

Abstract
Plexins are receptors for the axonal guidance molecules known as semaphorins, and the semaphorin 4D (Sema4D) receptor plexin-B1 induces repulsive responses by functioning as an R-Ras GTPase-activating protein (GAP). Here we characterized the downstream signalling of plexin-B1-mediated R-Ras GAP activity, inducing growth cone collapse. Sema4D suppressed R-Ras activity in hippocampal neurons, in parallel with dephosphorylation of Akt and activation of glycogen synthase kinase (GSK)-3β. Ectopic expression of the constitutively active mutant of Akt or treatment with GSK-3 inhibitors suppressed the Sema4D-induced growth cone collapse. Constitutive activation of phosphatidylinositol-3-OH kinase (PI(3)K), an upstream kinase of Akt and GSK-3β, also blocked the growth cone collapse. The R-Ras GAP activity was necessary for plexin-B1-induced dephosphorylation of Akt and activation of GSK-3β and was also required for phosphorylation of a downstream kinase of GSK-3β, collapsin response mediator protein-2. Plexin-A1 also induced dephosphorylation of Akt and GSK-3β through its R-Ras GAP activity. We conclude that plexin-B1 inactivates PI(3)K and dephosphorylates Akt and GSK-3β through R-Ras GAP activity, inducing growth cone collapse.
Keywords: plexin, semaphorin, R-Ras GAP, PI(3)K, Akt, GSK-3β
Introduction
Semaphorins are a large family of secreted and transmembrane molecules that function as repulsive axon guidance factors (Kolodkin et al, 1993). Plexin-B1 has been identified as a receptor for semaphorin 4D (Sema4D; Tamagnone et al, 1999). Signalling pathways of plexin-B1 have recently been extensively studied, and recent studies indicate that plexin-B1 forms a complex with PDZ-Rho guanine nucleotide exchange factor (GEF)/leukaemia-associated RhoGEF (LARG) through its carboxy-terminal PDZ-domain-binding motif (Swiercz et al, 2002). Sema4D induces growth cone collapse in hippocampal neurons (Swiercz et al, 2002; Oinuma et al, 2004a), and this action is, in part, mediated by RhoGEF-mediated RhoA activation. However, PDZ-domain-binding motif is restricted to the plexin-B subfamily. The small GTPase Rnd1, a constitutively active Rho family GTPase (Nobes et al, 1998), interacts directly with the cytoplasmic domain of plexin-B1 (Oinuma et al, 2003). We have recently shown that plexin-B1 functions as an R-Ras GTPase-activating protein (GAP) and directly and specifically downregulates R-Ras activity in response to Sema4D, inducing growth cone collapse in cultured hippocampal neurons. We have also shown that the expression of R-Ras GAP activity of plexin-B1 requires Rnd1 association with the receptor. Furthermore, R-Ras GAP activity of plexin-A1/Rnd1 complex has been shown to be required for Sema3A-induced repulsive response (Oinuma et al, 2004a; Toyofuku et al, 2005).
It has recently been demonstrated that activation of glycogen synthase kinase (GSK)-3β is required for Sema3A-induced growth cone collapse (Eickholt et al, 2002; Uchida et al, 2005) and that the activation of GSK-3β by Sema3A requires the activity of PTEN (phosphatase and tensin homologue deleted on chromosome ten; (Chadborn et al, 2005). GSK-3β has a high basal kinase activity, and its kinase activity is inhibited by phosphorylation at Ser 9. Inhibition of GSK-3β activity in neuronal cell lines increases the size of axonal growth cones (Owen & Gordon-Weeks, 2003). Collapsin response mediator protein 2 (CRMP2) is a microtubule-binding protein that promotes microtubule polymerization and stabilization. GSK-3β phosphorylates CRMP2 to suppress its ability to bind to microtubules (Brown et al, 2004; Cole et al, 2004; Yoshimura et al, 2005). Thus, activation of GSK-3β leads to inhibition of microtubule polymerization and stabilization, thereby inhibiting axonal elongation (Zumbrunn et al, 2001; Fukata et al, 2002). GSK-3β is inactivated by a phosphatidylinositol-3-OH kinase (PI(3)K)-dependent signalling (Cantley, 2002). PI(3)K is the predominant effector for R-Ras (Marte et al, 1997; Suire et al, 2002). In this study, we characterized the downstream signalling pathway of Sema4D/plexin-B1-mediated R-Ras GAP activity for growth cone collapse, and showed that Sema4D induces growth cone collapse by R-Ras GAP-mediated inactivation of PI(3)K and activation GSK-3β.
