

Abstract
Recent research has revealed that endogenous cannabinoid receptors (CB1 and CB2) react with the active ingredient of marijuana, Δ9-tetrahydrocannabinol. Two endogenous ligands activate these receptors. The principal one, anandamide (AEA), activates CB1. AEA and CB1 are localized to various neurons within the brain. Because Δ9-tetrahydrocannabinol inhibited prolactin (Prl) secretion following its intraventricular injection into male rats, we hypothesized that AEA would have a similar effect. Estrogen modifies many hormonal responses and is known to increase Prl secretion. Therefore, we hypothesized that responses to intraventricular AEA would change depending on the gonadal steroid environment. Consequently, we evaluated the effects of lateral cerebral ventricular microinjection of AEA (20 ng) into male, ovariectomized (OVX), and estrogen-primed (OVX-E) rats. AEA decreased plasma Prl in male rats, had little effect in OVX females, and increased Prl in OVX-E rats. The results were at least partially mediated by changes in dopaminergic turnover, altering the inhibitory dopaminergic control of Prl release by the anterior pituitary gland. Thus, dopamine turnover was increased in the male rats and decreased significantly in OVX and in OVX-E rats. The changes in Prl may be caused not only by altered dopamine input to the anterior pituitary gland but also by effects of AEA on other transmitters known to alter Prl release. Importantly, in OVX-E rats, the elevated Prl release and the response to AEA were blocked by the AEA antagonist, indicating that AEA is a synaptic transmitter released from neurons that decrease inhibitory control of Prl release.
Keywords: cannabinoid receptors‖estradiol‖dopamine ‖CB1 antagonist‖ dopamine receptors
The isolation and determination of structure of Δ9-tetrahydrocannabinol (THC), the principal psychoactive constituent of Cannabis sativa (1), made possible many investigations of the function and mechanism of action of THC (2). Two subtypes of cannabinoid receptors have been identified, the CB1 receptor, located mainly in the CNS (3–6), and the CB2 receptor, localized mainly in peripheral tissues (7). The identification of cannabinoid receptors led to the discovery of an endocannabinoid from porcine brain (8) arachidonylethanolamine, named anandamide (AEA), that binds specifically with high affinity to CB1 receptors. The presence of CB2 receptors in peripheral tissues led to the discovery of an additional peripheral ligand, 2-arachidonyl glycerol (9, 10).
Previously, it was shown that THC administered into the third cerebral ventricle of male rats lowered plasma prolactin (Prl) concentrations (11). This effect was exerted within the CNS, as THC did not alter the basal and thyroid-stimulating hormone (TSH)-releasing hormone-induced release of Prl from cultured anterior pituitary (AP) cells (11).
Prl release is tonically inhibited by dopamine (DA) released from the terminals of the tuberoinfundibular DAergic (TIDA) neurons in the external layer of the median eminence. The released DA diffuses into the hypophysial portal capillaries and is transported by the portal veins to the AP gland, where it acts on DA type 2 receptors (D2R) on lactotrophs to inhibit Prl release. It is well known that estrogens increase the secretion of Prl (12) and also can regulate the biosynthesis and release of DA (13). Cannabinoids also can affect brain DA concentrations and DA receptors (14). Furthermore, it has been shown that sex steroids can modulate the density and/or affinity of CB1 receptors in certain brain areas (15). Consequently, we hypothesized that the effects of AEA on Prl secretion might be altered by estrogen. Therefore, AEA was microinjected intracerebroventricularly (icv) into the lateral cerebral ventricle of normal adult male, ovariectomized (OVX) and OVX, estrogen-primed (OVX-E) female rats to determine the effects on plasma Prl concentrations and DAergic activity in the medial basal hypothalamus (MBH). Also, the possible colocalization of CB1 receptors on DAergic neurons in the MBH was examined by immunocytochemistry.
Materials and Methods
Animals.
Male and female Sprague–Dawley rats (220–250 g body weight) were kept in group cages in an animal room having a photoperiod of 14 h of light (7 a.m. to 9 p.m.) and room temperature of 22–24°C. Animals had free access to laboratory chow and tap water.
Male Rats.
In vivo studies.
