

Abstract
The eukaryotic translation initiation factor 4B (eIF4B) plays a critical role in recruiting the 40S ribosomal subunit to the mRNA. In response to insulin, eIF4B is phosphorylated on Ser422 by S6K in a rapamycin-sensitive manner. Here we demonstrate that the p90 ribosomal protein S6 kinase (RSK) phosphorylates eIF4B on the same residue. The relative contribution of the RSK and S6K modules to the phosphorylation of eIF4B is growth factor-dependent, and the two phosphorylation events exhibit very different kinetics. The S6K and RSK proteins are members of the AGC protein kinase family, and require PDK1 phosphorylation for activation. Consistent with this requirement, phosphorylation of eIF4B Ser422 is abrogated in PDK1 null embryonic stem cells. Phosphorylation of eIF4B on Ser422 by RSK and S6K is physiologically significant, as it increases the interaction of eIF4B with the eukaryotic translation initiation factor 3.
Keywords: crosstalk, eIF4B, phosphorylation, signaling, translation
Introduction
Translation initiation is the step at which the ribosome is recruited to the mRNA (Gingras et al, 1999; Hershey and Merrick, 2000). Multiple eukaryotic initiation factors (eIFs) are involved in this process. The heterotrimeric eIF4F consists of the cap-binding protein eIF4E, the scaffolding protein eIF4G, and the helicase eIF4A. eIF4F, through eIF4E, recognizes the mRNA 5′ cap structure. The eIF4A subunit is thought to unwind secondary structure in the mRNA 5′UTR to facilitate ribosome binding. The eukaryotic translation initiation factor 4B (eIF4B) stimulates eIF4F activity by potentiating the eIF4A RNA helicase activity (e.g. (Rozen et al, 1990; for reviews see (Gingras et al, 1999; Hershey and Merrick, 2000). eIF4G bridges the mRNA with the ribosome through its interaction with the eukaryotic translation initiation factor 3 (eIF3) (Etchison et al, 1982), which was demonstrated to interact directly with eIF4B (Methot et al, 1996; Vornlocher et al, 1999).
Initiation is a critical step and a checkpoint of translation. Translational control is exerted by many different types of extracellular stimuli, which activate various signaling pathways and nutrient-sensing modules (Raught et al, 2000). Signaling pathways regulate the activities of components of the translational machinery and stimulate ribosome biogenesis to coordinate the translational capacity of the cell with nutrient availability and mitogenic cues (Holland et al, 2004; Avruch et al, 2005). Two well-studied pathways that exhibit a paramount effect on translational regulation are the Ras–MAPK and PI3K/Akt/mTOR signaling modules.
Ras, through Raf, activates the dual threonine/tyrosine kinase MAPKKs, MEK1/2, which in turn phosphorylate and activate the ERK1/2 protein kinases, resulting in phosphorylation of multiple cytoplasmic (e.g. ribosomal protein S6 kinase (RSK), Mnk1/2) and nuclear (e.g. transcription factors) substrates (Roux and Blenis, 2004). PI3K phosphorylates the membrane-bound phosphatidylinositol 4,5-bisphosphate at position 3 to produce phosphatidylinositol 3,4,5-trisphosphate (PIP3). PIP3 serves as a membrane docking signal for PH domain-containing proteins such as the serine/threonine kinases Akt/PKB and PDK1 (phosphatidylinositol-dependent kinase 1). PDK1 activates Akt/PKB by phosphorylating Thr308 (in Akt1) within the T-loop of the catalytic domain (Alessi et al, 1996). PDK1 also phosphorylates the homologous site in multiple AGC family kinases (Williams et al, 2000). Among these are the different isoforms of S6K and RSK.
The highly homologous S6K1 and S6K2 proteins (>80% identity) are encoded by distinct genes. Both S6K1 and S6K2 are phosphorylated and activated in a rapamycin-sensitive manner by mTOR, which phosphorylates a threonine residue in the linker domain (Burnett et al, 1998; Volarevic and Thomas, 2001; Park et al, 2002), allowing phosphorylation by PDK1 in the catalytic domain (Alessi et al, 1998; Balendran et al, 1999).
The RSK family consists of four members (RSK1–4) (Blenis, 1993; Roux and Blenis, 2004). Activation of the RSKs requires coordinated input from the Ras/MAPK cascade (Blenis, 1993) and PDK1 (Jensen et al, 1999). The RSKs are involved in multiple processes in the cell, including transcriptional regulation, cell cycle control, protein synthesis, and feedback inhibition of the Ras/MAPK cascade via Sos phosphorylation (reviewed in Roux and Blenis, 2004). Here we identify RSK as an in vivo and in vitro eIF4B Ser422 kinase.
Results
Rapamycin-resistant eIF4B Ser422 phosphorylation is mediated by ERK1/2 MAPK signaling
Insulin-stimulated eIF4B phosphorylation at Ser422 was previously demonstrated to be rapamycin-sensitive, and the kinase responsible for Ser422 phosphorylation was identified as S6K (Raught et al, 2004; see also Figure 1A, compare lanes 9 and 10, upper panel). Interestingly, however, when HeLa cells are stimulated with serum, a significant fraction of Ser422 phosphorylation remains resistant to inhibition by rapamycin (Figure 1A, compare lane 6 to 5, upper panel).
Figure 1.