Results
Sema4D activates GSK-3β and phosphorylates CRMP2
Sema4D induces growth cone collapse in rat hippocampal neurons (Swiercz et al, 2002). We examined the effect of Sema4D stimulation on endogenous R-Ras activity in cultured hippocampal neurons by using a pull-down assay with the glutathione S-transferase (GST)-fused Ras-binding domain of c-Raf-1, which selectively isolates active R-Ras (van Triest et al, 2001). R-Ras activity was suppressed by Sema4D stimulation (Fig 1A), and Sema4D stimulation caused a marked decrease in phosphorylated (p)-Akt (Fig 1B) and p-GSK-3β, a downstream substrate of Akt that is activated by dephosphorylation (Fig 1C). Sema4D also induced phosphorylation of CRMP2 at Thr 514, the specific site for GSK-3β-mediated phosphorylation (Fig 1D).
Figure 1.
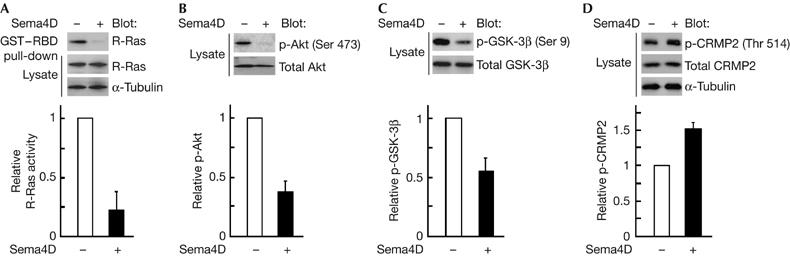
Sema4D suppresses R-Ras activity, dephosphorylates Akt and GSK-3β and phosphorylates CRMP2 in cultured hippocampal neurons. Neurons dissociated from rat embryos at embryonic day 18.5 were stimulated at 3 days in vitro (d.i.v.) with Sema4D for 1.5 h. (A) Activity of R-Ras with or without Sema4D stimulation. GTP-bound R-Ras isolated with GST–RBD was detected with an anti-R-Ras antibody. Relative activity of R-Ras was determined by the amount of R-Ras bound to GST–RBD normalized to the amount of R-Ras in cell lysates analysed by National Institutes of Health Image software. (B–D) Analysis of phosphorylated (p)-Akt, p-GSK-3β and p-CRMP2. Cell lysates were analysed by immunoblot analysis with the phospho-specific antibodies against Akt (Ser 473; B), GSK-3β (Ser 9; C) and CRMP2 (Thr 514; D). Results are the means±s.e.m. of three independent experiments. CRMP2, collapsin response mediator protein 2; GSK-3β, glycogen synthase kinase-3β; GST–RBD, glutathione S-transferase-fused Ras-binding domain; Sema4D, semaphorin 4D.
GSK-3β activation is required for Sema4D action
We next examined whether Sema4D-induced dephosphorylation of Akt and GSK-3β is involved in Sema4D-induced growth cone collapse in cultured hippocampal neurons. Sema4D-induced growth cone collapse was blocked by expression of R-Ras-QL, a constitutively active form of R-Ras, and by expression of myr-Akt, a constitutively active Akt mutant (Fig 2A). Constitutive activation of PI(3)K, an upstream kinase of Akt, by expression of p110α-CAAX (Berrier et al, 2000) also blocked the growth cone collapse. Furthermore, pretreatment with the GSK-3 inhibitors SB-216763 (100 μM) or LiCl (20 mM) also inhibited the Sema4D-induced growth cone collapse (Fig 2B), which suggests that inactivation of PI(3)K and Akt and activation of GSK-3β are required for Sema4D-induced growth cone collapse.
Figure 2.