An indwelling cannula was implanted into the lateral cerebral ventricle by using a stereotaxic instrument and coordinates from the atlas of Pellegrino et al. (16), while the rats were anesthetized with tribromoethanol (16). A week later, 24 h before the experiment, an indwelling catheter was placed into the right external jugular vein and advanced to the right atrium of anesthetized rats for collection of blood samples according to the technique of Harms and Ojeda (17).
On the day of the experiment, conscious, freely moving rats were divided into two groups. After the collection of the first blood sample (0.5 ml), the control group of rats was injected icv with 2 μl of 0.9% NaCl (saline). The experimental group of rats was injected icv with 20 ng of AEA in 2 μl of saline. All injections were made during a period of 1 min. The second blood sample was obtained 10 min later, and additional blood samples were taken every 10 min for 90 min for the determination of Prl. Each blood sample was immediately replaced with 0.5 ml of saline containing 50 IU of heparin per ml. After centrifugation, the plasma was stored frozen at −20°C until the determination of plasma Prl by RIA. Immediately after obtaining the last blood sample, following decapitation and removal of the brain, the MBH was dissected by making frontal cuts just behind the optic chiasm, extending dorsally 2 mm. A horizontal cut extended from this point caudally to just behind the pituitary stalk, where another frontal cut was made. Longitudinally, cuts were made 1 mm lateral to the midline bilaterally. The MBH was frozen at −70°C before measurements of DA and dihydroxyphenylacetic acid (DOPAC).
In vitro studies.
MBH explants from the brain immediately after decapitation of untreated normal male rats were preincubated in 0.5 ml of Krebs–Ringer bicarbonate-buffered medium (pH 7.4) containing 0.1% glucose for 30 min before replacement with 0.5 ml of fresh medium or medium containing AEA (10−7 or 10−9 M). The incubation was continued for 30 min followed by removal of the medium and tissues that were stored at −70°C. Tissue content of DA and DOPAC and DA released into the incubation medium were determined.
Female Rats.
OVX rats.
The rats were anesthetized using ether and bilaterally OVX. Two weeks later, they were implanted into the lateral cerebral ventricle with a cannula as described above for male rats. A week later and 24 h before the experiment, the indwelling jugular catheter was implanted as described above for male rats. On the day of the experiment, the rats were divided into two groups. After obtaining the first blood samples as described above, rats of the control group were microinjected icv with saline or AEA as described above. Thirty minutes later and every 30 min for 150 min, additional blood samples were drawn for determination of Prl. The MBH was dissected as described above and stored for measurements of DA and DOPAC content as described above.
OVX-E rats.
The rats were anesthetized using ether and bilaterally ovariectomized. Two weeks later, a cannula was implanted into the lateral cerebral ventricle as described above. Five days later and 48 h before the experiment, the rats were injected s.c. with estradiol-17 beta (E2) (10 μg/100 μl corn oil). Twenty-four hours later, 1 day before the experiment, an indwelling jugular catheter was implanted for obtaining blood samples from freely moving animals as described above. These rats were divided into four groups injected icv: (i) 2 μl of saline; (ii) 20 ng of AEA/2 μl of saline; (iii) 200 ng/2 μl of saline AM 251 (a CB1 antagonist); and (iv) AEA + AM251. AM251 was injected 30 min before AEA injection in this group. Blood samples for plasma Prl determinations were obtained as in the case of OVX rats. The MBH was dissected as described above for determination of DA and DOPAC.
The experimental procedures reported here were approved by the Animal Care Committee of the Center of Experimental Pharmacology and Botanials of the National Council for Research of Argentina and carried out in accord with the Declaration of Helsinki.
DA and DOPAC Determination.
DA and its metabolite DOPAC were extracted from the incubation media and tissue as previously described, and protein concentration was determined by the method of Lowry et al. (18). DA and DOPAC were determined by high pressure liquid chromatography with electrochemical detection (19).
Immunocytochemical Studies.
The rats were anesthetized with tribromoethanol and perfused with 4% paraformaldehyde and 0.17% picric acid in PBS (0.05 M, pH 7.4) for 30 min. After perfusion, the hypothalamic area was dissected and post-fixed. Transverse slices (20 μm) were cut in a cryostat and collected on gelatin-coated slides. For colocalization, tyrosine hydroxylase (TH) and CB1, a primary monoclonal antibody raised against TH (Chemicon, 1:1,000 in PBS), and anti-CB1 raised in rabbits (1:100 in PBS, kindly supplied by K. Mackie, University of Washington, Seattle) were used. Immunostaining of CB1 in the hippocampus was used as a positive control.