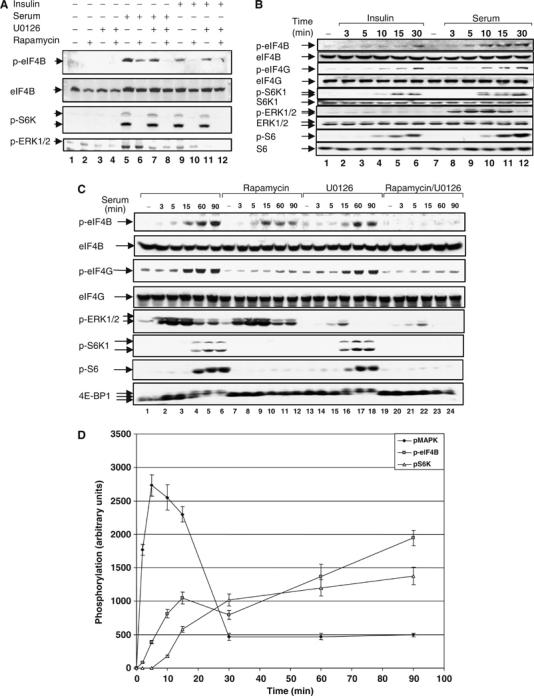
Rapamycin-resistant eIF4B Ser422 phosphorylation is mediated by ERK1/2 MAPK signaling. (A) HeLa cells were deprived of serum in the presence or absence of 20 nM rapamycin for 16–18 h. Cells were pretreated with 10 μM of U0126 for 2 h, and then stimulated with either 20% serum or insulin (100 nM) for 30 min. Total cell extracts were subjected to SDS–PAGE followed by immunoblotting with phospho-eIF4B S422, phospho-S6K1 T389, and phospho-ERK1/2 T202/Y204 antibodies and the membrane was reprobed with anti-eIF4B antiserum. (B) HeLa cells were starved for serum as in (A) and stimulated for the indicated times with either 20% serum or insulin (100 nM). Cell extracts were resolved by SDS–PAGE and immunoblotted with phospho-eIF4G S1108, phospho-eIF4B S422, phospho-S6K1 T389, phospho-ERK1/2 T202/Y204, phospho-S6 S240/244 antibodies and the indicated total proteins. (C) HeLa cells were deprived of serum in the presence or absence of 20 nM rapamycin for 16–18 h. Cells were pretreated with 10 μM of U0126 for 2 h, and then stimulated with 20% serum for the indicated times. Total cell extracts were resolved by SDS–PAGE, immunoblotted with phospho-eIF4G S1108, phospho-eIF4B S422, phospho-S6K1 T389, phospho-ERK1/2 T202/Y204, and phospho-S6 S240/244 antibodies and reprobed for the indicated proteins with pan-specific antibodies. (D) Sequential activation of signaling pathways involved in eIF4B Ser422 phosphorylation. HeLa cells were deprived of serum for 16–18 h. Cells were then stimulated with 20% serum for the indicated amounts of time. Protein extracts were resolved by SDS–PAGE and probed for phospho-eIF4B S422, phospho-ERK1/2 T202/Y204, and phospho-S6K T389.
In addition to the mTOR/PI3K pathway, the MAPK signaling module appears to play an important role in translational control (Rajasekhar et al, 2003; Naegele and Morley, 2004). It was thus pertinent to examine the contribution of this pathway to eIF4B phosphorylation. To determine whether the MAPK cascade is responsible for rapamycin-resistant eIF4B Ser422 phosphorylation, cells were treated with the MEK1/2/5 inhibitor U0126 (Duncia et al, 1998) before serum or insulin stimulation. To monitor for the efficiency of rapamycin and U0126 treatments, immunoblotting assays using phosphospecific antibodies raised against phosphorylated Thr389 of S6K, or active ERK1/2 (dually phosphorylated on Thr202 and Tyr204) were also carried out (Figure 1A). A rapamycin-resistant component of eIF4B Ser422 phosphorylation is observed in serum-stimulated but not in insulin-stimulated cells (compare lanes 6 and 10). This residual phosphorylation is abrogated by U0126 treatment (compare lanes 6 and 8). U0126 by itself has a minor effect on eIF4B phosphorylation in serum-stimulated cells, and no effect in insulin-stimulated cells, consistent with the lack of ERK activation by insulin (lanes 7 and 11, respectively). Total eIF4B protein levels were not affected by any of the treatments, as determined by reprobing the membrane with pan-eIF4B antibody. Thus, rapamycin-resistant phosphorylation of eIF4B Ser422 phosphorylation is mediated by ERK1/2 MAPK signaling. Experiments using specific inhibitors of p38 (SB203580) and JNK1/2 (JNK inhibitor II) ruled out an involvement of these MAP kinases in Ser422 phosphorylation, as these inhibitors failed to reduce serum-stimulated phosphorylation of Ser422 in rapamycin-pretreated cells (data not shown).
To study the differential sensitivity of eIF4B phosphorylation to rapamycin and U0126, a time-course experiment was carried out (Figure 1B). Both serum and insulin stimulated the phosphorylation of the PI3K/Akt/mTOR pathway substrates, eIF4G (Ser1108) and S6K1 (Thr389), with similar kinetics, although the insulin-induced S6K phosphorylation is somewhat delayed and less intense (compare lanes 4 and 10). A phosphorylation time course of the S6K substrates rpS6 (Ser240/244) and eIF4B (Ser422) is similar in insulin-stimulated cells. However, in serum-induced cells, eIF4B Ser422 phosphorylation appears faster than S6 phosphorylation (Figure 1B, lanes 9–12), and is detectable before S6K activation (compare lanes 3 and 9). Importantly, in contrast to serum, insulin is incapable of activating the MAPK ERK1/2 cascade in these cells (lanes 1–6). The inability of insulin to effect signaling through the MAPK module is the most likely explanation for the complete rapamycin sensitivity of eIF4B phosphorylation in insulin-stimulated cells.
To further characterize the biphasic pattern of eIF4B phosphorylation, a time-course experiment using serum in the presence or absence of U0126 or rapamycin was performed (Figure 1C). Activation of the MAPK cascade was monitored by immunoblotting with phosphospecific antibodies directed against activated ERK1/2. Cell extracts were also examined for phosphorylation of the rapamycin-sensitive substrates eIF4G and S6K1, using phosphospecific antibodies, and 4E-BP1 using a pan-specific antibody. Serum-induced MAPK phosphorylation is very rapid, detected as early as 3 min after serum stimulation, and reaches a peak at 5 min post-induction (Figure 1C). In contrast, S6K phosphorylation and activity (as determined by rpS6 Ser240/244 phosphorylation) are undetectable at these early time points, but are sustained for much longer (compare 60 and 90 min). These data demonstrate that eIF4B phosphorylation in response to serum is mediated by both the MAPK and PI3K/mTOR pathways. Importantly, eIF4B phosphorylation can be temporally divided into two phases: an early phase, which is sensitive to U0126, but not to rapamycin (compare the 15 min time points with the two inhibitors, lanes 10 and 16), and a late phase, which is inhibited by rapamycin (compare 60 and 90 min, lanes 11 and 12 versus 17 and 18). Simultaneous treatment of cells with both inhibitors abrogates eIF4B phosphorylation at all times (lanes 20–24). A detailed time-course experiment (Figure 1D) clearly demonstrates that serum-induced eIF4B phosphorylation is detectable before the activation of S6K (as judged by S6K1 T389 and rpS6 S240/244 phosphorylation).
