of the cell's signal transduction machinery (8, 9). When cell-generated forces or external stresses are applied to integrins, a local intracellular transduction response is activated that leads to focal adhesion assembly (10, 11) and associated cytoskeletal strengthening (6, 7) as well as activation of chemical signaling cascades and gene transcription (12–14). Given the importance of cell-generated forces for control of cell shape and function and the known effects of mechanical stress on tissue development, the mechanism by which cells sense mechanical cues and transduce these signals into an intracellular response within the focal adhesion has become a major focus of attention in cell biology.
Is mechanosensation a local event or a whole-cell process?
Probing a mechanochemical mechanism, rather than one that is purely molecular in nature, requires methods that are more familiar to the engineer than to the molecular cell biologist. Techniques used to apply controlled stresses to surface integrin receptors include use of magnetic microbeads in combination with applied magnetic fields (6), plastic beads manipulated with optical tweezers (7), and micropipettes pulled with a micromanipulator (14), all of which are precoated with ECM ligands. However, the problem of mechanosensation differs from other forms of stimulus–response coupling, because cells exist in a state of isometric tension and thus any external force is imposed on a preexisting force balance (1). For this reason, cells can display different responses to the same mechanical stimulus if cytoskeletal tension is altered (10, 11, 13, 14). It is therefore critical to measure cell-generated forces and to understand how they contribute to cellular mechanotransduction. In this issue of PNAS, Tan et al. (15) describe an elegant new microarray-based mechanosensor technology that permits direct manipulation and real-time analysis of mechanical interactions between living cells and ECM with subcellular resolution. In addition to quantitating the forces applied over focal adhesions, the authors explore whether mechanosensation is a local event or whether the whole cell processes the mechanical signal and integrates it with other information before eliciting a biochemical response.
The force exerted on ECM substrates by single cells has been estimated in the past by using traction force microscopy (2, 16, 17). Cells are cultured on flexible ECM-coated substrates, such as cross-linked polyacrylamide gels or silicone rubber, that contain small beads just beneath their surface. By quantitating bead displacements beneath a cell and the stiffness of the material, it is possible to approximate the traction applied by each cell. A related method utilizes a thin elastomeric substrate created by using microfabrication that contains micrometer-sized ECM islands distributed across its surface (10). Traction forces are estimated by measuring local displacement of fluorescently labeled focal adhesion components that are deposited by the cells. However, because deformations propagate on these continuous flexible substrates, the calculation of forces is computationally intensive. In fact, the complexity of the algorithms required to calculate the stresses in these studies is a major limitation, and thus only a few laboratories have these capabilities. An even more specialized technique uses a horizontally mounted cantilever etched into a silicon-based microelectromechanical system to measure cell-generated forces applied by cells that move across its surface (18). But this device measures only force exerted in a single direction and along one axis of the cell. A more complete understanding of how cell-generated forces influence focal adhesion structure and mechanotransduction therefore requires a new and simpler approach to directly quantitate traction applied through individual adhesive contacts beneath the entire surface of whole living cells in real time.
Tan et al. (15) accomplished this goal by culturing cells on a microfabricated postarray detector (mPAD) composed of a bed of flexible micrometer-scale posts or “microneedles” that were created by using a microfabrication strategy that involves soft lithography and replica molding. The desired 3D shape of the mPAD is etched into a silicon chip by using photolithography to create a master, and then an elastomeric polymer, polydimethylsiloxane (PDMS), is cast and cured on its surface. The polymerized PDMS substrate with a shape complementary to the master is peeled off and inverted, and then the mPAD is created by replica molding (curing and casting) another PDMS substrate on its surface. Once it is polymerized, peeled off the template, and inverted, ECM molecules are transferred to the top surface of each post by bringing a flat PDMS sheet coated with ECM in contact with the mPAD, much like a rubber stamp is used to transfer ink to paper. By varying the height, width, and shape of specific posts within the array, the mechanical stiffness and mechanical anisotropy in the substrate also can be varied to create well-defined mechanical landscapes with regional heterogeneity for cellular studies.