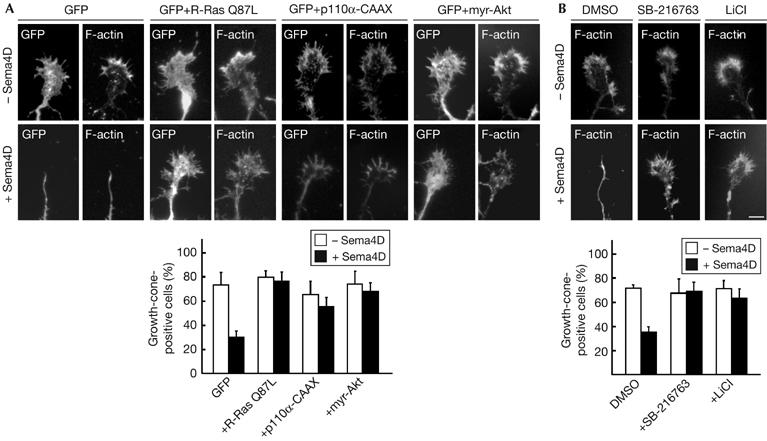
Constitutively active mutants of R-Ras, PI(3)K, Akt, and GSK-3 inhibitors suppress the Sema4D-induced growth cone collapse. (A) Inhibition of Sema4D-induced growth cone collapse by constitutive activation of R-Ras, PI(3)K and Akt. Neurons at 2 days in vitro (d.i.v.) were transfected with the indicated plasmids, and were fixed at 3 d.i.v. after stimulation with Sema4D for 1.5 h. F-actin was stained with rhodamine-conjugated phalloidin to visualize the lamellipodia and filopodia. (B) Inhibition of Sema4D-induced growth cone collapse by GSK-3 inhibitors. Neurons pretreated with inhibitors for GSK-3, SB-216763 and LiCl were used in the collapse assay. The results are the means±s.e.m. of three independent experiments in which at least 20 cells were counted. Scale bar, 10 μm. DMSO, dimethyl sulphoxide; GFP, green fluorescent protein; GSK-3, glycogen synthase kinase-3; PI(3)K, phosphatidylinositol-3-OH kinase; Sema4D, semaphorin 4D.
R-Ras GAP regulates Akt/GSK-3β/CRMP2 phosphorylation
COS-7 cells transfected with wild-type plexin-B1 and Rnd1, and the time course of the phosphorylation of Akt and GSK-3β after Sema4D stimulation were examined. Sema4D induced dephosphorylation of Akt and GSK-3β, the level reaching a minimum in 3 min (Fig 3A). This time course was correlated with morphological changes, and Sema4D-treated COS-7 cells showed transient reduction in cell area in 3 min (supplementary Fig 1 online). Sema4D-induced dephosphorylation of Akt or GSK-3β was not observed in the absence of Rnd1 (Fig 3B). We have recently reported that the extracellular-domain-deleted plexin-B1, plexin-B1Δect, shows constitutive, ligand-independent activity of R-Ras GAP (Oinuma et al, 2004b). In COS-7 cells, plexin-B1Δect decreased p-Akt and p-GSK-3β in the presence of Rnd1, but not in the absence of Rnd1 (Fig 3C). We further examined the effect of two mutants of plexin-B1Δect: plexin-B1Δect-GGA, lacking Rnd1 interacting activity, and plexin-B1Δect-RA, lacking R-Ras GAP activity. Both mutants failed to decrease p-Akt or p-GSK-3β. In addition, expression of R-Ras-QL antagonized the plexin-B1Δect/Rnd1-mediated decrease in p-Akt and p-GSK-3β (Fig 3D). Rnd1 interaction and R-Ras GAP activity of plexin-B1 were also required for phosphorylation of CRMP2 (Fig 3E). These results indicate that plexin-B1 dephosphorylates Akt and GSK-3β, and phosphorylates CRMP2 through R-Ras GAP activity. It has been reported that the Sema4D-mediated growth cone collapse requires PDZ-RhoGEF-mediated RhoA activation (Swiercz et al, 2002). However, inhibition of RhoA signalling by deletion of the PDZ-domain-binding motif of plexin-B1Δect had no effect on the plexin-B1Δect/Rnd1-mediated decrease in p-Akt and p-GSK-3β (Fig 3F). The extracellular-domain-deleted plexin-A1, plexin-A1Δect, is known to act as a constitutively active receptor (Takahashi & Strittmatter, 2001). Expression of plexin-A1Δect with Rnd1 caused a decrease in the level of both p-Akt and p-GSK-3β in COS-7 cells, and this effect was not observed with plexin-A1Δect-RA or plexin-A1Δect-GGA (Fig 3G). We also confirmed the involvement of endogenous Rnd1 in Sema3A- and Sema4D-mediated dephosphorylation of Akt and GSK-3β in cultured hippocampal neurons by expression of Rnd1-specific short interfering RNA (Oinuma et al, 2004a) using nucleofection technology (supplementary Fig 2 online). In addition, overexpression of the myristoylated GAP domain of R-RasGAP, which shows specific GAP activity towards R-Ras (Yamamoto et al, 1995), was sufficient for decreasing p-Akt and p-GSK-3β in COS-7 cells (Fig 3G). These results indicate that Rnd1-bound plexins show R-Ras GAP activity through their conserved R-Ras GAP domains and regulate phosphorylation of Akt and GSK-3β.
