

Abstract
The obligatory heterodimerization of the GABAB receptor (GBR) raises fundamental questions about molecular mechanisms controlling its signaling efficacy. Here, we show that NEM sensitive fusion (NSF) protein interacts directly with the GBR heterodimer both in rat brain synaptosomes and in CHO cells, forming a ternary complex that can be regulated by agonist stimulation. Inhibition of NSF binding with a peptide derived from GBR2 (TAT-Pep-27) did not affect basal signaling activity but almost completely abolished agonist-promoted GBR desensitization in both CHO cells and hippocampal slices. Taken with the role of PKC in the desensitization process, our observation that TAT-Pep-27 prevented both agonist-promoted recruitment of PKC and receptor phosphorylation suggests that NSF is a priming factor required for GBR desensitization. Given that GBR desensitization does not involve receptor internalization, the NSF/PKC coordinated action revealed herein suggests that NSF can regulate GPCR signalling efficacy independently of its role in membrane trafficking. The functional interaction between three bona fide regulators of neurotransmitter release, such as GBR, NSF and PKC, could shed new light on the modulation of presynaptic GBR action.
Keywords: desensitization, GABAB , heterodimer, NSF, PKC
Introduction
Ionotropic and metabotropic receptors mediate the action of the inhibitory neurotransmitter γ-amino-butyric acid (GABA) in the central nervous system. The metabotropic GABAB receptor (GBR) consists of an obligatory heterodimer between two seven transmembrane domain (7TM) receptors, GBR1 and GBR2 (Jones et al, 1998; Kaupmann et al, 1998; White et al, 1998; Kuner et al, 1999). In addition to playing a role in ER export (Couve et al, 1998; Margeta-Mitrovic et al, 2000), GBR1/GBR2 heterodimerization is required for the formation of a functional receptor. Indeed, whereas only GBR1 can bind GABA, GBR2 appears to engage the heterotrimeric G protein for downstream signaling (Galvez et al, 2001; Margeta-Mitrovic et al, 2001; Robbins et al, 2001). Such transactivation across two distinct 7TM receptors raises fundamental questions about the molecular mechanisms controlling their signaling efficacy.
Among the mechanisms controlling 7TM receptor activity, agonist-promoted desensitization is one of the best characterized at the molecular level. As for most receptors, sustained stimulation of GBR can lead to functional desensitization (Couve et al, 2002; Gonzalez-Maeso et al, 2003; Perroy et al, 2003; Tosetti et al, 2004). However, β-arrestin recruitment to the receptor and the ensuing endocytosis of the complex that are classically associated to desensitization do not appear to contribute to the regulation of GBR responsiveness (Perroy et al, 2003; Fairfax et al, 2004). We recently reported that a phosphorylation-independent mechanism involving the G protein receptor kinase 4 (GRK4) can regulate GBR activity in the cerebellum (Perroy et al, 2003). However, the restricted expression pattern of GRK4, mainly found in testes and cerebellum (Virlon et al, 1998; Sallese et al, 2000), suggests that other mechanisms may modulate GBR signaling efficacy in other tissues. There is in fact multiple evidences that different mechanisms may be at work since distinct desensitization and phosphorylation profiles were reported for different cellular systems (Couve et al, 2002; Gonzalez-Maeso et al, 2003; Tosetti et al, 2004). These discrepancies between different systems are most likely due to the relative expression levels of different protein partners that can influence receptor signaling efficacy. Among other factors, PKC activation has previously been shown to regulate GBR activity (Dutar and Nicoll, 1988; Thompson and Gahwiler, 1992), even if its direct role in agonist-promoted desensitization has not yet been documented.
In an effort to identify new proteins that could regulate GBR function, we performed a yeast two-hybrid screen using the GBR2 carboxyl tail (c-tail) as bait that revealed the N-ethylmaleimide (NEM) sensitive fusion (NSF) protein as a potential interacting partner. NSF belongs to the ‘ATPase Associated to various cellular Activities' (AAA) family and is classically devoted to the regulation of protein complexes supporting membrane fusion and trafficking events (Whiteheart and Matveeva, 2004). In this context, it promotes the disruption of soluble NSF-associated protein receptor (SNARE) coiled-coil interactions. Such NSF uncoiling activity is of particular interest when considering its interaction with GBR as receptor's c-tails are engaged in a coiled-coil interaction within the GBR1/GBR2 heterodimer (White et al, 1998; Kammerer et al, 1999).
In addition to the established role of NSF in controlling the assembly/disassembly of SNARE complexes, its direct interaction with cell surface receptors such as the AMPA receptor (Nishimune et al, 1998; Osten et al, 1998), the β2-adrenergic (β2AR) receptors (Cong et al, 2001), the dopaminergic receptors (Heydorn et al, 2004; Zou et al, 2005), and the adrenomedulin receptor (Bomberger et al, 2005) has been reported. In the three cases where the functional consequences were characterized, NSF binding was proposed to modulate the postendocytic sorting of these receptors (Noel et al, 1999; Cong et al, 2001; Hanley et al, 2002; Lee et al, 2004; Bomberger et al, 2005). Given that GBR does not undergo rapid agonist-promoted internalization in any of the systems tested (Perroy et al, 2003; Fairfax et al, 2004), we sought to investigate further the interaction between NSF and GBR and its potential role in regulating receptor function.
Results
NSF interacts directly with GBR subunits
A yeast two-hybrid screen (YTH) was performed with both GBR1 and GBR2 full-length c-tails (860I-961K and 741I-941L, respectively) against a human brain cDNA library. In addition to complementary GBR subunit (White et al, 2002) and transcription factor ATFx/CREB2 (White et al, 2000), we found NSF as a potential binding partner of GBR2 (Figure 1). Using shorter segments of the c-tail, the interacting region was narrowed down to the distal part of the GBR2 coiled-coil domain and more specifically to a 27 amino-acid peptide encompassing residues 799–825 (Pep27). As previously observed for the interaction between GluR2 and NSF (Nishimune et al, 1998), the integrity of the full-length ATPase appeared essential for its association with receptor c-tail. Indeed, none of the truncation mutants of NSF tested could bind to the receptor c-tail (data not shown).
Figure 1.
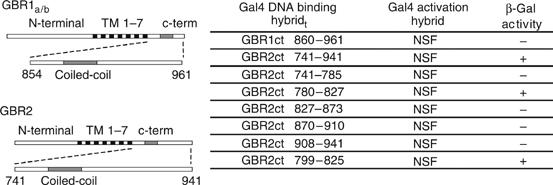
Interaction between NSF and GBR c-termini assessed in yeast two-hybrid system. GBR c-termini (see schematic representation) were used as bait in a yeast two-hybrid screen testing a human brain cDNA library. NSF was one of the positive clones obtained with the GBR2 c-terminus. To delineate the GBR2 minimal region of interaction with NSF, two-hybrid assay was performed between the indicated GBR2 c-tail mutants (see table) and NSF. β-Gal activity is determined (+/−) for each construct.
To further characterize the interaction between NSF and GBR2 c-tail (GBR2ct), we carried out in vitro binding assays using the receptor c-tail fused to a glutathione-S-transferase (GST) protein and purified His6-tagged NSF. GST pull-down performed with increasing amount of NSF in the presence of non-hydrolysable ATPγS (to inhibit ATPase activity) demonstrated a direct binding of NSF to GBR2ct (Figure 2A). This binding tends to saturate at ∼50 nM and reach half-saturation at 15 nM (Figure 2A, right panel), indicating a relatively high affinity of NSF for the receptor. The nucleotide dependence of NSF interaction was then determined (Figure 2B) by evaluating the impact of ATP and ATPγS under different salt conditions. Conditions favoring NSF ATPase activity (ATP and Mg2+) disrupted the association, whereas its inhibition (ATPγS+Mg2+, ATP+EDTA or ATP+Mg2++EDTA) favored the interaction with GBR2ct (Figure 2B). Such influence of the nucleotide state of NSF is reminiscent of other relevant interactions involving NSF (Sollner et al, 1993; Osten et al, 1998; Hanley et al, 2002). Consistent with YTH data presented above, the Pep27 region of the coiled-coil domain of GBR2 was sufficient to sustain NSF binding as shown by the efficient pull-down of NSF by a GST–Pep27 fusion protein (Figure 2C). Interestingly, Pep27 was as efficient as a previously described 10 amino-acid peptide corresponding to the GluR2 NSF binding site (Pep2m) (Nishimune et al, 1998; Osten et al, 1998; Song et al, 1998). In contrast to the results obtained in YTH experiments, NSF binding was not found to be restricted to GBR2ct and GBR1ct (854I-961K) also interacted selectively with NSF (Figure 2D).