When cells are cultured on this bed of microneedles, they spread and migrate selectively over the ECM-coated tips of the posts and form focal adhesions only in these regions, much as they do when they spread over microfabricated planar substrates that contain similar isolated ECM islands (Fig. 1). Importantly, when cells spread over the mPAD, the posts bend toward the center of the cell, and each post deflects independently of its neighbor. For small deflections, the posts behave like simple vertical cantilevers, and thus the deflection is directly proportional to the force applied by the cell. The mechanical stiffness of each post can be determined by measuring its deflection by using micropipettes with known spring constants. Hence, the subcellular spatial distribution of traction forces and their relation to individual focal adhesions can be simply calculated (i.e., without requiring computational assumptions) by measuring the deflection of each individual post by using microscopy.
Figure 1.
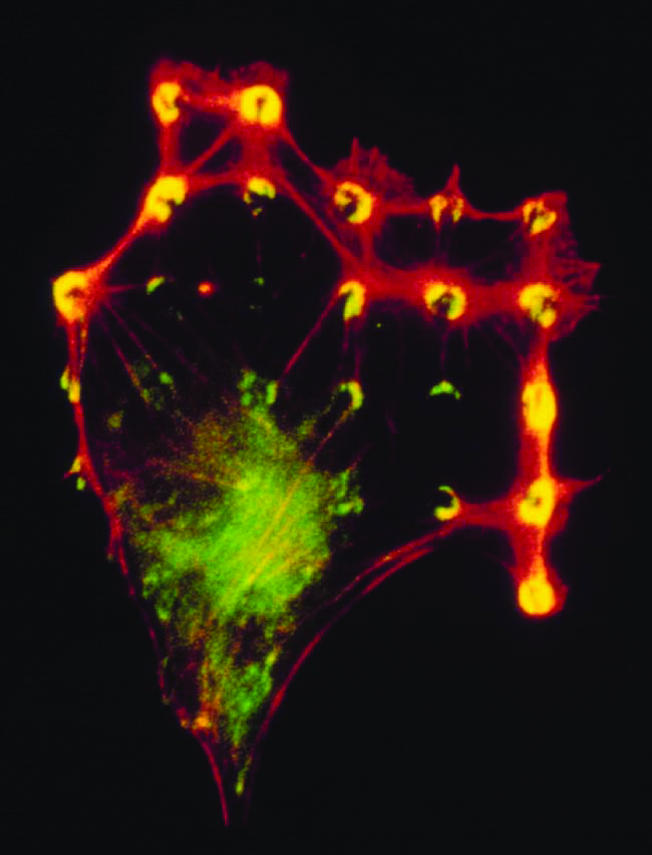
Fluorescence micrograph of an endothelial cell spread over a substrate containing a regular array of small (5-μm-diameter) circular ECM islands separated by nonadhesive regions created with a microcontact printing technique. Yellow rings and crescents indicate colocalization of vinculin (green) and F-actin (red) within focal adhesions that form only on the regularly spaced circular ECM islands (micrograph by J.-L. Alonso and D.E.I.).
Experiments carried out with vascular smooth muscle cells, endothelial cells, and fibroblasts led to some important insights into how cells interact mechanically with ECM. By measuring the magnitude and direction of deflections of individual posts, the origin of forces exerted by adherent cells was unequivocally localized to discrete points of cell–ECM contact on the top of each post, as previously demonstrated by using other techniques (3, 10). Cells exerted as much as 75 nN of force on individual posts, and the actin cytoskeleton was confirmed to be the source of these forces by using a myosin ATPase inhibitor. As expected, these local regions where traction was exerted generally corresponded to focal adhesions as defined by fluorescence staining for vinculin. Quantitative analysis, however, revealed a more complex relationship. Although the amount of force exerted on an individual ECM contact increased in direct proportion to focal adhesion size for contacts with >1 μm2 of vinculin staining, there was no clear relation between size and the magnitude of force exerted on smaller focal adhesions. This conflicts with the view that the presence of vinculin is an indicator of the cell's ability to exert force at adhesive contacts (10, 11); however, it is consistent with the finding that small adhesions near the leading edge of migrating cells transmit stronger propulsive tractions than larger focal adhesions (17). The smaller adhesions may correspond to newly formed focal complexes that progressively develop into larger focal adhesions over time in response to sustained stress (10, 11, 17). Alternatively, this finding may simply be an artifact of using vinculin accumulation as a measure of focal adhesion size because vinculin is not found in all focal adhesions (9); thus, other cytoskeletal linking proteins (e.g., α-actinin, talin, and paxillin) may be present in these regions.