eIF4B Ser422 phosphorylation persists in cells lacking S6K1 and S6K2
To further corroborate the existence of an eIF4B kinase that is distinct from S6K in cells other than HeLa, primary hepatocyte cultures from S6K1/2−/− double knockout (DKO) mice were used. The extent of eIF4B phosphorylation was similar in wild-type (wt) and S6K-deficient hepatocytes under serum-deprived conditions, and upon insulin or serum stimulation (Figure 2A). However, the wt and mutant cells differed in their sensitivity to rapamycin. Whereas rapamycin abrogated eIF4B phosphorylation following insulin stimulation, and partially after serum stimulation in wt cells, Ser422 phosphorylation was completely resistant to rapamycin treatment in S6K DKO hepatocytes. Thus, S6K phosphorylates eIF4B in insulin-stimulated hepatocytes, but an mTOR-independent kinase efficiently compensates for the S6K deletion. Consistent with the data in HeLa cells, serum activates an mTOR-independent mechanism that leads to phosphorylation of eIF4B in wt and S6K-deficient hepatocytes.
Figure 2.
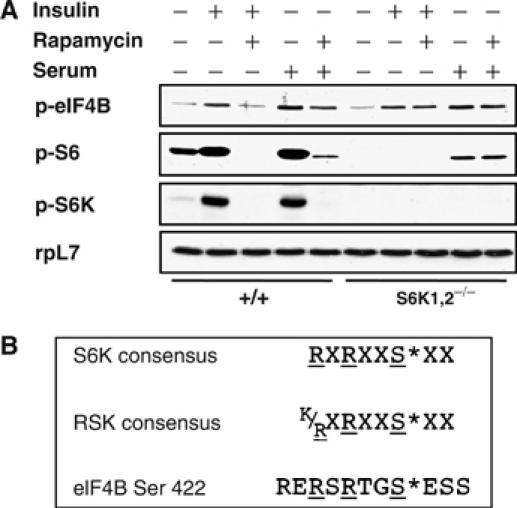
(A) eIF4B Ser422 phosphorylation persists in cells lacking S6K1 and S6K2. Hepatocytes derived from wt and S6K1/2 DKO animals were starved for nutrients and serum and stimulated with 1 μM insulin or 10% serum in the presence or absence of 20 nM rapamycin. Total cell lysates were immunoblotted with phospho-eIF4B S422, phospho-S6 S235/236, phospho-S6K1 T389, and rpL7 antibodies. (B) Substrate consensus sequences of S6K and RSK as compared to the eIF4B fragment encompassing the Ser422 phosphorylation site.
The amino-terminal kinase domain of the RSKs shares a high degree of homology with the S6K proteins, and the consensus phosphorylation sequence recognized by RSK1 (and presumably the highly homologous RSK2–4 isoforms) is almost identical to that of the S6K proteins (Leighton et al, 1995). Unlike S6K, which is activated by PDK1 (Alessi et al, 1998) and mTOR (Burnett et al, 1998), RSK activity is regulated by the ERK1/2 MAPKs (Frodin and Gammeltoft, 1999; Frodin et al, 2000) and PDK1 (Jensen et al, 1999). As the amino-acid sequence surrounding eIF4B Ser422 conforms to both the RSK and S6K consensus sequences (Figure 2B), and the RSKs are regulated by the ERK MAPKs, the RSKs appear to be the most likely candidates to effect the MAPK-dependent rapamycin-resistant phosphorylation of Ser422.
Ser422 is dephosphorylated in PDK1 null and PIF pocket mutant ES cells
Members of the AGC family of kinases are phosphorylated by PDK1 on the T-loop; this phosphorylation event is required for their full activation. PDK1 null cells are defective for both RSK and S6K activation (Mora et al, 2004). To determine whether Ser422 phosphorylation is affected in these cells, wt and PDK1 KO embryonic stem (ES) cells were serum starved for 16 h in the presence or absence of rapamycin, and then stimulated with serum for 15 min. Consistent with previously published data (Williams et al, 2000), phosphorylation of S6K T389 and rpS6 S240/244 is abrogated in PDK1 null cells (Figure 3A, lanes 5–8). Unlike their wt counterparts, PDK1 null ES cells exhibit no detectable eIF4B kinase activity upon serum stimulation (lanes 5–8). Similar to the data shown for HeLa cells (Figure 1C, lanes 4 and 10), rapamycin did not abrogate serum-induced eIF4B phosphorylation in wt ES cells (compare lane 4 to 3).
Figure 3.
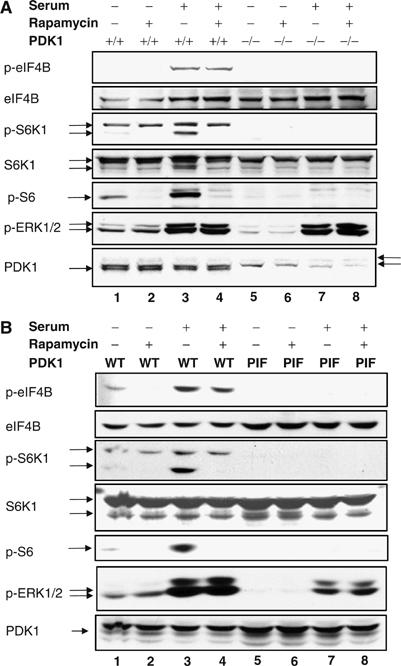
eIF4B Ser422 is dephosphorylated in PDK1 null and PDK1 PIF pocket mutant ES cells. Wt and PDK1−/− knockout (A) or PDK1 PIF pocket mutant (B) ES cells were starved for 16–18 h in the presence or absence of 20 nM rapamycin and then stimulated with 20% serum for 15 min. Total cell extracts were resolved by SDS–PAGE and proteins were detected by immunoblotting using phospho-eIF4B S422, phospho-S6K1 T389, phospho-S6 S240/244, and phospho-ERK1/2 T202/Y204 antibodies. Membranes were reprobed with antibodies against the indicated total proteins and against PDK1 (arrows on the right indicate nonspecific bands).