The slides were examined with a fluorescence microscope (Axioskop 40, Carl Zeiss), and the images were captured using an Axiovision 3.1 photographic system (Zeiss) or with a confocal microscope (Olympus BX61). The atlas of Paxinos and Watson (20) served to identify relevant brain structures.
Results
Male Rats.
Effect of AEA on plasma Prl.
Microinjection of 2 μl of saline icv did not alter plasma Prl for 30 min, after which a precipitous drop occurred, measured at 40 min, followed by a slower decline to a minimum at 80 min and a slight, but not significant rebound at the end of the experiment at 90 min (Fig. 1A). In contrast, AEA decreased plasma Prl significantly by 10 min, and the values continued to decline, reaching a nadir at 50 min. They rebounded slightly at 60 min and then declined gradually to the lowest value of the entire experiment at 90 min. The values in the AEA-injected animals were significantly lower than those of controls at 20, 30, and 90 min and reduced below those of the controls at all time intervals. The area under the curve of PRL secretion after AEA during 90 min was significantly decreased (P < 0.01) as compared with that of control rats injected with saline (Fig. 1B).
Figure 1.

(A) Effect on plasma Prl levels of AEA (20 ng/2 μl) or 2 μl of saline injected icv in male rats. The number of animals per group was seven to nine. *, P < 0.05; **, P < 0.01; vs. control at that time point. (B) Comparison of areas under the curve of Prl secretion during 90 min after the injections. **, P < 0.01 vs. control group.
Effect of AEA injected in vivo on DA concentration in MBH and AP.
At the end of the experiment (90 min) after the icv injection of AEA, the DA concentrations in MBH and AP were increased (P < 0.001 and P < 0.05, respectively) as compared with values in the control group injected with saline (Fig. 2).
Figure 2.

DA content in tissues 90 min after icv injection of AEA (20 ng/2 μl) in male rats. (A) DA content in MBH; ***, P < 0.001 vs. control (six MBH per group). (B) DA content in AP; *, P < 0.05 vs. control (six AP per group).
Effect of AEA on DA release and DOPAC/DA ratio of incubated MBH.
The presence of AEA at a concentration of 10−9 M increased highly significantly (P < 0.01) the release of DA from MBH incubated in vitro for 30 min (Fig. 3A) and decreased the DA tissue content (P < 0.01, Fig. 3B). The DOPAC/DA ratio in the MBH was also increased, indicating an increase in turnover of DA (Fig. 3C). Surprisingly, when the concentration of AEA was increased to 10−7 M, DA release and tissue content, plus the DOPAC/DA ratio, was unaffected in comparison with the values from control MBH (Fig. 3C).
Figure 3.

(A) Effect of AEA in vitro on DA release from MBH during 30 min. One-way ANOVA followed by Newman–Keuls' test (**, P < 0.01 vs. control and AEA 10−7 M groups). (B) Effect of AEA in vitro on DA content in MBH after 30 min. One-way ANOVA followed by Newman–Keuls' test (**, P < 0.01 vs. control and AEA 10−7 M groups). (C) Effect of AEA in vitro on DOPAC/DA ratio in MBH after 30 min. One-way ANOVA followed by Newman–Keuls' test (**, P < 0.01 vs. control and AEA 10−7 M groups).
Female Rats.
Effect of AEA on plasma PRL levels in OVX rats.
Microinjection of AEA (20 ng/2 μl saline) icv into OVX rats did not modify significantly plasma PRL levels in comparison with those of the control group injected with 2 μl of saline icv at any time after injection until the end of the experiment at 150 min (Fig. 4A). Prl concentrations in both groups were very similar, and they reached minimal values between 30 and 90 min followed by a slight return to values similar to the initial levels. The decline in Prl during the first 60 min in the control animals was significant, but the lesser decline in the AEA-injected animals at this time was not significantly different from that of the controls, so there was no overall effect of AEA in the OVX animals (Fig. 4A). The area under the curve of plasma Prl was also unchanged by AEA (Fig. 4B).