Figure 2.
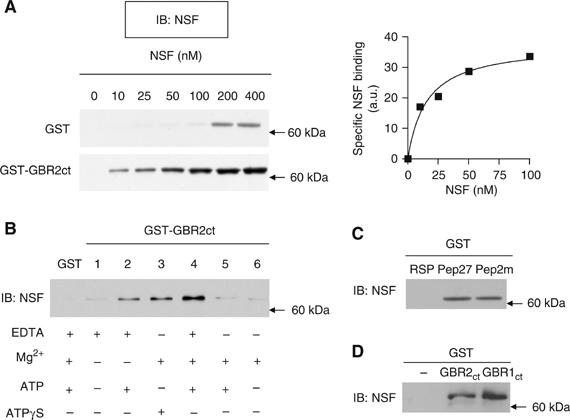
Influence of the ATPase state of NSF on its direct interaction with GBR c-termini. For all experiments, purified His6-NSF was incubated with GST or GST-hybrid proteins and bound NSF was revealed by Western blot analysis. In (A) GST pull-down assay was performed with increasing amount of His6-NSF and GST or GST-GBR2ct in the presence of ATPγS and MgCl2. The graphic representation of NSF binding to the c-terminus of GBR2 is shown on the right. (B) The binding of 50 nM NSF to GST or GST-GBR2ct was assessed under the indicated nucleotide and ionic conditions. (C) GST-Pep27, GST-Pep2m or GST-RSP were incubated with 50 nM NSF in the presence of ATP, MgCl2 and EDTA. (D) GST-GBR1ct or GST-GBR2ct were incubated with 50 nM NSF in the presence of ATP, MgCl2 and EDTA. In all cases, results are representative of two to three independent experiments.
Such partial discrepancy between YTH and GST pull-down experiments is not unusual. Nevertheless, to clarify whether the GBR1 could truly interact with NSF and to confirm that interaction between GBR2 and NSF can occur in living cells, immunoprecipitations were performed in CHO cells. Immunoprecipitation of GBR1 or GBR2 from cell expressing each of the receptor individually led to the co-sedimentation of NSF (Figure 3A and B). In the reverse configuration, NSF isolation revealed that it binds to two molecular species of GBR2 (Figure 3B, right panel), corresponding to ER-localized core-glycosylated precursor (∼100 kDa) and fully processed (∼120 kDa) forms of the receptor (Supplementary data and Supplementary Figure S1). These results suggest that NSF binds to distinct GBR2 species along the maturation path from the ER to the plasma membrane. In the case of GBR1, which cannot reach the cell surface and is retained in the ER when expressed alone (Couve et al, 1998), co-immunoprecipitation with NSF implies that interaction between these two molecules occurs in the ER. Taken together, these data may indicate that NSF could be involved in GBR transport to the cell surface, as has been previously suggested for other membrane proteins (Noel et al, 1999; Cong et al, 2001; Shi et al, 2001; Hanley et al, 2002).
Figure 3.
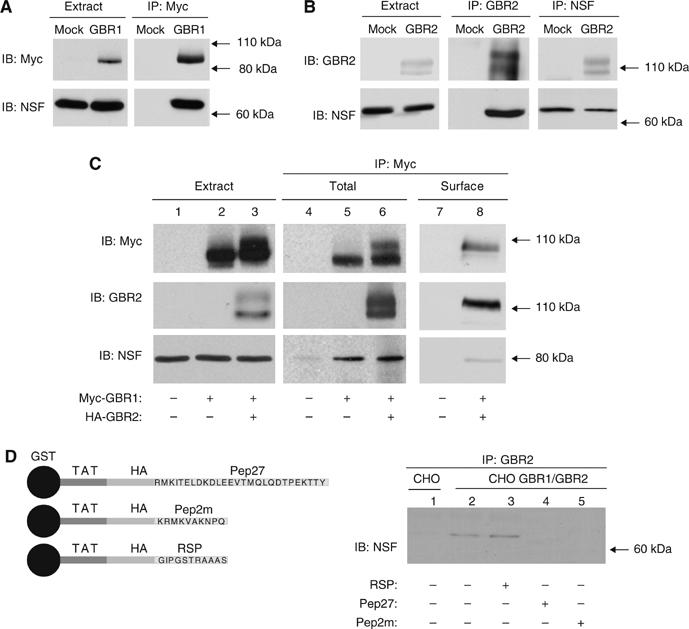
Association of NSF with the functional GBR heterodimer. Immunoprecipitations were performed on lysates from control CHO (Mock) or CHO cells stably expressing myc-GBR1b (GBR1, A), HA-GBR2 (GBR2, B), using either 9E10 anti-myc, anti-GBR2, or mouse anti-NSF antibodies. The presence of endogenous NSF or of receptors was confirmed by Western blot analysis using either 3F10 anti-myc, anti-GBR2, or rabbit anti-NSF antibodies. (C) CHO cells were transiently transfected with indicated plasmids. Cell lysates were then immunoprecipitated with 9E10 anti-myc antibody. For surface immunoprecipitation, attached cells were labeled with 9E10 anti-myc antibody before cell lysis. (D) GST-TAT hybrid proteins fused to the indicated peptide (see schematic representation) were used in order to inhibit the NSF/heterodimer interaction in CHO cells stably expressing or not expressing (Mock) myc-GBR1b/HA-GBR2. NSF/receptor complexes were immunoprecipitated with an anti-GBR2 antibody.
In agreement with the influence of NSF nucleotide binding state in GST-pull-down experiments, the functionality of its ATPase was found to be essential for its interaction with both GBR1 and GBR2 as a treatment with the alkylating agent, N-ethylmaleimide, inhibited its co-immunoprecipitation with these receptors (data not shown).
The active form of GBR associates with NSF
To determine if, in addition to its ability to interact with each receptor subtype individually, NSF can also bind to the functional GBR1/GBR2 heterodimer, we compared the amount of NSF co-immunoprecipitated with GBR1 between cells expressing and cells not expressing GBR2. Equivalent amount of NSF were recovered in both conditions (Figure 3C, lanes 5 and 6), suggesting that GBR1/GBR2 heterodimerization does not interfere with the NSF/GBR1 interaction. To confirm the existence of an NSF/GBR1/GBR2 ternary complex, we took advantage of the fact that GBR1 can be trafficked to the cell surface only in association with GBR2 (White et al, 1998). Hence, co-sedimentation of NSF following cell surface immunoprecipitation of GBR1 would indicate that at least part of the GBR interacting with NSF correspond to the GBR1/GBR2 heterodimer. As shown in Figure 3C (lanes 7 and 8), both NSF and GBR2 were co-precipitated with cell surface GBR1, suggesting the binding of the active GBR population to NSF. The occurrence of such an interaction between the functional heterodimer and NSF was also confirmed in vivo. Indeed, mass spectrometry analysis of the protein complex that co-sedimented with GBR1 following its immunoprecipitation from rat brain synaptosomes revealed the presence of NSF (Supplementary data and Supplementary Table 1).