Another member of the AGC family that phosphorylates a consensus sequence similar to that of the RSKs and S6Ks is Akt (Obata et al, 2000). Mutation of Leu155 to glutamate in the PDK1 substrate docking site, also known as the ‘PIF pocket' (for PDK1 interacting fragment), prevents PDK1 from interacting with and phosphorylating S6K and RSK, but does not affect its ability to activate Akt (Biondi et al, 2001; Collins et al, 2003). To determine whether Akt is able to phosphorylate eIF4B, we studied Ser422 phosphorylation in PDK1 PIF pocket mutant knock-in ES cells. Similar to PDK1 null cells, PDK1 PIF pocket mutant cells are devoid of Ser422 kinase activity (Figure 3B, compare lanes 5–8 to 1–4). These data, although consistent with the idea that both S6K and RSK are bona fide eIF4B kinases, do not completely rule out the participation of other AGC kinases in eIF4B phosphorylation. However, given that eIF4B phosphorylation is sensitive to pharmacological inhibitors that fail to inhibit other AGC kinases, it is very likely that the major kinases responsible for eIF4B phosphorylation are S6K and RSK.
Catalytically active RSK variants phosphorylate eIF4B in vitro and in vivo
To demonstrate that RSK can directly phosphorylate eIF4B, we examined eIF4B phosphorylation in an in vitro kinase assay. HeLa cells were transfected with plasmids encoding HA-tagged wt RSK1 and S6K1, as well as a kinase-inactive RSK1 mutant. Cells were serum starved for 16–18 h, and pretreated with U0126 or rapamycin before serum or insulin stimulation (Figure 4A). Immunocomplex kinase assays were then carried out with recombinant eIF4B as a substrate in vitro. Wt (but not kinase-dead) RSK elicited a 3.5-fold increase in eIF4B phosphorylation (Figure 4A), indicating that RSK, but not a co-purifying kinase activity, is responsible for the phosphorylation. Pretreatment of cells with U0126 abrogated RSK-mediated eIF4B phosphorylation in vitro. S6K immunoprecipitated from insulin-treated cells exhibited a five-fold increase in eIF4B phosphorylation relative to basal phosphorylation levels (an unstimulated sample expressing HA-S6K1). This phosphorylation was abrogated by rapamycin pretreatment (Figure 4A).
Figure 4.
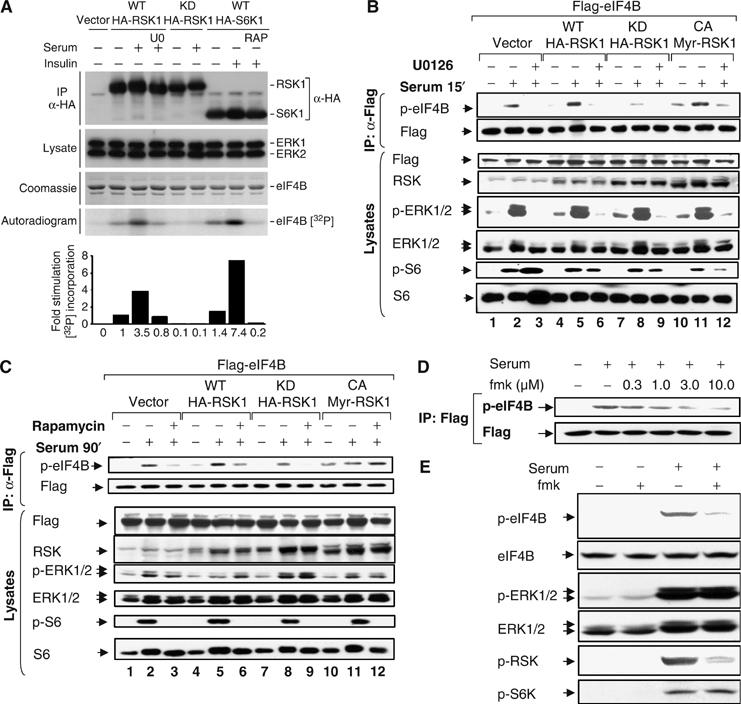
Catalytically active RSK variants phosphorylate eIF4B in vitro and in vivo. (A) Wt and kinase-dead HA-RSK- and wt HA-S6K1-transfected HeLa cells were serum starved for 16–18 h, pretreated with either U0126 (10 μM; U0) or rapamycin (20 nM; RAP) as indicated, and stimulated with either serum or insulin for 15 min. An aliquot of the total cell lysate was immunoblotted for ERK1/2. Another aliquot was used to immunoprecipitate RSK variants and S6K1 using anti-HA antibody. Immunoprecipitates were split. Half was subjected to SDS–PAGE and probed for HA and the remaining half was assayed for in vitro kinase activity by using recombinant eIF4B as substrate. Samples were resolved by SDS–PAGE, stained with Coomassie brilliant blue, and exposed to an X-ray film. 32P incorporation was quantified using a phosphorimager. A representative autoradiogram is shown. (B, C) HeLa cells cotransfected with Flag-tagged eIF4B together with wt, kinase-dead, and constitutively active RSK variants were serum starved for 16–18 h in the presence or absence of 10 μM U0126 (B) or 20 nM rapamycin (C) before serum stimulation for 15 min (B) or 90 min (C). Cell lysates were used to immunoprecipitate exogenous Flag-tagged eIF4B using anti-Flag (M2) antibody. Immune complexes were subjected to SDS–PAGE and probed with antibodies directed against phosphorylated eIF4B Ser422. Membranes were reprobed with anti-Flag antibody. Aliquots of total cell lysates were run on gel and probed with indicated antibodies. (D) HeLa cells were transfected with Flag-eIF4B. After 24 h, cells were deprived of serum in the presence or absence of increasing concentrations of RSK1/2 inhibitor fmk for 16–18 h. Cells were stimulated with 20% serum for 15 min. eIF4B was immunoprecipitated using anti-Flag antibody. Immune complexes were subjected to SDS–PAGE and Western blotting with phospho-eIF4B S422 antibody. The membrane was stripped and reprobed with Flag antibody. (E) HeLa cells were deprived of serum in the presence or absence of 10 μM RSK1/2 inhibitor fmk for 16–18 h. Cells were stimulated with 20% serum for 15 min. Total cell extracts were subjected to SDS–PAGE followed by immunoblotting with phospho-eIF4B S422, phospho-ERK1/2 T202/Y204, phospho-RSK S380, and phospho-S6K1 T389 antibodies and then reprobed for total eIF4B and ERK1/2.