Figure 4.

(A) Effect on plasma Prl levels of AEA injected icv (20 ng/2 μl) or 2 μl of saline in OVX rats. The number of animals per group was seven to nine. (B) Comparison of areas under the curve of Prl secretion in OVX rats during 150 min after the injections.
Effect of AEA injected in vivo on DA and DOPAC in OVX rats.
At 150 min after icv injection of AEA, the DA content in the MBH was increased (P < 0.01; Fig. 5A). There was no change in DOPAC content, but the DOPAC/DA ratio was decreased (P < 0.01, Fig. 5 B and C).
Figure 5.

(A) Effect of AEA (20 ng/2 μl) or 2 μl of saline injected icv into OVX rats on DA content in MBH after 150 min. **, P < 0.01 vs. control (Student's t test, six MBH per group). (B) Lack of effect of AEA (20 ng/2 μl) or 2 μl of saline injected icv on DOPAC content in MBH after 150 min. (C) Effect of AEA (20 ng/2 μl) or 2 μl of saline injected icv on DOPAC/DA ratio in MBH after 150 min. **, P < 0.01 vs. control (Student's t test, six MBH per group).
Effect of AEA on plasma Prl levels in OVX-E rats.
Microinjection of AEA icv into OVX-E rats increased plasma Prl levels (Fig. 6A). The increase was present by 30 min, but not significant. The values plateaued between 30 and 90 min, but then rose to reach a maximum at 150 min. The previous icv injection of AM251, the CB1 receptor antagonist, prevented this increase until 90 min. However, after this time, the values rose to equal those in the animals injected only with AEA at 120 and 150 min. The icv injection of AM251 alone decreased plasma Prl levels by 30 min, and values progressively declined to a minimum of 150 min, at which time they were highly significantly lower (P < 0.01) than those of the saline-injected controls.
Figure 6.

(A) Effect on plasma Prl concentrations of AEA (20 ng/2 μl), AM251 (200 ng/2 μl), AEA and AM251, or 2 μl of saline for the control group injected icv in OVX-E rats. The number of animals per group was seven to nine. (B) Comparison of areas under the curve of Prl secretion during 150 min after the injections into the rats in A. One way-ANOVA followed by Newman–Keuls' test. (***, P < 0.001 vs. control group).
The areas under the Prl secretion curves were evaluated statistically (Fig. 6B) and confirmed the conclusions drawn from looking at the individual values. There was a significant increase in Prl area after AEA injection in comparison with the control groups (P < 0.001), and the injection of AM251 before AEA injection prevented this increase (P < 0.001). The injection of the AEA inhibitor, AM251, alone decreased plasma Prl levels (P < 0.001) in comparison with those of the control group.
Effect of AEA on DA and DOPAC in OVX-E rats.
The DA content of MBH 150 min after icv AEA injection was not changed in comparison with that of the control group (Fig. 7A). There was a decrease (P < 0.05) in DOPAC content (Fig. 7B), and the DOPAC/DA ratio was also decreased (P < 0.05, Fig. 7C).
Figure 7.

(A) Effect of AEA (20 ng/2 μl) or 2 μl of saline injected icv on DA content in MBH after 150 min (six MBH per group, Pns). (B) Effect of AEA icv (20 ng/2 μl) or 2 μl of saline injected icv on DOPAC content in MBH after 150 min. *, P < 0.05 vs. control (Student's t test, six MBH per group). (C) Effect of AEA (20 ng/2 μl) or 2 μl of saline injected icv on DOPAC/DA ratio in MBH after 150 min. *, P < 0.05 vs. control (Student's t test; six MBH per group).
Colocalization of CB1 receptors on TH neurons in the hypothalamus.
TH is the rate-limiting enzyme in the synthesis of catecholamines (DA, norepinephrine, and epinephrine). There are few, if any, catecholamine neurons in the basal tuberal region other than DA neurons. Consequently, the TH neurons identified herein are considered to be DAergic neurons. Immunoreactive TH (ir-TH) and ir-CB1 cells were found distributed throughout the hypothalamus. Colocalization of TH and CB1 was observed in the hypothalamic areas, especially in the periventricular hypothalamic nucleus. Staining of CB1 receptors showed a punctated pattern (green; Fig. 8 Middle). TH staining showed a homogeneous distribution in the neuronal cytoplasm (red; Fig. 8 Top). The superposition of the two labels appears in yellow (Fig. 8 Bottom). Indeed, 7 of the TH neurons colocalized with CB1 receptors and 8 did not, whereas of the 10 neurons exhibiting CB1 receptors, 7 colocalized with TH neurons and 3 did not. Thus, nearly half of the TH neurons colocalized with CB1 receptors, and a majority of CB1 neurons localized to TH neurons.