Figure 3.
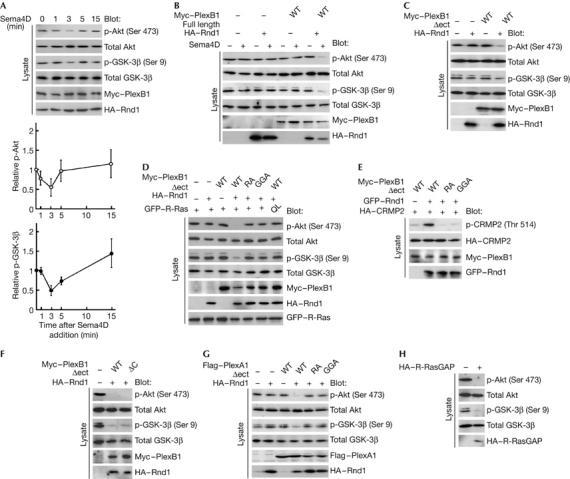
Sema4D dephosphorylates Akt and GSK-3β and phosphorylates CRMP2 through the conserved R-Ras GAP domain within plexin-B1. (A) Time course of Sema4D-induced dephosphorylation of Akt and GSK-3β. COS-7 cells expressing full-length plexin-B1 and Rnd1 were stimulated with Sema4D for the indicated times. The cell lysates at the indicated times were analysed by immunoblot analysis with the phospho-specific antibodies against Akt and GSK-3β. (B) Requirement of Rnd1 in Sema4D-induced dephosphorylation of Akt and GSK-3β. COS-7 cells expressing the indicated plasmids were stimulated with Sema4D for 3 min, and the cell lysates were analysed by immunoblot analysis with phospho-specific antibodies against Akt and GSK-3β. (C–F) Effects of plexin-B1Δect mutants on phosphorylation of Akt, GSK-3β and CRMP2. Lysates from COS-7 cells expressing the indicated plasmids were analysed by immunoblot analysis with phospho-specific antibodies against Akt, GSK-3β and CRMP2. (G) Plexin-A1-mediated dephosphorylation of Akt and GSK-3β. Lysates from COS-7 cells expressing plexin-A1Δect mutants and Rnd1 were analysed by immunoblot analysis with phospho-specific antibodies against Akt and GSK-3β. (H) Dephosphorylation of Akt and GSK-3β by suppression of R-Ras activity. Lysates from COS-7 cells expressing myr-R-RasGAP were analysed by immunoblot analysis with phospho-specific antibodies against Akt and GSK-3β. The results shown are representative of two or three experiments. CRMP2, collapsin response mediator protein 2; GAP, GTPase-activating protein; GFP, green fluorescent protein; GSK-3β, glycogen synthase kinase-3β; HA, haemagglutinin; Sema4D, semaphorin 4D; WT, wild type.
R-Ras GAP–GSK-3β induces collapse
We next confirmed whether suppression of the downstream molecules of R-Ras is sufficient to induce growth cone collapse in cultured hippocampal neurons. As shown in Fig 4, ectopic expression of myr-R-RasGAP, an effector loop mutant of R-Ras that impairs the ability of R-Ras to activate PI(3)K (R-Ras D64A; Oertli et al, 2000), a kinase-dead form of p110α or a constitutively active mutant of GSK-3β (GSK-3β S9A; Yoshimura et al, 2005), by itself, induced growth cone collapse. In addition, treatment with a pharmacological PI(3)K inhibitor, LY294002, promoted growth cone collapse. These results demonstrate that inhibition of R-Ras-mediated PI(3)K activation or constitutive activation of GSK-3β is sufficient to induce growth cone collapse in cultured hippocampal neurons.
Figure 4.