To confirm the specificity of interaction between NSF and GBR, we assessed the ability of the Pep27 peptide to inhibit the association of NSF to GBR2 in cells co-expressing GBR1 and GBR2. For this purpose, we inserted an HIV TAT sequence (that allows the diffusion of the hybrid protein through the plasma membrane (Becker-Hapak et al, 2001)) attached to an HA epitope within the GST-Pep27 protein in order to generate a GST-TAT-HA-Pep27 (TAT-Pep27) fusion protein. The ability of this peptide to penetrate plasma membrane was confirmed by immunofluorescence using an anti-HA antibody (data not shown). As expected for a specific interaction, treatment performed with the TAT-Pep27 abolished NSF/receptor association (Figure 3D), whereas a treatment with a random sequence peptide (TAT-RSP) had no effect. Interestingly, TAT-Pep2m also impaired NSF co-precipitation with the receptor, indicating that GluR2- and GBR2-derived peptides share a unique binding site on NSF.
Classically, NSF is described as a cytoplasmic protein (Morgan and Burgoyne, 2004). To further document a possible interaction between GBR and NSF at cellular surface, their co-localization was assessed using confocal immunofluorescence microscopy. For this purpose, cells expressing GBR1 and GBR2 (Figure 4A) were first labeled using antibodies directed against the N-terminal epitopes displayed by each of the receptor (myc for GBR1 (panel a) and HA for GBR2 (panel b)). Following extensive washing, cells were then permeabilized and NSF labeled with a specific antibody (panel c). As can be seen in the overlay panels e and f, both GBR1 and GBR2 (that were as well co-localizing; panel d, signal in turquoise) are detected in close apposition with NSF at the plasma membrane, as illustrated by co-localization signals in magenta (panel e) and yellow (panel f), respectively. Similar results were obtained using green fluorescent protein-tagged-NSF (avoiding the need for permeabilization), indicating that the co-localization between NSF and both receptors did not result from a permeabilization artifact (data not shown). Moreover, this co-localization did not result from a massive redistribution of NSF upon expression of the GBR as the extent of co-localization between NSF and GFP-GRK5, a protein constitutively associated to the plasma membrane (Thiyagarajan et al, 2004), was not noticeably affected by GBR expression (Supplementary Figure S2). This indicates that NSF/GBR interaction may occur in specialized membrane domains.
Figure 4.
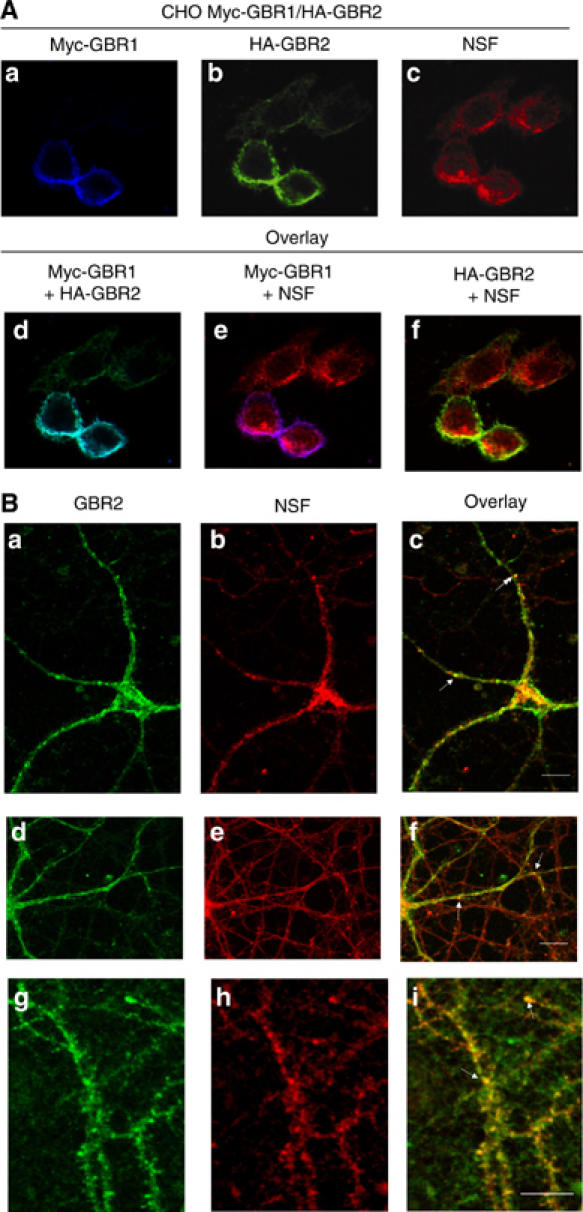
NSF co-localizes with both GBR subunits at the plasma membrane. (A) CHO cells stably expressing myc-GBR1b/HA-GBR2 were used. Myc-GBR1b, GBR2 and NSF were respectively labeled for immunofluorescence experiments with the secondary antibodies coupled to Alexa633 (Blue) (a), Oregon green (b) and Texas red (c) fluorophore respectively. GBR1 and GBR2 were surface labeled before cell fixation and permeabilization was then carried out in order to mark NSF. Overlay panels (d, e and f) correspond to the superposition of the a-b, a-c and b-c images, respectively. (B) GBR2 (green, a, d and g) and NSF (red, b, e and h) co-localizations (c, f and i) were determined in primary cultures of cortical neurons. Arrows indicate synaptic structures where both proteins are co-localized. White bars represent the scale of the image (20 μm).
To confirm that co-localization of GBR and NSF can occur in native tissue, we examined the subcellular localization of GBR2 and NSF in primary cultures of rat cortical neurons. Co-localized immunoreactivity for GBR2 and NSF was observed in both neuronal cell bodies (Figure 4B (a–f)) and dendritic extensions (Figure 4B (a–i), where they display a punctuated labeling pattern (white arrows). This is reminiscent of the co-localization between NSF and GluR2 AMPA receptor previously observed at the synaptic junctions of hippocampal neurons (Song et al, 1998).
GBR activation destabilize the NSF/heterodimer complex
As NSF was found to bind to the heterodimer, we wondered if GBR activation could modulate this association. As shown in Figure 5A, stimulation with GABA promoted the disruption of the ternary complex, as indicated by the time-dependent decrease in the amount of NSF co-immunoprecipitated with GBR1. This effect was mimicked by baclofen, a selective GBR agonist (Figure 5B). Pharmacological selectivity of GABA action is further supported by the observation that the level of co-immunoprecipitated NSF with GBR2 expressed alone (the subunit that does not bind agonists) was insensitive to agonist pre-incubation (Figure 5C). In contrast, agonist effect was recovered when GBR2 was expressed with GBR1 (the subunit harboring the GABA/baclofen binding site) (Figure 5C). Interestingly, similar results were obtained if GBR2 was co-expressed with GBR1a or GBR1b, two common splice variants of GBR1. In addition to confirming that NSF binds the GBR1/GBR2 heterodimer at cellular surface (where GABA acts), these results laid the foundation to explore the potential functional implications of this interaction.
Figure 5.