Figure 8.

Colocalization of ir-CB1 receptors and TH cells in the periventricular hypothalamic nucleus, Bregma −1.8 mm, according to the Paxinos–Watson atlas. (Top) ir-TH cells (red) around the third ventricle. (Middle) ir-CB1 receptors (green) in the same area. (Bottom) Overlay of Top and Middle (400 μm). Arrows indicate two examples of colocalization of TH cells with CB1 receptors. There were a total of seven neurons in the section that had CB1 receptors colocalized to TH cells. Eight TH cells did not have CB1 receptors localized to them; asterisks indicate examples. There were 10 neurons that had CB1 receptors, and only three were not colocalized with TH neurons (arrowhead). (Inset) Magnification of two cells containing ir-TH and ir-CB1 receptors.
Discussion
The present results in conscious male rats employing the endogenous cannabinoid, AEA-injected icv confirmed our earlier experiment with THC, the principal active ingredient of marijuana (11). AEA acted rapidly, within 10 min, to suppress Prl release, and the effect persisted for the 90-min duration of the experiment. Prl concentrations declined slowly in saline-injected controls, but the effect was not significant until at 40 min, and the area under the plasma Prl curve was highly significantly decreased below that of control rats in the AEA-injected rats. The initial Prl concentrations in both groups were elevated, probably because of the stress of connection of the catheter and removal of the initial blood sample. Thereafter in the saline-injected rats, the values declined slowly as the response to this initial stress dissipated. At the end of the experiment, there was an increase in DA content both in the MBH and AP, indicating that AEA increased synthesis and release of DA from TIDA neurons. DA was released into the portal vessels, increasing AP DA concentrations that presumably inhibited Prl release. These effects of AEA on DA in the MBH are similar to acute effects of THC on DAergic activity in several other brain areas of the rat (21). The presumed effect of AEA to increase synthesis and release of DA from the MBH was confirmed by incubation of MBH explants in the presence of AEA (10−9 M), as release of DA was increased in the face of an increase in the DOPAC/DA ratio, indicating increased DA turnover. Surprisingly, increasing the concentration of AEA to 10−7 M abolished all of these effects, suggesting that there may be a bell-shaped dose-response curve of this response.
In contrast to the inhibitory effect of AEA on Prl release mediated by increased DA release into the hypophyseal portal veins in male rats, there was little or no overall effect of AEA on plasma Prl concentrations of OVX rats. At 150 min, DA content in the MBH was slightly elevated, but DOPAC values were unchanged, resulting in a decreased DA turnover in AEA rats that may have resulted in decreased release of DA into portal blood that did not alter Prl levels from those in saline-injected OVX rats. Thus, in the OVX rats, AEA decreased DA turnover and yet plasma Prl was not altered.
The results in the OVX-E rats were dramatically different from those in male or OVX rats. Prl was elevated significantly on first measurement at 30 min, remained on a plateau until 90 min, and then rose rapidly to a maximum at 150 min four times greater than the initial concentrations. The CB1 receptor antagonist completely blocked the action of AEA from 30 to 60 min. Thereafter, the values rose at 90 min to equal those in AEA-treated rats at 120 min, indicating that the action of the CB1 receptor blocker had dissipated during this time and could no longer antagonize the effect of AEA.