These findings extend work that similarly shows that cells respond to local changes in ECM compliance or to external stresses applied to integrins by increasing focal adhesion formation, promoting stress fiber assembly, and increasing adhesive strength, in addition to activating various chemical signaling cascades (6, 7, 10–14). On the basis of these observations, integrins have come to be viewed as mechanoreceptors and focal adhesions as discrete mechanosensory organelles (1, 6, 7, 9–11, 14). Thus, the common view of how cells sense physical properties of the substrate through focal adhesions is that the cytoskeleton generates a level of tension against the ECM that is proportional to ECM stiffness at the site of integrin binding. Tan et al. (15) explored this mechanism by examining whether changes in the size of the whole cell could modulate the magnitude of forces exerted locally when cells sense an individual ECM-coated post of defined mechanical stiffness. Cell spreading was restricted by printing ECM onto small sets of neighboring posts (e.g., 2 × 2 vs. 5 × 5 arrays) surrounded by nonadhesive posts. In the presence of serum, decreasing the area that a cell could extend resulted in a progressive decrease in the total amount of traction exerted on each post. When small cells that were serum-starved for 1 d were stimulated with the contractile agonist, lysophosphatidic acid (LPA), they did not increase traction force over their basal levels. Moreover, this block in contractile signaling was overcome by transfecting the cells with a constitutively active form of the small GTPase RhoA that promotes tension generation and focal adhesion assembly through its downstream effectors, Rho-associated kinase and mDia1 (14). This contradicts a past study that found that LPA activation of Rho in the first hour after plating neither requires nor is enhanced by cell adhesion (19). Thus, once cell spreading over ECM comes to a steady-state, cells appear to use a distinct LPA signaling mechanism.
The mPAD studies suggest that cell shape modulates LPA signaling to Rho and thereby controls the set-point for cell contractility: the same soluble agonist produces a larger effect on traction in large vs. small cells. Vascular smooth muscle cell contractility in response to endothelin-1 can be similarly controlled through modulation of cell shape by altering ECM adhesivity (20). How cell distortion can modulate the contractile response remains unclear; however, it likely involves higher-order changes in cytoskeletal structure given the central role the cytoskeleton plays in control of shape-dependent control of growth, apoptosis, and motility (5, 21, 22). Actin bundles assemble along the tension field lines that stretch between different focal adhesions, and thus stress fiber length increases in cells that spread over multiple ECM islands (Fig. 1). When individual cells are cultured on large square ECM islands on planar substrates, they reorient their actin bundles along their long (diagonal) axes and preferentially form focal adhesions in their corners, even in the absence of soluble stimuli (5). Thus, cell shape can influence local focal adhesion assembly induced by ECM as well as soluble factors. Shape-dependent alterations in microtubule organization also may contribute to these responses, because microtubule depolymerization releases bound nucleotide exchange factor GEF-H1 and thereby activates Rho (23). In fact, cell shape distortion has been shown to regulate the membrane targeting of Rho as well as Rac (which controls small focal complex assembly; ref. 11) by altering microtubule polymerization (24). Importantly, although cell shape governs the overall level of traction force cells apply in response to binding to ECM, the amount of traction exerted on each focal adhesion is fine-tuned locally. This was demonstrated by using the mPAD: differences were observed in the magnitude of traction from post to post as well as in the temporal and spatial dynamics of activation after LPA stimulation (15). These regional variations in force transmission may be mediated by local changes in microtubules that grow into adhesion sites in response to tension (25) and retard or reverse focal adhesion development (26), potentially through inhibition of Rho via local sequestration of GEF-H1 (23). Thus, cells use both coarse and fine controls to respond to local changes in ECM mechanics: the cell acts locally, but it thinks globally.