To further demonstrate that RSK can phosphorylate eIF4B in vivo, HeLa cells were cotransfected with various RSK1 mutants and Flag-tagged eIF4B. Cells were stimulated with serum for 15 (Figure 4B) or 90 min (Figure 4C), following pretreatment with U0126 or rapamycin, respectively. Whereas catalytically active wt RSK and MyrRSK (a constitutively active, membrane-targeted form) potently phosphorylated Ser422, the kinase-dead RSK variant not only failed to do so, but actually suppressed serum-induced eIF4B phosphorylation (compare Figure 4B, lane 8 to lanes 2 and 5). Ser422 phosphorylation was readily detectable in unstimulated cells transfected with MyrRSK (Figure 4B and C). This basal phosphorylation was increased after 15 min of serum stimulation, but not when cells were treated with U0126 (Figure 4B). A fraction of the Ser422 phosphorylation induced by MyrRSK is not inhibited by U0126. Consistent with the earlier report, MyrRSK retains some activity even in the presence of MEK inhibitors (Roux et al, 2004). Flag-eIF4B phosphorylation in cells stimulated with serum for 90 min exhibited rapamycin sensitivity, unless coexpressed with MyrRSK (Figure 4C).
Additional evidence for an in vivo contribution of RSK to eIF4B phosphorylation was obtained through the use of a recently designed and characterized fluoromethylketone (fmk), which potently and selectively inactivates RSK1 and RSK2 in mammalian cells (Cohen et al, 2005). The inhibitor targets the C-terminal kinase domain of RSK1 and RSK2, preventing autophosphorylation on S380 and S386 of human RSK1 and RSK2, respectively. This phosphorylation enables PDK1 docking, which then phosphorylates and activates the RSK N-terminal kinase domain (Jensen et al, 1999; Frodin et al, 2000). To determine the optimal inhibitory concentration of fmk in HeLa cells, a dose–response experiment was carried out. HeLa cells were transfected with Flag-tagged eIF4B, then deprived of serum in the presence of increasing concentrations of fmk for 16–18 h. Cells were then stimulated with serum for 15 min, and eIF4B was immunoprecipitated using anti-Flag antibody and subjected to SDS–PAGE and Western blotting with the phosphospecific eIF4B Ser422 antibody. Fmk addition resulted in a dose-dependent inhibition of serum-induced eIF4B phosphorylation, reaching a plateau at 3 μM (Figure 4D). To determine whether endogenous eIF4B phosphorylation is also sensitive to fmk treatment, HeLa cells were starved of serum in the presence or absence of 10 μM fmk for 16 h, before serum stimulation for 15 min. Fmk strongly reduced serum-stimulated phosphorylation of eIF4B and RSK1, whereas S6K and MAPK activation remained unaffected (Figure 4E). In conclusion, RSK phosphorylates Ser422 both in vitro and in vivo. The early phase of eIF4B phosphorylation is more dependent on RSK activity than at later times.
RNAi of the RSK1 and RSK2 isoforms leads to reduced eIF4B Ser422 phosphorylation and inhibits cap-dependent translation
To further substantiate the involvement of RSK in eIF4B Ser422 phosphorylation, HeLa S3 cells were cotransfected with small interfering RNAs (siRNAs) targeting RSK1 and RSK2, or with mock siRNA. The siRNA treatment resulted in a >90% knockdown of both RSK1 and RSK2 expression. At 24 h post-transfection, cells were starved of serum, pretreated with inhibitors, and then stimulated with either serum or insulin for 15 min. Both serum and insulin elicited phosphorylation of eIF4B on Ser422 in mock siRNA-transfected cells (Figure 5A, lanes 2 and 6). As expected, serum-induced phosphorylation of eIF4B was sensitive to U0126 (lanes 2 and 4), whereas insulin-stimulated eIF4B phosphorylation was sensitive to rapamycin (lanes 6 and 7). Serum-induced (compare lanes 2 and 3 to lanes 9 and 10), but not insulin-induced (compare lanes 6 and 13), eIF4B Ser422 phosphorylation was prevented by RNAi directed against RSK1/2.
Figure 5.