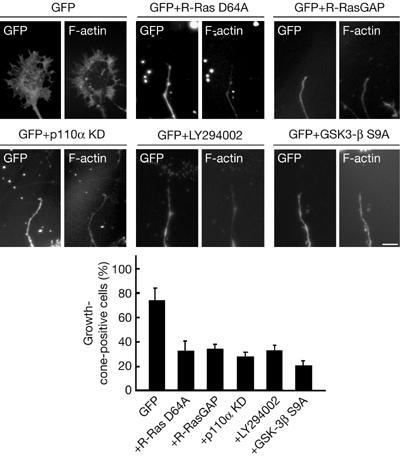
Inhibition of R-Ras-mediated PI(3)K activation or constitutive activation of GSK-3β is sufficient to induce growth cone collapse. Neurons were transfected with the indicated plasmids at 2 days in vitro (d.i.v.) and neurons at 3 days in vitro (d.i.v.) were fixed. Alternatively, cells were treated with LY294002 for 1.5 h before fixation. F-actin was stained with rhodamine-conjugated phalloidin to visualize the lamellipodia and filopodia. The results are the means±s.e.m. of three independent experiments in which at least 20 cells were counted. Scale bar, 10 μm. GAP, GTPase-activating protein; GFP, green fluorescent protein; GSK-3β, glycogen synthase kinase-3β; KD, kinase dead.
Discussion
We have recently reported that Sema4D receptor plexin-B1 induces growth cone collapse by functioning as a GAP for R-Ras, a member of the Ras family implicated in neurite outgrowth. In this study, we observed the downstream signalling of plexin-B1-mediated R-Ras GAP activity and showed that plexin-B1 inactivates PI(3)K and dephosphorylates Akt and GSK-3β, and induces growth cone collapse through the R-Ras GAP activity.
Plexin-B1 activates RhoA through direct association with PDZ-RhoGEF/LARG by means of its C-terminal PDZ-domain-binding motif, and this activation is in part involved in the repulsive functions of plexin-B1 (Swiercz et al, 2002). However, a plexin-B1 mutant, plexin-B1-ΔC, lacking the PDZ-domain-binding motif, shows a decrease in p-Akt and p-GSK-3β, indicating that the RhoA activation signal of plexin-B1 is not involved in dephosphorylation of Akt and GSK-3β. Conversely, plexin-B1-RA, lacking the catalytic activity of R-Ras GAP, does not inhibit dephosphorylation of Akt and GSK-3β. In addition to plexin-B1, plexin-A1 also functions as a GAP towards R-Ras (Toyofuku et al, 2005), and we showed that both plexin-B1- and plexin-A1-mediated dephosphorylation of Akt and GSK-3β required Rnd1 binding to the receptor. It has been reported that mere overexpression of the active form of plexin-A1, plexin-A1Δect, induces the collapse of COS-7 cells (Takahashi & Strittmatter, 2001). Moreover, another report has shown that Sema4D elicits the collapse of COS-7 cells transfected with plexin-B1 only (Barberis et al, 2004). In our experiments, prolonged expression of plexin-A1Δect (>24 h) in COS-7 cells could indeed induce a partial collapse response (data not shown). Therefore, we suppose that a functional response might be seen in COS-7 cells even in the absence of Rnd1 overexpression, although with a different activation threshold. The lipid phosphatase PTEN has been recently shown to be required for Sema3A-mediated dephosphorylation of Akt and GSK-3β (Chadborn et al, 2005). We speculate that inhibition of PI(3)K activation by R-Ras GAP activity and dephosphorylation of phosphatidylinositol-3-phosphate (PIP3) by elevated phosphatase activity of PTEN act together to induce plexin-mediated repulsive responses. Sema3A also induces phosphorylation of CRMP2, a downstream substrate of GSK-3β (Cole et al, 2004; Uchida et al, 2005). CRMP2 binds to tubulin heterodimers, promoting microtubule assembly (Fukata et al, 2002), and phosphorylation of CRMP2 by GSK-3β lowers its activity in tubulin interaction. We have shown that activation of GSK-3β by Sema4D/plexin-B1-mediated R-Ras GAP activity regulates phosphorylation of CRMP2. Phosphorylation of microtubule interacting proteins, CRMP2 and adenomatous polyposis coli (APC), by active GSK-3β inhibits microtubule polymerization and stabilization, thereby suppressing axonal elongation (Zumbrunn et al, 2001). It is inferred that R-Ras GAP activity of plexin regulates microtubule dynamics through phosphorylation of CRMP2 and APC.