The ultimate challenge in cell and developmental biology is to understand how cells sense physical and chemical cues in their microenvironment, process this information, and respond appropriately. In biology, we tend to emphasize linear thinking and to focus on local molecular binding and assembly events. However, if all mechanosensing were carried out locally within subcellular microdomains, then cells would be continuously activated by subtle variations in ECM structure within living tissues that are normally exposed to physiological stresses, including tension, compression, pressure, and flow. Thus, the stability of tissue form may be optimally maintained by lowering the sensitivity of the contractile machinery in a compact cell within a normally confluent tissue structure. Once there is a large-scale change in ECM mechanics due to proteolysis or regional tissue distortion that alters cell shape, then the cell becomes sensitive to soluble contractile agonists as well as growth and motility factors. The cell also now becomes more responsive to mechanical cues within local microdomains. Differentials in growth across a few cell diameters are responsible for bud formation during epitheliogenesis. Localized mechanical changes at the subcellular level may influence the direction and speed of cell movement (2, 5, 7) and thereby facilitate extension of branching structures, such as capillary sprouts and nerve cell processes, that are known to be stimulated by localized tensional stress (27, 28). Cell shape-dependent changes in the sensitivity of the contractile machinery also may ensure “compliance matching” in smooth muscle and blood vessels, so that the level of tension exerted by the cell precisely balances the mechanical stress transmitted through the surrounding ECM in response to tissue distortion. The corollary is that loss of this mechanical form of tissue homeostasis may lead to disease, as seen, for example, in patients with systemic or pulmonary hypertension.
Microfabricated materials are being used more frequently in cell biology because the size, shape, and mechanical properties of their surface features can be tailored specifically for use with living cells. As seen in studies with mPADs, substrates created with soft lithography are particularly useful because they are biocompatible, optically clear, and pliant. These substrates also offer additional advantages. For example, they may be integrated with microfluidics systems containing micromixers and microactuators in the future to create microsensors that contain living cells (“biochips”) for pharmaceutical applications or biopathogen detection. A device of this type containing mPADs could potentially provide a massively parallel screening approach for new therapies for hypertension, asthma, intestinal dysfunction, cardiac failure, and other diseases in which altered cell contractility contributes to their etiology. On the basis of the fundamental role cell tractional forces play in cell and developmental regulation, this micromechanical array-based screening method also could be appropriate for studying many other disease processes, including angiogenesis and cancer metastasis. Thus, this elegant technique devised to attack a simple biological question is a prime example of how merging engineering and biological sciences may both advance our understanding of fundamental biological mechanisms and change the way in which medicine is practiced in the future.
Footnotes
See companion article on page 1484.
References
- 1.Ingber D E. Curr Opin Cell Biol. 1991;3:841–848. doi: 10.1016/0955-0674(91)90058-7. [DOI] [PubMed] [Google Scholar]
- 2.Lo C M, Wang H B, Dembo M, Wang Y L. Biophys J. 2000;79:144–152. doi: 10.1016/S0006-3495(00)76279-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chen C S, Mrksich M, Huang S, Whitesides G, Ingber D E. Science. 1997;276:1425–1428. doi: 10.1126/science.276.5317.1425. [DOI] [PubMed] [Google Scholar]