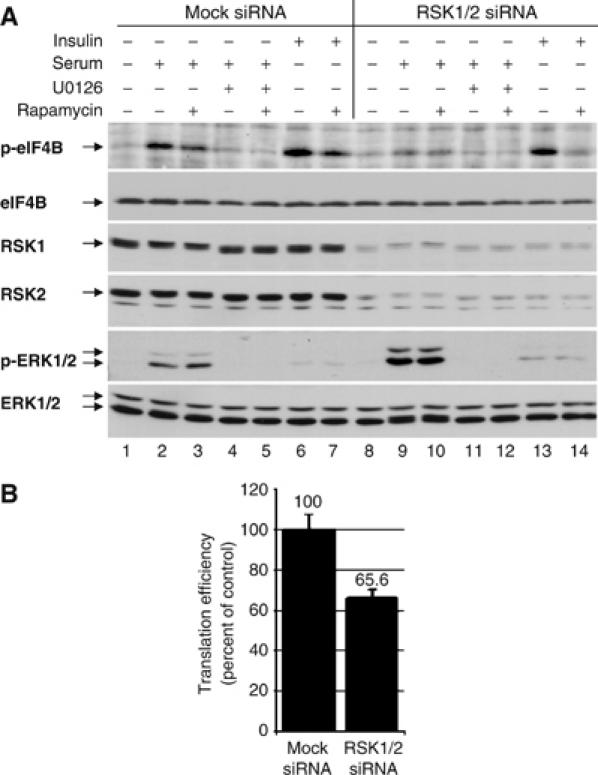
RNAi-mediated silencing of RSK1 and RSK2 isoforms expression leads to reduced eIF4B Ser422 phosphorylation and inhibition of cap-dependent translation. (A) HeLa cells were subjected to RNAi using synthetic oligos nonspecific (Mock) or specific to RSK1 and RSK2 isoforms. At 24 h post-transfection, cells were serum starved for 16–18 h in the presence or absence of rapamycin, then indicated samples were treated with U0126 and stimulated with serum or insulin as shown. Total cell extracts were immunoblotted with phospho-eIF4B S422 and phospho-ERK1/2 T202/Y204 antibodies followed by reprobing for the corresponding total proteins. RSK1 and RSK2 Western blots were also carried out to demonstrate the efficiency of the knockdown. (B) HEK293 cells were transfected with the bicistronic luciferase construct and indicated siRNAs. After 48 h, cells were harvested and assayed for Renilla (RL) and firefly (FL) luminescence. Results are presented as average of RL/FL ratio±standard error from three independent experiments carried out in triplicate.
To assess the effect of RSK1/2 RNAi on cap-dependent translation, HEK293 cells were cotransfected with RSK1 and RSK2 targeting siRNAs and bicistronic Renilla-HCV IRES-firefly luciferase reporter (see Figure 5B). After 48 h, cells were harvested and analyzed for luciferase activity. The data suggest that RSK1/2 RNAi leads to ∼34% inhibition in cap-dependent translation. These results provide further evidence that RSK proteins are playing an important role in regulating cap-dependent translation in part through eIF4B Ser422 phosphorylation.
Phosphorylation of eIF4B on Ser422 enhances its affinity for the eIF3 complex
eIF4B co-purifies with eIF3 through several purification steps (e.g. Brown-Luedi et al, 1982), eIF3 can be immunoprecipitated with eIF4B (Methot et al, 1996), and eIF4B directly interacts with eIF3 in yeast and mammalian cells (Methot et al, 1996; Vornlocher et al, 1999). To examine whether the association of eIF3 with eIF4B is affected by the phosphorylation state of eIF4B, a co-immunoprecipitation experiment was carried out. Cells coexpressing Flag-eIF4B and various mutants of RSK were starved of serum overnight, pretreated with U0126 for 2 h, and then stimulated with serum for 15 min. Immunoprecipitates were subjected to SDS–PAGE and probed with phosphospecific-Ser422 eIF4B and anti-eIF3a (p170 subunit) antibodies. To monitor for the amount of Flag-tagged eIF4B loaded on the gel, the membrane was stripped and reprobed with anti-Flag antibody (Figure 6A). Serum stimulation strongly enhanced the interaction between eIF4B and eIF3 in cells expressing wt and constitutively active RSK variants (lanes 2 and 8, 3.5- and 7.5-fold, respectively), but not in cells expressing the kinase-dead RSK (lane 5). Thus, phosphorylated eIF4B is enriched in a complex containing eIF3 as compared to its hypophosphorylated counterpart.
Figure 6.
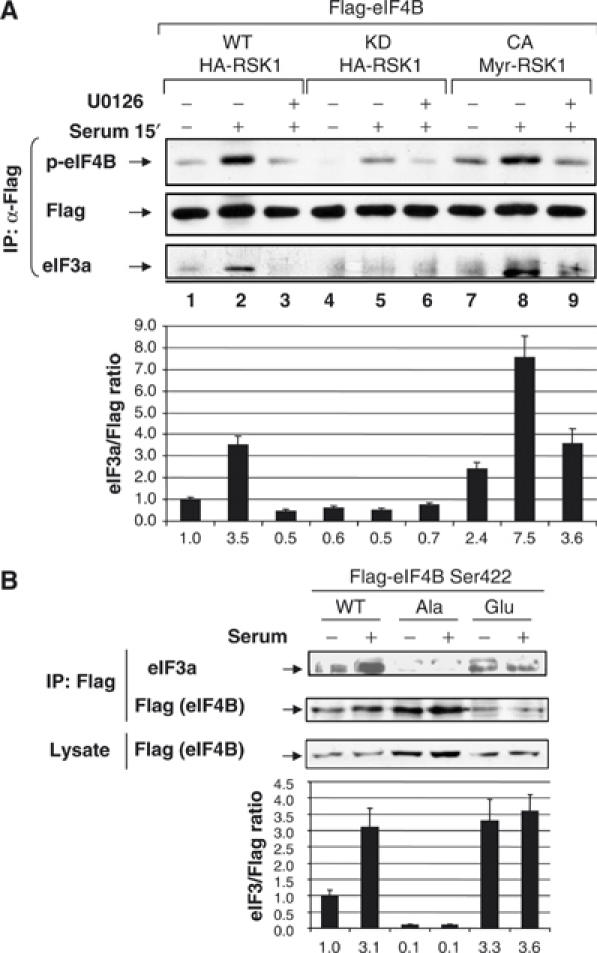
eIF4B Ser422 phosphorylation results in enhanced interaction between eIF4B and a complex containing eIF3. (A) HeLa cells cotransfected with Flag-tagged eIF4B and wt, kinase-dead, and constitutively active RSK variants were starved for 16–18 h in the presence or absence of 10 μM U0126 before serum stimulation for 15 min. Immunoprecipitation of Flag-tagged eIF4B was carried out using anti-Flag (M2) antibody. Immune complexes were subjected to SDS–PAGE and probed with a phosphospecific eIF4B S422 antibody and an eIF3a (p170) antibody. Membranes were reprobed with anti-Flag antibody. (B) HeLa cells were transfected with Flag-tagged wt eIF4B and Ser422 point mutants: Ser422Ala and Ser422Glu. After 16–18 h of serum starvation, cells were stimulated with serum for 15 min, and samples were processed as in (A).