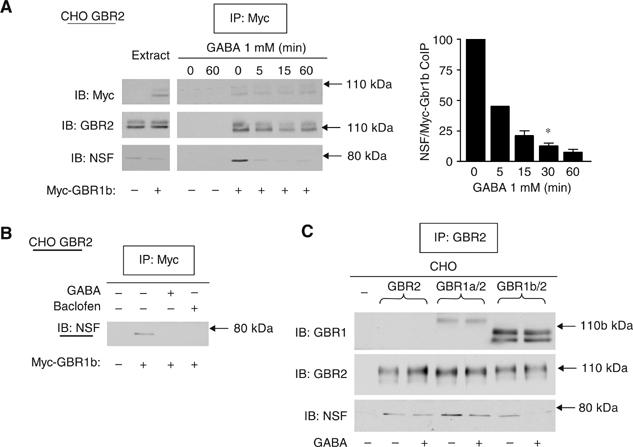
GABA stimulation disrupts the NSF/GBR complex. (A) CHO cells stably expressing HA-GBR2 were transfected or not transfected with myc-GBR1b and were treated with GABA for the indicated times. Following immunoprecipitation with 9E10 anti-myc, the amount of precipitated NSF was revealed by Western blot and quantified (bar graph). These results represent the means±s.e.m. of three to five experiments performed independently. (B) Cells were treated either with 1 mM GABA or with 0.1 mM baclofen for 30 min. In (C) control CHO cells (−) or CHO cells stably expressing HA-GBR2 alone (GBR2), GBR1a/GBR2 or myc-GBR1b/HA-GBR2 (GBR1b/GBR2) were used and treated or not treated with GABA 1 mM for 30 min. GBR2 was immunoprecipitated and the presence of endogenous NSF and receptors revealed by Western blot.
Preventing NSF binding preserves the GBR/G protein coupling following chronic stimulation
The observation that NSF is released from GBR complex following stimulation leads us to test whether NSF could be implicated in the regulation of the GABA-mediated G protein activation of the receptor. Given the ability of native GBR to interact with NSF in rat brain, quantification of the receptor-promoted GDP/GTP exchange was assessed by GTPγ[35S] binding assays in both CHO cells and hippocampal slices (a tissue known to endogenously express functional GBR (Lopez-Bendito et al, 2004)). Treatment of hippocampal slices with TAT-Pep27 (to impair the GBR/NSF interaction) or the corresponding TAT-RSP control protein did not alter the ability of the receptor to promote GTPγS35 binding. Indeed, the peptides were without effect on either baclofen maximal efficacy or EC50 to stimulate GTPγS35 binding (Table I). Previous studies demonstrated that GBR G protein coupling activity wanes over time, following sustained agonist stimulation in CHO cells, cerebellar granule, or rat hippocampal cells (Couve et al, 2002; Gonzalez-Maeso et al, 2003; Perroy et al, 2003; Tosetti et al, 2004). Consistent with these findings, we observed that a 30-min prestimulation with baclofen led to a 36±2% reduction of the maximal baclofen-stimulated GTPγS binding (Figure 6A and B) in hippocampal slices. Treatment with TAT-Pep27 completely blocked this baclofen-promoted desensitization (−3±9%), whereas TAT-RSP was without effect (45±5%; Figure 6A and B). These results, which indicate a role of NSF in the agonist-promoted desensitization of GBR in hippocampus, were recapitulated in CHO cells (Figure 6C) where the 39±8% desensitization promoted by a 30 min pretreatment with GABA was also blocked by TAT-Pep27 (8±4%). As NSF was previously implicated in the membrane sorting of GluR2 and β2AR (Cong et al, 2001; Shi et al, 2001; Hanley et al, 2002), we assessed whether receptor prestimulation with GABA affected cell surface expression of GBR1 or GBR2 in these cells. As recently reported in hippocampal neurons (Fairfax et al, 2004), ELISA analysis revealed that the cell surface receptor density was stable in the presence of its ligand (Supplementary Figure S2). NSF, however, has previously been shown to modulate both forward trafficking and endocytosis of membrane proteins (Cong et al, 2001; Shi et al, 2001; Hanley et al, 2002; Lee et al, 2004). Therefore, as NSF was found to interact with GBR immature species, one cannot formally exclude the possibility that NSF affects both endocytosis and insertion of de novo synthesized receptors such that the steady-state concentration of GBR at the cell surface remained unaffected. This is however unlikely given the lack of surface labeled GBR internalization following a 30 min agonist stimulation in CHO cells (data not shown); a behavior also observed in HEK293 cells (Perroy et al, 2003), COS cells, and hippocampal neurons (Fairfax et al, 2004). It follows that, in the absence of agonist-promoted internalization, insertion of de novo synthesized receptors should lead to an increase in the steady-state receptor level detected by ELISA. As this was not the case, the above results suggest that the role that NSF could play in GBR forward trafficking does probably not impact on the short-term events contributing to rapid desensitization.
Table 1.
Functional characterization of native hippocampal GBR following treatment with TAT-peptides
| Condition | Basal GTPγS35 binding (% of control) | Maximal GTPγS35 binding (% of control) | EC50 (M) | n | R2 |
|---|---|---|---|---|---|
| Vehicle | 100 | 100±7 | 2.48 10−5 | 3 | 0.909 |
| RSP | 105±8 | 85,5±6 | 2.03 10−5 | 3 | 0.909 |
| Pep27 | 103±18 | 96±9 | 1.3 10−5 | 3 | 0.857 |
| Rat hippocampal slices were treated for 1 h with the indicated TAT-peptide and baclofen-stimulated [35S]GTPγS binding was measured in membranes derived from these slices expressing native GBR. The results were expressed in percentage of the vehicle condition for basal and maximal GTPγS binding. EC50 was derived from dose–response curves. These results are the mean±s.e.m. of three independent experiments performed in triplicate. | |||||
Figure 6.
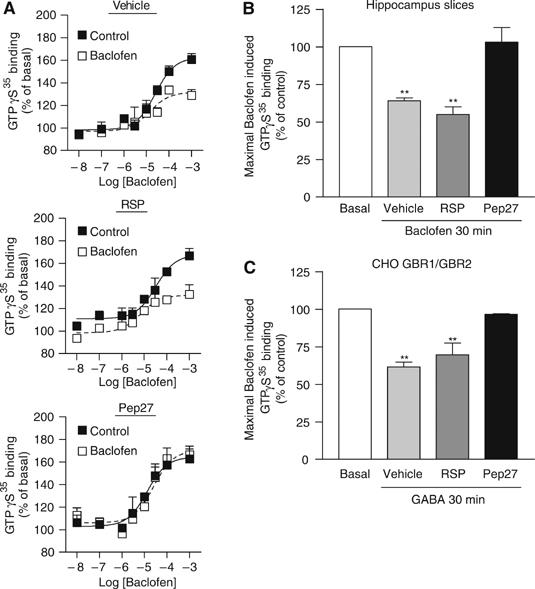
Preventing NSF binding preserves GBR activity following GABA prestimulation. (A) Hippocampus slices were treated for 1 h with vehicle or the indicated TAT-peptide and then with 0.1 mM baclofen (empty square) or not (full square) for an additional 30 min. Curves represent the specific GTPγS binding obtained for increasing amount of baclofen. (B) Bar graph representation of the maximal baclofen induced GTPγS binding obtained from the curves in (A). (C) Similar experiments were performed in CHO cells stably expressing GBR1a and GBR2. Cells were stimulated for 30 min with 1 mM GABA.
The agonist-promoted desensitization of GBR is a PKC-dependent mechanism
Given the proposed role of PKC in the regulation of GBR signaling efficacy in rat hippocampus (Dutar and Nicoll, 1988; Thompson and Gahwiler, 1992; Tosetti et al, 2004), we investigated the contribution of this kinase in the NSF-mediated desensitization of the receptor in CHO cells. First, we studied the ability of GABA to promote PKC plasma membrane translocation in cells expressing a GFP-tagged PKCα (GFP-PKC) construct. In agreement with what was previously observed in hippocampal neurons (Tremblay et al, 1995), treatment with GABA induced membrane recruitment of GFP-PKC (Figure 7A), reaching its maximum at 5 min. This translocation was comparable to the one promoted by phorbol 12-myristate 13-acetate (PMA), a molecule directly activating PKC (Blumberg, 1991). Inhibiting PKC activation with GFX completely blocked the GABA-promoted attenuation of baclofen-stimulated GTPγS binding (Figure 7B, right panel), suggesting a role for this protein in the desensitization process. Consistent with such a role, direct PKC activation with PMA reduced the maximal baclofen response by 48±3%, thus mimicking the desensitizing effects of GABA (40±3%; Figure 7B). Also, consistent with a role for PKC is the observation that the GABA-promoted phosphorylation of GBR1 (154±9%; Figure 7C) and GBR2 (data not shown) was blocked by GFX whereas PMA strongly stimulated it (278±19%). Interestingly, PKC recruitment occurred concomitantly with the release of NSF from the receptor (50% of NSF being released from GBR following 5 min of stimulation, a time at which the recruitment of PKC is maximal; Figure 5), thus suggesting that these two events act in a coordinated manner to promote agonist-mediated desensitization of GBR.