When the CB1 antagonist was injected alone, it lowered plasma Prl below the concentrations of control OVX-E rats within 30 min, and the difference became greater with time and maximal by the end of the experiment at 150 min, indicating that endogenous AEA is acting as a neurotransmitter maintaining plasma Prl concentration in the OVX-E animals. Because Prl release was suppressed by the CB1 receptor antagonist for the duration of the experiment, the results indicate that it was still active at 150 min to block the effect of endogenously released AEA, but not that of the combined action of exogenous AEA plus endogenous AEA by the end of the experiment. These findings support the hypothesis that estrogen is inducing the release of AEA from AEA neurons in the MBH, which acts on AEA receptors on TIDA neurons to inhibit the release of DA, thereby removing DA inhibitory tone on Prl release that results in increased Prl release from the lactotropes of the AP gland. This hypothesis is supported by our finding of colocalization of CB1 receptors on the cell bodies of periventricular TH-containing neurons, presumably DA neurons that are known to project not only to the intermediate lobe of the pituitary but also to the median eminence, where the released DA is transported to the AP by the portal vessels (22), there to inhibit Prl release. The DA concentration in the MBH was increased above that in OVX rats, but the turnover of DA estimated by the DOPAC/DA ratio was decreased; these results are consistent with removal of DA inhibitory tone on the lactotropes.
Changes in CB1 receptor affinity and/or density in some brain areas related to estrogenic status have been reported (17). These could be involved in the different responses observed in the present study. It was shown previously that administration of estrogen for a week decreased TH mRNA content in TIDA neurons and also lowered TH activity (23). On the other hand, the colocalization of CB1 receptors with dopaminergic pathways in other regions of the brain suggests functional interactions of DA with AEA in those regions similar to those in the hypothalamus (24).
Because Prl release from the AP is controlled by many factors, it is likely that some of these may also have played a role in the results. For example, oxytocin is a potent Prl-releasing factor, and γ-aminobutyric acid (GABA) is a Prl-inhibitory factor. It was reported recently that CB1 is expressed in a subpopulation of GABAergic interneurons that also expresses cholecystokynin in the hippocampus and neocortex (25). Therefore, it is likely that there is an interaction between AEA and γ-aminobutyric acid (26). Furthermore, there are at least 14 factors that alter Prl release from the AP, 4 inhibitors and 10 stimulators of the release of this hormone (12). In view of the ubiquitous presence of CB1 receptors on neurons in the hypothalamus and elsewhere in the CNS and effects of E2 on many neuronal systems, it is clear that more research is necessary to understand fully the roles of AEA and E2 in control of Prl release.
In conclusion, the results of this research clearly show that AEA inhibits Prl release from the pituitary of male rats by action on CB1 receptors on dopaminergic neurons in the MBH, activating DA release into the portal vessels that inhibits Prl release (Fig. 9 Top). In OVX rats, the action changed and there was little or no effect of AEA on Prl release, even though DA turnover was decreased, which should have decreased DA inhibitory control of Prl release (Fig. 9 Middle). Treatment of OVX rats with estrogen increased Prl levels and reversed the inhibitory effect of AEA on Prl release, possibly in part by decreasing DA inhibitory tone on the lactotropes. Endogenous release of AEA acting through CB1 receptors on DA, and probably other neurons controlling Prl release as well, plays a physiological stimulatory role as a synaptic transmitter in estrogen-induced Prl release because the CB1 receptor blocker decreased plasma Prl in the OVX-E-treated rats (Fig. 9 Bottom).
Figure 9.