Finally, to demonstrate that eIF4B Ser422 phosphorylation is important for its interaction with eIF3, we examined the ability of eIF4B point mutants to bind eIF3. HeLa cells were transfected with plasmids encoding Flag-tagged wt and Ser422Ala (non-phosphorylatable) and Ser422Glu (phosphomimetic) point mutants of eIF4B. Cells were serum starved for 16–18 h before serum stimulation for 15 min (Figure 6B). Cells were lysed and immune complexes precipitated with anti-Flag antibody were subjected to SDS–PAGE and Western blot analysis using anti-Flag and anti-eIF3a antibodies. As shown above, wt eIF4B exhibited enhanced interaction with eIF3a upon serum stimulation (∼3.1-fold increase; Figure 6B). Strikingly, the non-phosphorylatable Ser422Ala mutant showed a decreased interaction with eIF3 under both serum-starved and serum-stimulated conditions (∼10% of unstimulated wt control). A phosphomimetic mutant of eIF4B (Ser422Glu) exhibited a constitutive high level of interaction between eIF4B and eIF3 (∼3.3- to 3.6-fold increase as compared to wt control). These data thus indicate that the interaction between eIF4B and eIF3 is regulated through the phosphorylation of eIF4B on Ser422. Similar results were recently published by Holz et al (2005).
Discussion
Here we demonstrate that two major signaling pathways involved in translational control converge to phosphorylate eIF4B on Ser422 (Figure 7). This conclusion is based on the following results: (a) Ser422 phosphorylation is sensitive to both a pharmacological inhibitor of MEK, U0126, and the mTOR inhibitor rapamycin, (b) Ser422 phosphorylation is observed in S6K1/2 DKO cells, (c) eIF4B phosphorylation is dependent upon functional PDK1, and serum-induced Ser422 phosphorylation requires active RSK protein, and (d) RSK directly phosphorylates eIF4B in vitro. In addition, we show that eIF4B phosphorylation results in an enhanced interaction between eIF4B and eIF3. Importantly, the expression of a phosphomimetic Ser422Asp mutant of eIF4B in cells resulted in increased translation (Holz et al, 2005). Moreover, RNAi against RSK1/2 resulted in reduced eIF4B phosphorylation and inhibited cap-dependent translation. Thus, the temporal serum-induced biphasic phosphorylation of Ser422, first by the MAPK signaling module, and subsequently by the PI3K/Akt/mTOR cascade (Figure 1D), is likely to be of biological significance.
Figure 7.
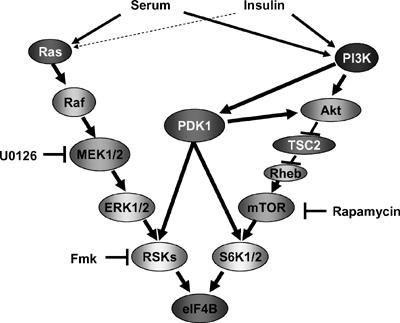
Signaling pathways involved in eIF4B Ser422 phosphorylation. Growth factor-activated MAPK and PI3K cascades activate RSK and S6K proteins correspondingly and converge at the level of eIF4B phosphorylation. In systems where insulin is a marginal activator (dashed arrow) of MAPK cascade, insulin-induced eIF4B phosphorylation is absolutely sensitive to rapamycin. PDK1 protein plays a central role in activation of both RSK and S6K proteins and is indispensable for eIF4B phosphorylation.
Consistent with our data, the recovery of translation in human kidney cells after hypertonic stress-induced translational shut off requires phosphorylation of downstream substrates of both the ERK1/2 MAPK and PI3K signaling modules (Naegele and Morley, 2004). Also, activation of the MAPK and PI3K signaling pathways results in the recruitment of a large number of mRNAs (∼200) to polysomes (Rajasekhar et al, 2003). It is noteworthy that cytokine-driven mitogenesis is also dependent on two temporally distinct phases of signaling; the first is the ERK1/2 MAPK cascade activity, and the second is the PI3K pathway (Jones and Kazlauskas, 2001; Mirza et al, 2004). IL-2-induced hematopoietic cell proliferation is dependent on MAPK effectors (c-myc, c-fos, and c-jun) and rapamycin-sensitive bcl-2 expression (Miyazaki et al, 1995). It is, however, unlikely that the two converging signaling cascades have a redundant role in eIF4B phosphorylation and compensate for each other's function, because of the transient nature of the ERK1/2 MAPK cascade-mediated eIF4B phosphorylation as opposed to a later, more sustained phosphorylation mediated by the PI3K–mTOR-dependent pathway. Kinetics of mitogen-stimulated ERK1/2 MAPK cascade activation in cells is typically faster than PI3K–mTOR module activation, and thus allows for a more precise regulation and an immediate response (e.g. transcription, translation, etc.). Thus, it is plausible that ERK1/2 MAPK-mediated transient phosphorylation of eIF4B fills the temporal gap that exists between mitogenic stimuli and PI3K–mTOR pathway activation to more closely orchestrate mitogenic cues and rates of translation.
In addition to eIF4B, the S6K and RSK family members phosphorylate upstream components of the signaling pathways that lead to eIF4B phosphorylation. These include TSC2 and Sos by RSK, and mTOR and IRS1 by S6K. Inactivation of Ras-GAP results in robust phosphorylation of S6 through Ras-mediated PI3K/mTOR pathway activation (Dasgupta et al, 2005). Also, ERK1/2 phosphorylates TSC2, leading to the dissociation of TSC2 from TSC1 and subsequent inactivation of the complex (Ma et al, 2005) and derepression of mTOR activity. This complex pattern of phosphorylation is a hallmark of all signaling pathways, as it engenders essential signal amplification and establishes checkpoints and feedback regulation loops. The RSKs have previously been implicated in translational control: activated RSK translocates to polysomes, where it stimulates the phosphorylation of several ribosome-associated proteins (Angenstein et al, 1998). The RSKs also phosphorylate and inactivate GSK3 to stimulate translation (Eldar-Finkelman et al, 1995; Torres et al, 1999). Both S6K and RSK phosphorylate and inhibit elongation factor 2 kinase (Wang et al, 2001).