Figure 7.
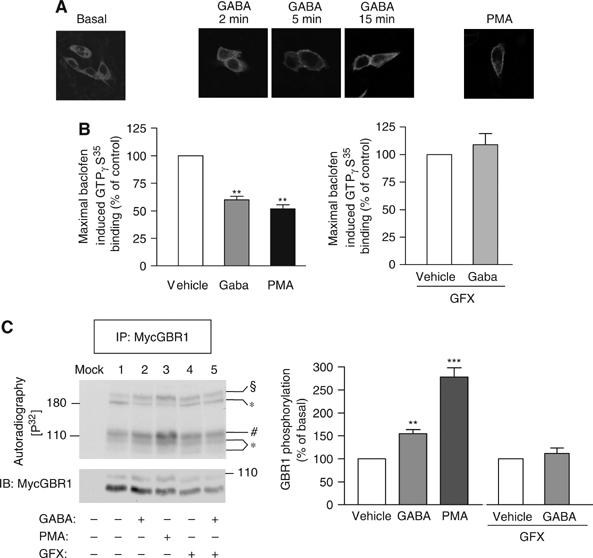
PKC is implicated in the desensitization process of the GBR. (A) CHO cells stably expressing GBR1a/GBR2 were transiently transfected with GFP-PKCα construct. Cells were treated as indicated before their fixation and PKC localization was visualized by confocal microscopy. These results are representative of two independent experiments. (B) Maximal baclofen-stimulated [35S]GTPγS binding was measured in membranes derived from CHO cells stably expressing GBR1a/GBR2. The effect of agonist prestimulation on receptor activity was measured in the absence or presence of 0.5 μM GFX and compared to the one induced by PMA. These results represent the mean±s.e.m. of three independent experiments. (C) CHO cells stably expressing myc-GBR1b/HA-GBR2 were labeled with [32P]Pi. GBR1 was immunoprecipitated (IP, 9E10 anti-myc) and the level of phosphorylation analyzed by autoradiography. Bands corresponding to the GBR1 monomer (mature and immature species) and homodimer are indicated by (*), GBR2 monomer by (#) and GBR1/GBR2 heterodimer by (§). Results were normalized by quantifying the amount of precipitated receptor in each condition (IB). Histograms represent the mean±s.e.m. of five independent experiments.
NSF primes GBR for its PKC-dependent desensitization
To further explore the PKC/NSF link in the agonist-promoted desensitization process, we assessed the influence of the NSF/GBR interaction blocking peptide, TAT-Pep27, on the GABA-promoted recruitment of PKC and the ensuing phosphorylation of GBR. As observed in Figure 8A, pretreatment with TAT-Pep27 completely abolished the GABA-induced recruitment of GFP-PKC to the plasma membrane. Consistent with these results, TAT-Pep27 also prevented the agonist-dependent increase in GBR phosphorylation (Figure 8B). Overall, these results indicate that NSF/GBR interaction is critical to the PKC-mediated processes involved in the blunting of the receptor signaling efficacy.
Figure 8.
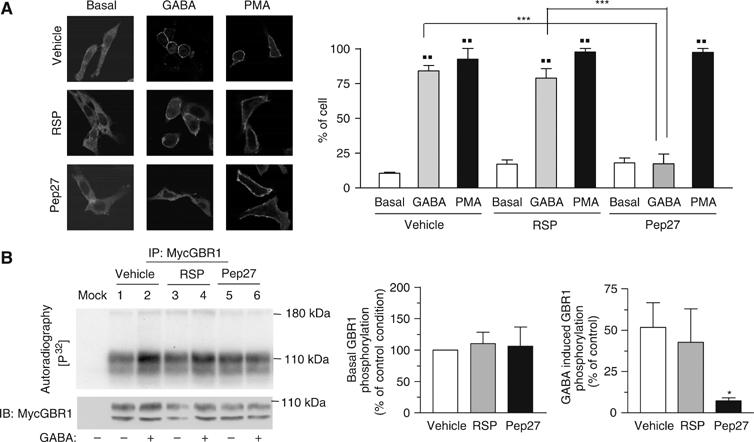
GBR/NSF interaction is essential to PKC recruitment and GBR phosphorylation upon agonist stimulation. CHO cells stably expressing GBR1a/GBR2 were transiently transfected with GFP-PKC construct. (A) Cells were treated as indicated before their fixation and PKC localization was visualized by confocal microscopy. The histogram represents the percentage of cells displaying membrane PKC recruitment in each treatment condition and is the result of two independent experiences. In each experience, an average of 30 cells was considered per condition. (B) CHO cells stably expressing myc-GBR1b/HA-GBR2 were labeled with [32P]Pi and treated as indicated. GBR1 was immunoprecipitated (9E10 anti-myc, IP) and phosphorylation signal was analyzed by autoradiography. Histograms represent the mean±s.e.m. of four independent experiments.
PKC induces the dissociation of NSF from the activated GBR
As we previously observed that NSF dissociates from the receptor following agonist stimulation, we assessed the role of PKC in this process. As shown in Figure 9, GFX strongly inhibited the disruption of the NSF/receptor complex that follows the activation of the receptor. These results indicate that the GABA-stimulated dissociation of NSF from the receptor involves PKC activation. Consistently, PMA treatment was sufficient to promote NSF release (Figure 9).
Figure 9.
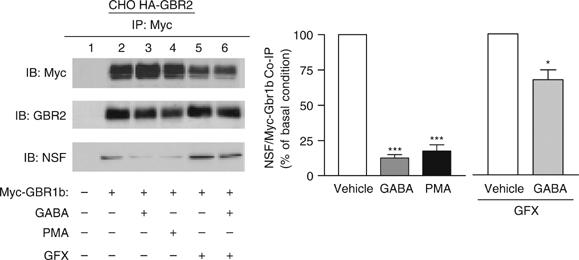
PKC modulates the NSF/GABAB receptor interaction. Cells stably expressing HA-GBR2 and transiently transfected or not transfected with myc-GBR1b were used. Following treatments to modulate receptor and/or PKC activities, GBR1b was immunoprecipitated (9E10 anti-myc antibody) and the presence of endogenous NSF was revealed by western blot (IB) and quantified (bar graph). These results represent the mean±s.e.m. of three independent experiments.
Discussion
In the present study, we showed that NSF/GBR interaction regulates the signaling efficacy of this heterodimeric receptor via a PKC-dependent mechanism.
NSF was found to interact directly with both GBR1 and GBR2 in a noncompetitive manner as its binding to a given subunit was not inhibited by the co-expression of the other. This contrasts with other proteins such as 14-3-3 (Couve et al, 2001), CREB2 (White et al, 2000) and Marlin-1 (Couve et al, 2004), which were found to interact only with GBR1 and to compete for the GBR1/GBR2 interaction. This association of NSF with the functional heterodimeric population of the receptor is illustrated by the regulation of its binding upon GBR stimulation. As previously reported for the NSF/AMPA receptor interaction (Hanley et al, 2002), a peptide corresponding to the interacting domain of GluR2 with NSF was sufficient to block the interaction. Interestingly, peptides derived from both GluR2 (Pep2 m) and GBR2 (Pep27) subunits inhibited the association of NSF to GBR. This indicates that these peptides share a unique binding site on NSF, which may represent a common interaction domain for GBR and GluR2 subunits.