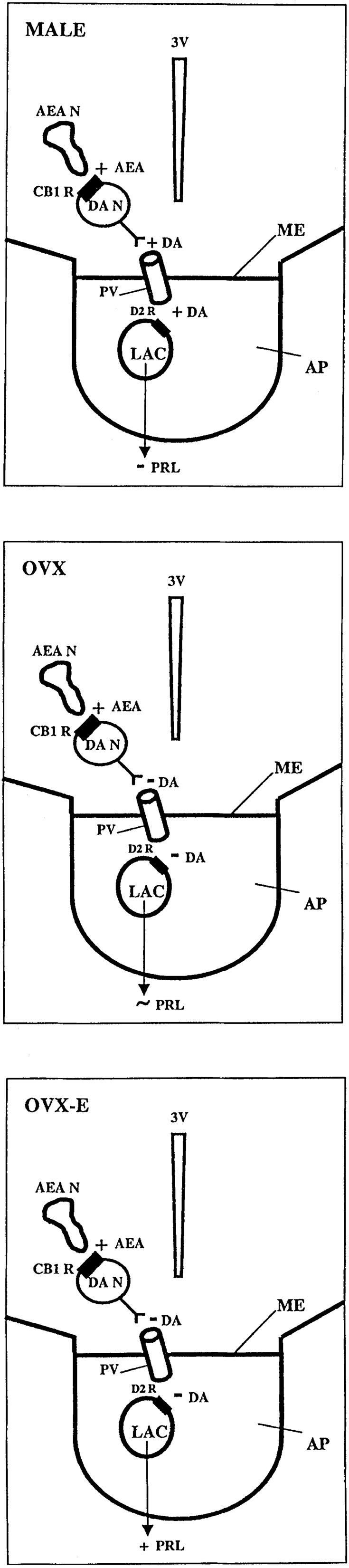
Diagrammatic representation of the differential control of Prl release by AEA in male, OVX, and OVX-E rats. As can be seen, AEA acts on CB1 receptors on TIDA neurons to increase release of DA into portal vessels, which reaches the lactotropes to inhibit Prl release by action on D2 DA receptors in male rats (A), whereas in OVX animals, AEA also acts on the DA neurons, but this time the action on the CB1 receptor causes inhibition of DA release slightly decreasing inhibitory DA control over Prl release, but with little effect on Prl release. In OVX-E animals, AEA neurons activate CB1 receptors on DA neurons to inhibit DA release, resulting in a reduction in DA inhibitory tone on the lactotrope; however, because the effect on Prl is not matched by a larger decrease in DA tone, other transmitters are probably partially responsible for the elevated Prl in the OVX-E animals under resting or AEA-stimulated conditions. The inhibitor of AEA action lowered basal Prl and blocked the action of AEA in the OVX-E animals, indicating that AEA is a synaptic transmitter that acts on CB1 receptors to increase plasma Prl in OVX-E rats. 3V, third ventricle; AEAN, AEA neuron; CB1 R, CB1 receptors; DAN, dopaminergic neuron; PV, hypophyseal portal veins; ME, median eminence; D2 R, DA type 2 receptors; LAC, lactotropes; +, increase; −, decrease; ∼, little or no change.
Acknowledgments
We thank Dr. Mario Ravaglia for his photographic assistance, Ana Inés Casella for her administrative help, and Judy Scott and Nicole Mestayer for their excellent secretarial assistance. This work was supported by Ministerio de Salud: Beca Ramón Carrillo-Arturo Oñativia, Agencia Nacional de Promoción Científica y Tecnológica, Argentina Grant PICT 99#5-6117, and National Institutes of Mental Health Grant MH51853 (to S.M.M.).
Abbreviations
- THC
Δ9-tetrahydrocannabinol
- AEA
anandamide
- Prl
prolactin
- OVX
ovariectomized
- OVX-E
OVX, estrogen-primed
- AP
anterior pituitary
- DA
dopamine
- TIDA
tuberoinfundibular DAergic
- icv
intracerebroventricularly
- MBH
medial basal hypothalamus
- DOPAC
dihydroxyphenylacetic acid
- TH
tyrosine hydroxylase
- ir
immunoreactive
References
- 1.Gaoni Y, Mechoulam R. J Am Chem Soc. 1964;86:1646–1647. [Google Scholar]
- 2.Breivogel C S, Childers S T. Neurobiol Dis. 1998;5:417–431. doi: 10.1006/nbdi.1998.0229. [DOI] [PubMed] [Google Scholar]
- 3.Howlett A C, Fleming R M. Mol Pharmacol. 1984;26:532–538. [PubMed] [Google Scholar]
- 4.Devane W A, Dysarz F A, Johnson M, Melvin L S, Howlett A C. Mol Pharmacol. 1988;34:605–613. [PubMed] [Google Scholar]
- 5.Herkenham M, Lynn A B, Little M D, Johnson M R, Melvin L S, De Costa B R, Rice K C. Proc Natl Acad Sci USA. 1990;87:1932–1936. doi: 10.1073/pnas.87.5.1932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pertwee R G. In: Cannabinoid Receptors. Pertwee R G, editor. London: Academic; 1995. pp. 1–34. [Google Scholar]