- 4.Dike L, Chen C S, Mrkisch M, Tien J, Whitesides G M, Ingber D E. In Vitro Cell Dev Biol Anim. 1999;35:441–448. doi: 10.1007/s11626-999-0050-4. [DOI] [PubMed] [Google Scholar]
- 5.Parker K K, Brock A L, Brangwynne C, Mannix R J, Wang N, Ostuni E, Geisse N A, Adams J C, Whitesides G M, Ingber D E. FASEB J. 2002;16:1195–1204. doi: 10.1096/fj.02-0038com. [DOI] [PubMed] [Google Scholar]
- 6.Wang N, Butler J P, Ingber D E. Science. 1993;260:1124–1127. doi: 10.1126/science.7684161. [DOI] [PubMed] [Google Scholar]
- 7.Choquet D, Felsenfeld D P, Sheetz M P. Cell. 1997;88:39–48. doi: 10.1016/s0092-8674(00)81856-5. [DOI] [PubMed] [Google Scholar]
- 8.Miyamoto S, Teramoto H, Coso O A, Gutkind J S, Burbelo P D, Akiyama S K, Yamada K M. J Cell Biol. 1995;131:791–805. doi: 10.1083/jcb.131.3.791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Plopper G, McNamee H, Dike L, Bojanowski K, Ingber D E. Mol Biol Cell. 1995;6:1349–1365. doi: 10.1091/mbc.6.10.1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Balaban N Q, Schwarz U S, Riveline D, Goichberg P, Tzur G, Sabanay I, Mahalu D, Safran S, Bershadsky A, Addadi L, Geiger B. Nat Cell Biol. 2001;3:466–472. doi: 10.1038/35074532. [DOI] [PubMed] [Google Scholar]
- 11.Galbraith C G, Yamada K M, Sheetz M P. J Cell Biol. 2002;159:695–705. doi: 10.1083/jcb.200204153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Meyer C J, Alenghat F J, Rim P, Fong J H-J, Fabry B, Ingber D E. Nat Cell Biol. 2000;2:666–668. doi: 10.1038/35023621. [DOI] [PubMed] [Google Scholar]
- 13.Chen J, Fabry B, Schiffrin E L, Wang N. Am J Physiol. 2001;280:C1475–C1484. doi: 10.1152/ajpcell.2001.280.6.C1475. [DOI] [PubMed] [Google Scholar]
- 14.Riveline D, Zamir E, Balaban N Q, Schwarz U S, Ishizaki T, Narumiya S, Kam Z, Geiger B, Bershadsky A D. J Cell Biol. 2001;153:1175–1186. doi: 10.1083/jcb.153.6.1175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tan J L, Tien J, Pirone D M, Gray D S, Bhadriraju K, Chen C S. Proc Natl Acad Sci USA. 2003;100:1484–1489. doi: 10.1073/pnas.0235407100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Roy P, Rajfur Z, Pomorski P, Jacobson K. Nat Cell Biol. 2002;4:E91–E96. doi: 10.1038/ncb0402-e91. [DOI] [PubMed] [Google Scholar]
- 17.Beningo K A, Dembo M, Kaverina I, Small J V, Wang Y L. J Cell Biol. 2001;153:881–888. doi: 10.1083/jcb.153.4.881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Galbraith C G, Sheetz M P. Proc Natl Acad Sci USA. 1997;94:9114–9118. doi: 10.1073/pnas.94.17.9114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ren X D, Kiosses W B, Schwartz M A. EMBO J. 1999;18:578–585. doi: 10.1093/emboj/18.3.578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lee K-M, Tsai K, Wang N, Ingber D E. Am J Physiol. 1997;274:H76–H82. doi: 10.1152/ajpheart.1998.274.1.H76. [DOI] [PubMed] [Google Scholar]
- 21.Huang S, Chen C S, Ingber D E. Mol Biol Cell. 1998;9:3179–3193. doi: 10.1091/mbc.9.11.3179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Flusberg D A, Numaguchi Y, Ingber D E. Mol Biol Cell. 2001;12:3087–3094. doi: 10.1091/mbc.12.10.3087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Krendel M, Zenke F T, Bokoch G M. Nat Cell Biol. 2002;4:294–301. doi: 10.1038/ncb773. [DOI] [PubMed] [Google Scholar]
- 24. Putnam, A. J., Cunningham, J. J., Pillemer, B. B. & Mooney, D. J. (2003) Am. J. Physiol., in press. [DOI] [PubMed]
- 25.Kaverina I, Krylyshkina O, Beningo K, Anderson K, Wang Y L, Small J V. J Cell Sci. 2002;115:2283–2291. doi: 10.1242/jcs.115.11.2283. [DOI] [PubMed] [Google Scholar]
- 26.Kaverina I, Krylyshkina O, Small J V. J Cell Biol. 1999;146:1033–1044. doi: 10.1083/jcb.146.5.1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Korff T, Augustin H G. J Cell Sci. 1999;112:3249–3258. doi: 10.1242/jcs.112.19.3249. [DOI] [PubMed] [Google Scholar]
- 28.Bray D. Dev Biol. 1984;102:379–389. doi: 10.1016/0012-1606(84)90202-1. [DOI] [PubMed] [Google Scholar]