PI(3)K has emerged as the predominant effector for R-Ras, and R-Ras is a more potent activator of PI(3)K than Ras (Marte et al, 1997; Suire et al, 2002). Activated R-Ras induces increased cell adhesion and cell migration by activating β1 integrins (Zhang et al, 1996), and R-Ras-induced cell migration is sensitive to PI(3)K inhibitors (Keely et al, 1999). In addition, GSK-3β is a multi-tasking kinase involved in a variety of cellular responses including directed cell migration (Etienne-Manneville & Hall, 2003). Further work is required to delineate the role of inhibition of PI(3)K and Akt, and activation of GSK-3β, downstream of plexin-mediated R-Ras GAP activity in a wide range of semaphorin-mediated cellular responses, such as neurite remodelling and cell migration.
Methods
DNA constructs. Mouse plexin-A1 complementary DNA and human plexin-B1 cDNA were from H. Fujisawa (Nagoya University, Nagoya, Japan) and L. Tamagnone (Torino University, Torino, Italy), respectively. Amino-terminal Flag-tagged p110α constructs were from T. Katada (Tokyo University, Tokyo, Japan). Details on other DNA constructs used in this study are described in the supplementary information online.
Antibodies and reagents. Antibodies used in this study are described in the supplementary information online. Inhibitors for GSK-3, SB-216763 (Biomol, Plymouth Meeting, PA, USA) and LiCl (Wako, Osaka, Japan) were dissolved in dimethyl sulphoxide (DMSO). Neurons were incubated for 1 day at the concentration of 100 μM (SB-216763) or for 1.5 h at 20 mM (LiCl) before Sema4D stimulation. LY294002 (Calbiochem, San Diego, CA, USA) dissolved in DMSO was added to the culture medium 1.5 h before fixation at 100 μM. In every experiment, application of DMSO for the corresponding incubation times was performed as control experiments to exclude the effect of the solvent. A soluble form of Sema4D fused to human IgG1-Fc was from H. Kikutani (Osaka University, Osaka, Japan), and Sema4D stimulation was performed by replacing the culture medium with Sema4D-containing conditioned medium, which contains about 1.3 nM Sema4D-Fc. We used different exposure times of Sema4D in COS-7 cells (3 min) and in hippocampal neurons (1.5 h) for the analysis of changes in Akt and GSK-3β phosphorylation, as COS-7 cells showed quick reduction in cell area in 3 min (supplementary Fig 2A online), whereas 1.5 h was required for full collapse of growth cones (data not shown) and short time (∼15 min) exposure to Sema4D had no effect on Akt and GSK-3β phosphorylation (supplementary Fig 2B online). The soluble form of Sema3A was a generous gift from Y. Goshima (Yokohama City University) and Sema3A stimulation was performed by replacing the culture medium with Sema3A-containing medium, which contains 2 nM Sema3A. Sema3A and Sema4D were purified by using affinity columns, eluted in glycine/HCl solution and dialysed in PBS. An estimate of Sema3A and Sema4D concentration and purity was performed by SDS–polyacrylamide gel electrophoresis and Coomassie brilliant blue staining (comparing with BSA standards) or by immunoblotting with an anti-Fc antibody (and comparing with recombinant Fc protein standards). Although the eluted solution was not 100% pure, these methods nevertheless allowed us to quantify the amount of Sema3A and Sema4D present.
Cell culture and transfection. COS-7 cells were cultured and transfected as described previously (Oinuma et al, 2004a). Primary hippocampal neurons were dissociated from rats at embryonic day 18.5 and were plated onto poly-D-lysine-coated and laminin-coated (from Sigma, St Louis, MO, USA) coverslips (circular, 13 mm in diameter) or plastic dishes (60 mm in diameter), at a density of 3.5 × 104 cells/cm2. After 2 days in culture, the medium was changed to OPTI-MEM (Invitrogen, Carlsbad, CA, USA) supplemented with 2% B-27 (Invitrogen), and the neurons were transfected using Lipofectamine 2000. After one more day in culture (3 d.i.v.), neurons were stimulated with Sema4D-Fc, fixed with 4% paraformaldehyde in PBS and processed for immunohistochemistry. Tips of the longest neurites were analysed, and growth-cone-positive cells were defined as those that have lamellipodia and filopodia. Co-transfection efficiency of each plasmid and green fluorescent protein was greater than 90% as shown by immunostaining (data not shown).