- 7.Munro S, Thomas K L, Abu-Shaar M. Nature. 1993;365:61–65. doi: 10.1038/365061a0. [DOI] [PubMed] [Google Scholar]
- 8.Devane W A, Hanus L, Breuer A, Pertwee R G, Stevenson L A, Griffin G, Gibson D, Mandelbaum A, Etinger A, Mechoulam R. Science. 1992;258:1946–1949. doi: 10.1126/science.1470919. [DOI] [PubMed] [Google Scholar]
- 9.Mechoulam R, Ben-Shabah S, Hanus L, Ligumsky M, Kaminski N E, Schatz A R, Gopher A, Almog S, Martin B R, Compton D R, et al. Biochem Pharmacol. 1995;50:83–90. doi: 10.1016/0006-2952(95)00109-d. [DOI] [PubMed] [Google Scholar]
- 10.Sugiura T, Kondo S, Sukagawa A, Nakane S, Shinoda A, Itoh K, Yamashita A, Waku K. Biochem Biophys Res Commun. 1995;215:89–97. doi: 10.1006/bbrc.1995.2437. [DOI] [PubMed] [Google Scholar]
- 11.Rettori V, Wenger T, Snyder G, Dalterio S, McCann S M. Neuroendocrinology. 1988;47:498–503. doi: 10.1159/000124961. [DOI] [PubMed] [Google Scholar]
- 12.Neill J D, Nagy G M. The Physiology of Reproduction. New York: Raven; 1994. pp. 1148–1453. [Google Scholar]
- 13.Jones E E, Naftolin F. Brain Res. 1990;510:84–91. doi: 10.1016/0006-8993(90)90730-y. [DOI] [PubMed] [Google Scholar]
- 14.Tanda G, Pontieri F E, Chiara G. Science. 1997;276:2048–2050. doi: 10.1126/science.276.5321.2048. [DOI] [PubMed] [Google Scholar]
- 15.Rodriguez de Fonseca F, Cebeira M, Ramos J A, Martin M, Fernandez-Ruiz J J. Life Sci. 1994;54:159–170. doi: 10.1016/0024-3205(94)00585-0. [DOI] [PubMed] [Google Scholar]
- 16.Pellegrino L J, Pellegrino A S, Ashman A J. A Stereotaxic Atlas of the Rat Brain. 2nd Ed. New York: Plenum; 1979. [Google Scholar]
- 17.Harms P, Ojeda S R. J Appl Physiol. 1974;36:391–392. doi: 10.1152/jappl.1974.36.3.391. [DOI] [PubMed] [Google Scholar]
- 18.Lowry O, Rosenbrough A, Farr A, Randall R. J Biol Chem. 1951;193:265–275. [PubMed] [Google Scholar]
- 19.De Laurentiis A, Pisera D, Caruso C, Candolfi M, Mohn C, Rettori V, Seilicovich A. Neuroimmunomodulation. 2002;10:30–39. doi: 10.1159/000064412. [DOI] [PubMed] [Google Scholar]
- 20.Paxinos G, Watson C. The Rat Brain in Stereotaxic Coordinates. San Diego: Academic; 1997. [DOI] [PubMed] [Google Scholar]
- 21.Rodriguez de Fonseca F, Fernadez-Ruiz J J, Murphy L L, Cebeira M, Steger R W, Bartke A, Ramos J A. Pharmacol Biochem Behav. 1992;42:269–275. doi: 10.1016/0091-3057(92)90526-l. [DOI] [PubMed] [Google Scholar]
- 22.Freeman M E, Kanycska B, Lerant A, Nagy G. Physiol Rev. 2000;80:1523–1631. doi: 10.1152/physrev.2000.80.4.1523. [DOI] [PubMed] [Google Scholar]
- 23.Arbogast L A, Voogt J L. Neuroendocrinology. 1993;58:501–510. doi: 10.1159/000126583. [DOI] [PubMed] [Google Scholar]
- 24.DeMaria J E, Livingstone J D, Freeman M E. Brain Res. 2000;879:139–147. doi: 10.1016/s0006-8993(00)02763-3. [DOI] [PubMed] [Google Scholar]
- 25.Pertwee R G. Pharmacol Ther. 1997;74:129–180. doi: 10.1016/s0163-7258(97)82001-3. [DOI] [PubMed] [Google Scholar]
- 26.Wilson R I, Nicoll R A. Science. 2002;296:678–682. doi: 10.1126/science.1063545. [DOI] [PubMed] [Google Scholar]