Also reminiscent of GluR2/NSF interaction, the nucleotide binding status of NSF was found to regulate its interaction with GBR subunits. This is important when considering that classical functions of NSF are dictated by its nucleotide binding state and its ATPase activity. For instance, NSF-promoted SNARE complex uncoiling involved in membrane fusion events requires ATP hydrolysis that leads to NSF dissociation (Sollner et al, 1993). Interestingly, GBR stimulation led to a reduction of NSF co-immunoprecipitation, indicative of an agonist-promoted dissociation of the enzyme. As NSF interacts within the coiled-coil region of GBR2, it is tempting to draw a parallel with the typical uncoiling action of NSF and propose that this protein regulates GBR c-tail association. However, an uncoiling of the GBR c-tails alone would not be sufficient to explain the NSF-mediated agonist-promoted desensitization. Indeed, it was previously shown that the entire deletion of the GBR coiled-coil domain does not affect the receptor activity (Margeta-Mitrovic et al, 2001; Grunewald et al, 2002). Thus, an additional factor needs to be invoked to explain how NSF contributes to the desensitization of GBR. Overall, our data suggest that this additional factor could be PKC.
Consistent with this hypothesis, inhibition of the NSF/GBR interaction by TAT-Pep27 blocked both PKC recruitment and GBR phosphorylation, as well as the agonist-promoted desensitization. Taken with the observation that PMA-mediated activation of PKC promoted both GBR phosphorylation and its functional uncoupling and that inhibiting PKC with GFX prevented desensitization, these data clearly support the existence of a coordinated role of NSF and PKC in the regulation of GBR activity. As PKC-dependent phosphorylation of NSF has been shown to regulate its activity (Matveeva et al, 2001), a role of NSF phosphorylation in the regulation of GBR cannot be excluded. This is, however, unlikely as no change in the NSF phosphorylation state was observed following GBR stimulation or PMA treatment (data not shown).
The coordinated action of NSF and PKC in the regulation of GBR activity is further supported by the observation that the agonist-promoted release of NSF from the receptor requires PKC activation and that PMA treatment is sufficient to induce this event. The pre-association of NSF with GBR could hence be seen, as a priming event required for the engagement of PKC that in turn would promote receptor desensitization and terminate the regulatory cycle by favoring NSF release.
A functional interaction between PKC and NSF also emerged from several studies on the regulation of ionotropic GluR2-containing AMPA receptor. In this case, however, NSF was shown to stabilize the receptor at synaptic membrane and prevent its removal by a combined action of PKC and the protein interacting with C kinase (PICK-1) (Perez et al, 2001; Hanley et al, 2002), thus preventing the occurrence of long-term depression. In this scenario, NSF has been proposed to modulate the lateral diffusion of GluR2 in the plasma membrane (Steinberg et al, 2004). Given that GBR activation does not promote endocytosis and does not affect steady-state receptor density at cellular surface, the role of NSF in GBR desensitization does not appear to involve removal or insertion of receptor from the plasma membrane. Therefore, the hypothesized role of NSF in membrane protein lateral diffusion may be a more appealing mechanism to explain the regulatory influence of NSF on GBR function, especially when considering the presence of GBR in raft microdomains and its potential role in the control of receptor signaling activity (Becher et al, 2004).
Taken together, our results demonstrate the existence of a concerted regulation of GBR signaling efficacy involving both NSF and PKC. Based on our observations, we propose a model whereby the preassociation of NSF with the receptor is a prerequisite for the occurrence of the PKC-promoted desensitization (Figure 10). In the absence of GBR/NSF interaction (e.g. in the presence of TAT-Pep27), GBR stimulation fails to promote PKC recruitment and phosphorylation of the receptor that becomes refractory to desensitization (Figure 10A). In contrast, preassociation of NSF primes the receptor such that agonist stimulation results in the recruitment of PKC and the ensuing phosphorylation of GBR leading to agonist-promoted desensitization (Figure 10B). In addition to promote desensitization, PKC also terminates the regulatory process by favoring NSF dissociation from the heterodimer (Figure 10C). Although the precise mechanism by which NSF/GBR association primes the desensitization process cannot be firmly established, two nonmutually exclusive mechanisms can be imagined: (1) through its uncoiling activity, NSF could unmask phosphorylation sites that are implicated in desensitization; (2) the presence of NSF could favor the engagement of a signaling pathway required for PKC activation. Also remaining to be established is whether NSF dissociation occurs before or after the completion of the PKC-dependent desensitization and/or contributes to the regulation of signaling efficacy.
Figure 10.
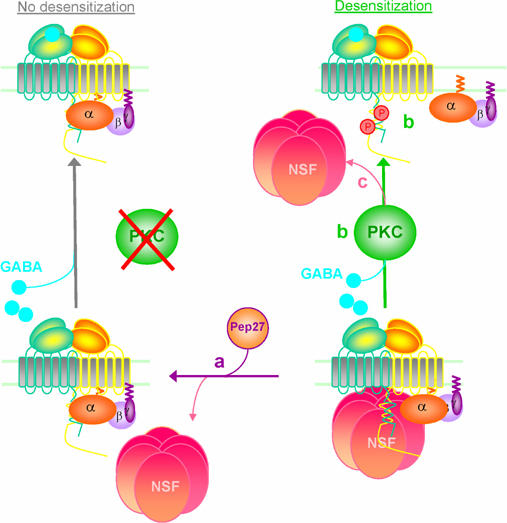
Coordinated action of NSF and PKC regulates GABAB receptor signaling efficacy. (a) In the presence of TAT-Pep27, NSF dissociates from GBR and agonist-stimulation fails to promote desensitization. (b) Preassociation of NSF, however, primes the receptor such that agonist stimulation results in the recruitment of PKC and the ensuing phosphorylation of GBR leading to agonist-promoted desensitization. (c) PKC terminates the regulatory process by favoring NSF dissociation from the heterodimer.
The role of NSF and PKC in the desensitization of GBR should also be placed in the context of other mechanisms that have been shown to contribute to this process. For instance, a phosphorylation-independent contribution of GRK4 (Perroy et al, 2003) as well as a negative influence of PKA-mediated phosphorylation of GBR (Couve et al, 2002) on its desensitization have been reported. The relative contribution of these various mechanisms most likely depends on the relative expression levels of the different regulatory proteins. Noticeably, no GRK4 expression could be detected in CHO cells, suggesting that the NSF/PKC may represent the dominant mechanism in these cells. This process also appear to be relevant for GBR desensitization in at least some native tissues as the NSF blocking peptide TAT-Pep27 abolished agonist-promoted desensitization in hippocampal slices. Interestingly, the GBR desensitization previously observed in rat hippocampal slices was shown to concern pre-synaptic GBR that regulates neurotransmitter release (Tosetti et al, 2004). Taken with the role of both NSF and PKC in the regulation of synaptic exocytosis (Lin and Scheller, 2000; Barclay et al, 2003), our demonstration of their implication in GBR desensitization raises the intriguing possibility of the existence of an integrated process controlling the GBR-mediated inhibition of neurotransmitter release.
Materials and methods
A detailed description of the different materials and plasmids used in this manuscript may be found in Supplementary data.
YTH
This protocol has already been described (White et al, 2000), but a detailed description can be found in Supplementary data.
In vitro protein interaction assay
Protocols to purify the different proteins and describing the assay are detailed in the Supplementary methods appended to this manuscript.
Cell culture and rat hippocampal slice preparation
The protocols are described in detail in the Supplementary data section.
Cell treatments
Treatments were performed at 37°C on CHO cells or at RT for hippocampal slices. Prestimulations of cells with 1 mM GABA or 0.1 mM baclofen were performed for 30 min when not specified or for the indicated time. To demonstrate the role of PKC, cells were incubated with vehicle or 0.5 μM GFX for 30 min before the prestimulation with GABA or with 1 μM PMA for 10 min. To inhibit NSF/GBR interaction, cells were incubated with TAT-Pep27 400 nM or TAT-Pep2 m or RSP 800 nM, for 1 h before any treatment.