Measurement of R-Ras activity. Measurement of R-Ras activity in cells was performed as described previously (Oinuma et al, 2004a), and details are given in the supplementary information online.
Detection of Akt, GSK-3β and CRMP2 phosphorylation. For analysis of p-Akt and p-GSK-3β, COS-7 cells were maintained in DMEM with 5% fetal bovine serum for 16 h, and for analysis of p-CRMP2, cells were maintained in DMEM with 10% fetal bovine serum for 2 days after transfection. Cells were directly lysed on dishes with 1 × Laemmli sample buffer and analysed by SDS–polyacrylamide gel electrophoresis and immunoblotting, using phospho-specific antibodies against Akt, GSK-3β and CRMP2. For analysis of p-CRMP2, cells were pretreated with 0.2% Triton X-100 on ice for 10 min before lysis.
Supplementary information is available at EMBO reports online (http://www.emboreports.org).
Supplementary Material
supplementary information
Acknowledgments
We thank L. Tamagnone, H. Fujisawa, Y. Goshima, H. Kikutani and T. Katada for providing reagents. We also thank T. Uemura (Kyoto University, Kyoto, Japan) for experimental help with the Nucleofector® technology. This work was supported in part by grants-in-aid for scientific research from the Ministry of Education, Science, Sports, and Culture of Japan.
References
- Barberis D, Artigiani S, Casazza A, Corso S, Giordano C, Love CA, Jones EY, Comoglio PM, Tamagnone L (2004) Plexin signaling hampers integrin-based adhesion, leading to Rho-kinase independent cell rounding, and inhibiting lamellipodia extension and cell motility. FASEB J 18: 592–594 [DOI] [PubMed] [Google Scholar]
- Berrier AL, Mastrangelo AM, Downward J, Ginsberg M, LaFlamme SE (2000) Activated R-Ras, Rac1, PI3-kinase and PKCɛ can each restore cell spreading inhibited by isolated integrin β1 cytoplasmic domains. J Cell Biol 151: 1549–1560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown M, Jacobs T, Eickholt B, Ferrari G, Teo M, Monfries C, Qi RZ, Leung T, Lim L, Hall C (2004) α2-Chimaerin, cyclin-dependent kinase 5/p35, and its target collapsin response mediator protein-2 are essential components in semaphorin 3A-induced growth-cone collapse. J Neurosci 24: 8994–9004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantley LC (2002) The phosphoinositide 3-kinase pathway. Science 296: 1655–1657 [DOI] [PubMed] [Google Scholar]
- Chadborn NH, Ahmed AI, Holt MR, Prinjha R, Dunn GA, Jones GE, Eickholt BJ (2005) PTEN couples Sema3A signaling to growth cone collapse. J Cell Sci 119: 951–957 [DOI] [PubMed] [Google Scholar]
- Cole AR, Knebel A, Morrice NA, Robertson LA, Irving AJ, Connolly CN, Sutherland C (2004) GSK-3 phosphorylation of the Alzheimer epitope within collapsin response mediator protein regulates axon elongation in primary neurons. J Biol Chem 279: 50176–50180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eickholt BJ, Walsh FS, Doherty P (2002) An active pool of GSK-3 at the leading edge of growth cone is implicated in semaphorin 3A signaling. J Cell Biol 157: 211–217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Etienne-Manneville S, Hall A (2003) Cdc42 regulates GSK-3β and adenomatous polyposis coli to control cell polarity. Nature 421: 753–756 [DOI] [PubMed] [Google Scholar]
- Fukata Y et al. (2002) CRMP-2 binds to tubulin heterodimers to promote microtubule assembly. Nat Cell Biol 4: 583–591 [DOI] [PubMed] [Google Scholar]
- Keely PJ, Rusyn EV, Cox AD, Parise LV (1999) R-Ras signals through specific integrin α cytoplasmic domains to promote migration and invasion of breast epithelial cells. J Cell Biol 145: 1077–1088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolodkin AL, Matthes DJ, Goodman CS (1993) The semaphorin genes encode a family of transmembrane and secreted growth cone guidance molecules. Cell 75: 1389–1399 [DOI] [PubMed] [Google Scholar]
- Marte BM, Rodriguez-Viciana P, Wennstrom S, Warne PH, Downward J (1997) R-Ras can activate the phosphoinositide 3-kinase but not the MAP kinase arm of the Ras effector pathways. Curr Biol 7: 63–70 [DOI] [PubMed] [Google Scholar]
- Nobes CD, Lauritzen I, Mattei MG, Paris S, Hall A, Chardin P (1998) A new member of the Rho family, Rnd1, promotes disassembly of actin filament structures and loss of cell adhesion. J Cell Biol 141: 187–197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oertli B, Han J, Marte BM, Sethi T, Downward J, Ginsberg M, Hughes PE (2000) The effector loop and prenylation site of R-Ras are involved in the regulation of integrin function. Oncogene 19: 4961–4969 [DOI] [PubMed] [Google Scholar]
- Oinuma I, Katoh H, Harada A, Negishi M (2003) Direct interaction of Rnd1 with Plexin-B1 regulates PDZ-RhoGEF-mediated Rho activation by Plexin-B1 and induces cell contraction in COS-7 cells. J Biol Chem 278: 25671–25677 [DOI] [PubMed] [Google Scholar]
- Oinuma I, Ishikawa Y, Katoh H, Negishi M (2004a) The semaphorin 4D receptor Plexin-B1 is a GTPase activating protein for R-Ras. Science 305: 862–865 [DOI] [PubMed] [Google Scholar]
- Oinuma I, Katoh H, Negishi M (2004b) Molecular dissection of the semaphorin 4D receptor Plexin-B1-stimulated R-Ras GTPase-activating protein activity and neurite remodeling in hippocampal neurons. J Neurosci 24: 11473–11480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owen R, Gordon-Weeks PR (2003) Inhibition of glycogen synthase kinase 3β in sensory neurons in culture alters filopodia dynamics and microtubule distribution in growth cones. Mol Cell Neurosci 23: 626–637 [DOI] [PubMed] [Google Scholar]
- Suire S, Hawkins P, Stephens L (2002) Activation of phosphoinositide 3-kinase γ by Ras. Curr Biol 12: 1068–1075 [DOI] [PubMed] [Google Scholar]
- Swiercz JM, Kuner R, Behrens J, Offermanns S (2002) Plexin-B1 directly interacts with PDZ-RhoGEF/LARG to regulate RhoA and growth cone morphology. Neuron 35: 51–63 [DOI] [PubMed] [Google Scholar]
- Takahashi T, Strittmatter SM (2001) Plexin-A1 autoinhibition by the plexin sema domain. Neuron 29: 429–439 [DOI] [PubMed] [Google Scholar]
- Tamagnone L et al. (1999) Plexins are large family of receptors for transmembrane, secreted, and GPI-anchored semaphorins in vertebrates. Cell 99: 71–80 [DOI] [PubMed] [Google Scholar]
- Toyofuku T, Yoshida J, Sugimoto T, Zhang H, Kumanogoh A, Hori M, Kikutani H (2005) FARP2 triggers signals for Sema3A-mediated axonal repulsion. Nat Neurosci 12: 1712–1719 [DOI] [PubMed] [Google Scholar]
- Uchida Y et al. (2005) Semaphorin3A signaling is mediated via sequential Cdk5 and GSK3β phosphorylation of CRMP2: implication of common phosphorylating mechanism underlying axon guidance and Alzheimer's disease. Genes Cells 10: 165–179 [DOI] [PubMed] [Google Scholar]
- van Triest M, de Rooij J, Bos JL (2001) Measurement of GFP-bound Ras-like GFPases by activation-specific probes. Methods Enzymol 333: 343–348 [DOI] [PubMed] [Google Scholar]
- Yamamoto T, Matsui T, Nakafuku M, Iwamatsu A, Kaibuchi K (1995) A novel GTPase-activating protein for R-Ras. J Biol Chem 270: 30557–30561 [DOI] [PubMed] [Google Scholar]
- Yoshimura T, Kawano Y, Arimura N, Kawabata S, Kikuchi A, Kaibuchi K (2005) GSK-3β regulates phosphorylation of CRMP-2 and neuronal polarity. Cell 120: 137–149 [DOI] [PubMed] [Google Scholar]
- Zhang Z, Vuori K, Wang H, Reed JC, Ruoslahti E (1996) Integrin activation by R-Ras. Cell 85: 61–69 [DOI] [PubMed] [Google Scholar]
- Zumbrunn J, Kinoshita K, Hyman AA, Nathke IS (2001) Binding of the adenomatous polyposis coli protein to microtubules increase microtubule stability and is regulated by GSK-3β phosphorylation. Curr Biol 11: 44–49 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
supplementary information