Immunoprecipitation
The protocol is described in detail in the Supplementary data section.
Whole phosphorylation assay
This was performed previously described (Perroy et al, 2003), but see details in Supplementary methods.
[35S]GTPγS binding assay
This protocol has been performed as previously described (Perroy et al, 2003) and details can be found in Supplementary materials.
Immunofluorescence
The protocol is described in detailed in the Supplementary data section.
Mathematical and statistical analysis
For GTPγS binding, dose–response curve experiments were analyzed by nonlinear regression using Prism program (GraphPad software, San Diego, CA) (Figure 6A). For other GTPγS binding studies, basal GTPγS binding obtained without stimulation was subtracted to the maximal GTPγS binding obtained in the presence of 0.1 mM baclofen. Every condition was expressed in the percentage of the corresponding control condition. The statistical significance of results obtained in GTPγS binding, co-immunoprecipitation, or PKC recruitment experiments was determined using a one-way ANOVA analysis followed by a Bonferroni's multiple comparison test. Statistical significances between the control condition and the condition of interest are represented as follows: * when P<0.05, ** when P<0.01 and *** when P<0.001.
Supplementary Material
Supplementary Figures S1, S2 and S3
Supplementary Table 1
Supplementary methods
Acknowledgments
We are grateful to Monique Lagacé for her constant fruitful discussion and her careful reading of the manuscript. We are also grateful to Billy Breton for the kind gift of the GFP-GRK5 plasmid. This work was supported by a CIHR/Rx&D Grant and sponsored by GlaxoSmithKline. NL holds a CIHR studentship. MB is the recipient of the Canada Research Chair in Signal Transduction and Molecular Pharmacology.
References
- Barclay JW, Craig TJ, Fisher RJ, Ciufo LF, Evans GJ, Morgan A, Burgoyne RD (2003) Phosphorylation of Munc18 by protein kinase C regulates the kinetics of exocytosis. J Biol Chem 278: 10538–10545 [DOI] [PubMed] [Google Scholar]
- Becher A, Green A, Ige AO, Wise A, White JH, McIlhinney RA (2004) Ectopically expressed gamma-aminobutyric acid receptor B is functionally down-regulated in isolated lipid raft-enriched membranes. Biochem Biophys Res Commun 321: 981–987 [DOI] [PubMed] [Google Scholar]
- Becker-Hapak M, McAllister SS, Dowdy SF (2001) TAT-mediated protein transduction into mammalian cells. Methods 24: 247–256 [DOI] [PubMed] [Google Scholar]
- Blumberg PM (1991) Complexities of the protein kinase C pathway. Mol Carcinog 4: 339–344 [DOI] [PubMed] [Google Scholar]
- Bomberger JM, Parameswaran N, Hall CS, Aiyar N, Spielman WS (2005) Novel function for receptor activity-modifying proteins (RAMPs) in post-endocytic receptor trafficking. J Biol Chem 280: 9297–9307 [DOI] [PubMed] [Google Scholar]
- Cong M, Perry SJ, Hu LA, Hanson PI, Claing A, Lefkowitz RJ (2001) Binding of the beta2 adrenergic receptor to N-ethylmaleimide-sensitive factor regulates receptor recycling. J Biol Chem 276: 45145–45152 [DOI] [PubMed] [Google Scholar]
- Couve A, Filippov AK, Connolly CN, Bettler B, Brown DA, Moss SJ (1998) Intracellular retention of recombinant GABAB receptors. J Biol Chem 273: 26361–26367 [DOI] [PubMed] [Google Scholar]
- Couve A, Kittler JT, Uren JM, Calver AR, Pangalos MN, Walsh FS, Moss SJ (2001) Association of GABA(B) receptors and members of the 14-3-3 family of signaling proteins. Mol Cell Neurosci 17: 317–328 [DOI] [PubMed] [Google Scholar]
- Couve A, Restituito S, Brandon JM, Charles KJ, Bawagan H, Freeman KB, Pangalos MN, Calver AR, Moss SJ (2004) Marlin-1, a novel RNA-binding protein associates with GABA receptors. J Biol Chem 279: 13934–13943 [DOI] [PubMed] [Google Scholar]
- Couve A, Thomas P, Calver AR, Hirst WD, Pangalos MN, Walsh FS, Smart TG, Moss SJ (2002) Cyclic AMP-dependent protein kinase phosphorylation facilitates GABA(B) receptor-effector coupling. Nat Neurosci 5: 415–424 [DOI] [PubMed] [Google Scholar]
- Dutar P, Nicoll RA (1988) Pre- and postsynaptic GABAB receptors in the hippocampus have different pharmacological properties. Neuron 1: 585–591 [DOI] [PubMed] [Google Scholar]
- Fairfax BP, Pitcher JA, Scott MG, Calver AR, Pangalos MN, Moss SJ, Couve A (2004) Phosphorylation and chronic agonist treatment atypically modulate GABAB receptor cell surface stability. J Biol Chem 279: 12565–12573 [DOI] [PubMed] [Google Scholar]
- Galvez T, Duthey B, Kniazeff J, Blahos J, Rovelli G, Bettler B, Prezeau L, Pin JP (2001) Allosteric interactions between GB1 and GB2 subunits are required for optimal GABA(B) receptor function. EMBO J 20: 2152–2159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Maeso J, Wise A, Green A, Koenig JA (2003) Agonist-induced desensitization and endocytosis of heterodimeric GABAB receptors in CHO-K1 cells. Eur J Pharmacol 481: 15–23 [DOI] [PubMed] [Google Scholar]
- Grunewald S, Schupp BJ, Ikeda SR, Kuner R, Steigerwald F, Kornau HC, Kohr G (2002) Importance of the gamma-aminobutyric acid(B) receptor C-termini for G-protein coupling. Mol Pharmacol 61: 1070–1080 [DOI] [PubMed] [Google Scholar]
- Hanley JG, Khatri L, Hanson PI, Ziff EB (2002) NSF ATPase and alpha-/beta-SNAPs disassemble the AMPA receptor-PICK1 complex. Neuron 34: 53–67 [DOI] [PubMed] [Google Scholar]
- Heydorn A, Sondergaard BP, Hadrup N, Holst B, Haft CR, Schwartz TW (2004) Distinct in vitro interaction pattern of dopamine receptor subtypes with adaptor proteins involved in post-endocytotic receptor targeting. FEBS Lett 556: 276–280 [DOI] [PubMed] [Google Scholar]
- Jones KA, Borowsky B, Tamm JA, Craig DA, Durkin MM, Dai M, Yao WJ, Johnson M, Gunwaldsen C, Huang LY, Tang C, Shen Q, Salon JA, Morse K, Laz T, Smith KE, Nagarathnam D, Noble SA, Branchek TA, Gerald C (1998) GABA(B) receptors function as a heteromeric assembly of the subunits GABA(B)R1 and GABA(B)R2. Nature 396: 674–679 [DOI] [PubMed] [Google Scholar]
- Kammerer RA, Frank S, Schulthess T, Landwehr R, Lustig A, Engel J (1999) Heterodimerization of a functional GABAB receptor is mediated by parallel coiled-coil alpha-helices. Biochemistry 38: 13263–13269 [DOI] [PubMed] [Google Scholar]
- Kaupmann K, Malitschek B, Schuler V, Heid J, Froestl W, Beck P, Mosbacher J, Bischoff S, Kulik A, Shigemoto R, Karschin A, Bettler B (1998) GABA(B)-receptor subtypes assemble into functional heteromeric complexes. Nature 396: 683–687 [DOI] [PubMed] [Google Scholar]
- Kuner R, Kohr G, Grunewald S, Eisenhardt G, Bach A, Kornau HC (1999) Role of heteromer formation in GABAB receptor function. Science 283: 74–77 [DOI] [PubMed] [Google Scholar]
- Lee SH, Simonetta A, Sheng M (2004) Subunit rules governing the sorting of internalized AMPA receptors in hippocampal neurons. Neuron 43: 221–236 [DOI] [PubMed] [Google Scholar]
- Lin RC, Scheller RH (2000) Mechanisms of synaptic vesicle exocytosis. Annu Rev Cell Dev Biol 16: 19–49 [DOI] [PubMed] [Google Scholar]
- Lopez-Bendito G, Shigemoto R, Kulik A, Vida I, Fairen A, Lujan R (2004) Distribution of metabotropic GABA receptor subunits GABAB1a/b and GABAB2 in the rat hippocampus during prenatal and postnatal development. Hippocampus 14: 836–848 [DOI] [PubMed] [Google Scholar]
- Margeta-Mitrovic M, Jan YN, Jan LY (2000) A trafficking checkpoint controls GABA(B) receptor heterodimerization. Neuron 27: 97–106 [DOI] [PubMed] [Google Scholar]
- Margeta-Mitrovic M, Jan YN, Jan LY (2001) Function of GB1 and GB2 subunits in G protein coupling of GABA(B) receptors. Proc Natl Acad Sci USA 98: 14649–14654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matveeva EA, Whiteheart SW, Vanaman TC, Slevin JT (2001) Phosphorylation of the N-ethylmaleimide-sensitive factor is associated with depolarization-dependent neurotransmitter release from synaptosomes. J Biol Chem 276: 12174–12181 [DOI] [PubMed] [Google Scholar]
- Morgan A, Burgoyne RD (2004) Membrane traffic: controlling membrane fusion by modifying NSF. Curr Biol 14: R968–R970 [DOI] [PubMed] [Google Scholar]
- Nishimune A, Isaac JT, Molnar E, Noel J, Nash SR, Tagaya M, Collingridge GL, Nakanishi S, Henley JM (1998) NSF binding to GluR2 regulates synaptic transmission. Neuron 21: 87–97 [DOI] [PubMed] [Google Scholar]
- Noel J, Ralph GS, Pickard L, Williams J, Molnar E, Uney JB, Collingridge GL, Henley JM (1999) Surface expression of AMPA receptors in hippocampal neurons is regulated by an NSF-dependent mechanism. Neuron 23: 365–376 [DOI] [PubMed] [Google Scholar]
- Osten P, Srivastava S, Inman GJ, Vilim FS, Khatri L, Lee LM, States BA, Einheber S, Milner TA, Hanson PI, Ziff EB (1998) The AMPA receptor GluR2 C terminus can mediate a reversible, ATP-dependent interaction with NSF and a. Neuron 21: 99–110 [DOI] [PubMed] [Google Scholar]
- Perez JL, Khatri L, Chang C, Srivastava S, Osten P, Ziff EB (2001) PICK1 targets activated protein kinase Calpha to AMPA receptor clusters in spines of hippocampal neurons and reduces surface levels of the AMPA-type glutamate receptor subunit 2. J Neurosci 21: 5417–5428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perroy J, Adam L, Qanbar R, Chenier S, Bouvier M (2003) Phosphorylation-independent desensitization of GABA(B) receptor by GRK4. EMBO J 22: 3816–3824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robbins MJ, Calver AR, Filippov AK, Hirst WD, Russell RB, Wood MD, Nasir S, Couve A, Brown DA, Moss SJ, Pangalos MN (2001) GABA(B2) is essential for g-protein coupling of the GABA(B) receptor heterodimer. J Neurosci 21: 8043–8052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sallese M, Salvatore L, D'Urbano E, Sala G, Storto M, Launey T, Nicoletti F, Knopfel T, De Blasi A (2000) The G-protein-coupled receptor kinase GRK4 mediates homologous desensitization of metabotropic glutamate receptor 1. FASEB J 14: 2569–2580 [DOI] [PubMed] [Google Scholar]
- Shi S, Hayashi Y, Esteban JA, Malinow R (2001) Subunit-specific rules governing AMPA receptor trafficking to synapses in hippocampal pyramidal neurons. Cell 105: 331–343 [DOI] [PubMed] [Google Scholar]
- Sollner T, Bennett MK, Whiteheart SW, Scheller RH, Rothman JE (1993) A protein assembly-disassembly pathway in vitro that may correspond to sequential steps of synaptic vesicle docking, activation, and fusion. Cell 75: 409–418 [DOI] [PubMed] [Google Scholar]
- Song I, Kamboj S, Xia J, Dong H, Liao D, Huganir RL (1998) Interaction of the N-ethylmaleimide-sensitive factor with AMPA receptors. Neuron 21: 393–400 [DOI] [PubMed] [Google Scholar]
- Steinberg JP, Huganir RL, Linden DJ (2004) N-ethylmaleimide-sensitive factor is required for the synaptic incorporation and removal of AMPA receptors during cerebellar long-term depression. Proc Natl Acad Sci USA 101: 18212–18216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiyagarajan MM, Stracquatanio RP, Pronin AN, Evanko DS, Benovic JL, Wedegaertner PB (2004) A predicted amphipathic helix mediates plasma membrane localization of GRK5. J Biol Chem 279: 17989–17995 [DOI] [PubMed] [Google Scholar]
- Thompson SM, Gahwiler BH (1992) Comparison of the actions of baclofen at pre- and postsynaptic receptors in the rat hippocampus in vitro. J Physiol 451: 329–345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tosetti P, Bakels R, Colin-Le Brun I, Ferrand N, Gaiarsa JL, Caillard O (2004) Acute desensitization of presynaptic GABAB-mediated inhibition and induction of epileptiform discharges in the neonatal rat hippocampus. Eur J Neurosci 19: 3227–3234 [DOI] [PubMed] [Google Scholar]
- Tremblay E, Ben Ari Y, Roisin MP (1995) Different GABAB-mediated effects on protein kinase C activity and immunoreactivity in neonatal and adult rat hippocampal slices. J Neurochem 65: 863–870 [DOI] [PubMed] [Google Scholar]
- Virlon B, Firsov D, Cheval L, Reiter E, Troispoux C, Guillou F, Elalouf JM (1998) Rat G protein-coupled receptor kinase GRK4: identification, functional expression, and differential tissue distribution of two splice variants. Endocrinology 139: 2784–2795 [DOI] [PubMed] [Google Scholar]
- White JH, McIllhinney RA, Wise A, Ciruela F, Chan WY, Emson PC, Billinton A, Marshall FH (2000) The GABAB receptor interacts directly with the related transcription factors CREB2 and ATFx. Proc Natl Acad Sci U S A 97: 13967–13972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- White JH, Wise A, Main MJ, Green A, Fraser NJ, Disney GH, Barnes AA, Emson P, Foord SM, Marshall FH (1998) Heterodimerization is required for the formation of a functional GABA(B) receptor. Nature 396: 679–682 [DOI] [PubMed] [Google Scholar]
- White JH, Wise A, Marshall FH (2002) Heterodimerization of gamma-aminobutyric acid B receptor subunits as revealed by the yeast two-hybrid system. Methods 27: 301–310 [DOI] [PubMed] [Google Scholar]
- Whiteheart SW, Matveeva EA (2004) Multiple binding proteins suggest diverse functions for the N-ethylmaleimide sensitive factor. J Struct Biol 146: 32–43 [DOI] [PubMed] [Google Scholar]
- Zou S, Li L, Pei L, Vukusic B, Van Tol HH, Lee FJ, Wan Q, Liu F (2005) Protein-protein coupling/uncoupling enables dopamine D2 receptor regulation of AMPA receptor-mediated excitotoxicity. J Neurosci 25: 4385–4395 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figures S1, S2 and S3
Supplementary Table 1
Supplementary methods