eIF4B stimulates the helicase activity of eIF4A (Lawson et al, 1989; Rozen et al, 1990), and interacts with eIF3 (Methot et al, 1996). This interaction is presumably required for stabilization of the bridge between the mRNA and eIF3 through eIF4G. Here, we present evidence that eIF4B phosphorylation on Ser422 stimulates the interaction between eIF4B and eIF3. Although we have not demonstrated this, it is likely to stimulate the direct interaction between eIF4B and eIF3. The eIF4B–eIF3 interaction correlates with increased translation rates in cells upon eIF4B phosphorylation. It is also possible that Ser422 phosphorylation increases the stimulatory effect of eIF4B exerted on the eIF4A-mediated helicase activity. Recently, Dmitriev et al (2003)showed that eIF4B is obligatory for 48S ribosome initiation complex formation on mRNAs, which possess even a relatively low complexity in their 5′UTRs. They reported that recombinant eIF4B protein poorly substituted for the native factor, suggesting that a post-translational modification, which is absent in bacteria (e.g. phosphorylation), is important for eIF4B function. Importantly, as aforementioned, Holz et al (2005) demonstrated recently that phosphorylated eIF4B stimulates cap-dependent translation in vivo (Holz et al, 2005). Although these results are in contrast to earlier reports, which showed an inhibition of translation by eIF4B overexpression (Naranda et al, 1994; Raught et al, 2004), it is possible that the discrepancies are due to different expression levels of the exogenous eIF4B. Highly overexpressed eIF4B (25- to 50-fold) can be inhibitory to translation due to its potential interference with endogenous complexes by creating inactive pools of physiological eIF4B interacting partners (eIF4A, eIF3, PABP, etc.).
MAPK and PI3K signaling pathways stimulate translation by increasing the rates of translation initiation and elongation, and by stimulating ribosome biogenesis (Holland et al, 2004). Cooperation between these two major signaling pathways results in preferential increase in ribosome recruitment of mRNAs that encode oncogenic proteins in glial cells (Rajasekhar et al, 2003). In light of the importance of eIF4B phosphorylation for its function, this report presents a new paradigm for the interaction between PI3K/Akt/mTOR and Ras/MAPK cascades in controlling translation.
Materials and methods
Constructs
Flag-tagged eIF4B in a pcDNA3 vector was previously described (Raught et al, 2004). Plasmids encoding the HA-tagged wt S6K, HA-tagged wt and kinase-dead avian RSK1, and constitutively active myristoylated avian RSK1 were described elsewhere (Roux et al, 2004). The bicistronic Renilla-HCV IRES-firefly luciferase plasmid was published (Kruger et al, 2001).
Cell culture/transfections
Human cervical carcinoma-derived HeLa R19 cells were maintained in Dulbecco's modified Eagle's medium (DMEM; Sigma) supplemented with 10% fetal bovine serum (FBS; Gibco). Transfections were carried out using Lipofectamine 2000 (Invitrogen) following the manufacturer's instructions. Cells were grown to 80–90% confluence before overnight serum withdrawal. Cells were treated with 20 nM rapamycin (Sigma) or 10 μM fmk (Cohen et al, 2005) overnight, or 10 μM U0126 (Promega) for 2 h before stimulation with 20% serum or 100 nM insulin (Sigma) as indicated in the figure legends. Murine PDK1+/+, PDK1−/−, and PDK PIF pocket mutant cells were a kind gift of Dr Alessi. ES cells were grown on gelatinized plasticware in KnockOut DMEM containing 15% KnockOut serum (Gibco) supplemented with 0.1 mM non-essential amino acids, antibiotics (100 U penicillin G, 100 μg/ml streptomycin), 2 mM L-glutamine, 0.1 mM β-mercaptoethanol, and 1000 U/ml ESGRO (Leukemia inhibitory factor, used to prevent differentiation of ES cells) (Gibco). Cells were grown to 80% confluence, serum starved for 16 h in the presence or absence of 20 nM rapamycin, and stimulated with 20% serum.
S6K mutant mice and primary cell cultures
The generation of S6K1- and S6K2-deficient mice was previously described (Shima et al, 1998; Pende et al, 2004). Adult male mice in a mixed C57Bl/6-129Ola genetic background were used. Primary hepatocytes from 12- to 14-week-old male mice were isolated by liver perfusion as described previously (Pende et al, 2004). After 3 h of adhesion, cells were incubated for 2 days in serum-free M-199 medium containing 1 mg of BSA/ml. Cells were incubated overnight in amino acid- and glucose-free media. The next day, the hepatocytes were pretreated for 30 min with 20 nM rapamycin and stimulated for 1 h with growth factors (10% FBS or 1 μM insulin).
Antibodies/immunoprecipitation/Western blotting
Anti-Flag (M2) and anti-HA mouse monoclonal antibodies were from Sigma. Anti-4E-BP1, anti-RSK1, and anti-RSK2 rabbit polyclonal antibodies were from Zymed. Anti-avian RSK1 antibody was previously characterized (Roux et al, 2004). Anti-eIF4G and anti-eIF4B rabbit antisera were described before (Methot et al, 1996; Ferraiuolo et al, 2005). A monoclonal antibody against eIF3a p170 was a kind gift of Dr Altmann (Mengod and Trachsel, 1985). All other antibodies were purchased from Cell Signaling Technology (Beverly, MA). Flag-tagged eIF4B was immunoprecipitated from 1 mg cell lysate protein extracted from transiently transfected HeLa cells. The samples were incubated at 4°C overnight with 4 μg of anti-Flag (M2) antibody and immune complexes were collected for two additional hours by 20 μl of protein G-Sepharose beads. Resultant pellets were washed three times with 1 ml of RIPA buffer (50 mM Tris–HCl, pH 7.4, 1% NP-40, 150 mM NaCl, 1 mM EDTA, 1 mM PMSF, 10 μg/ml each of aprotinin, leupeptin, pepstatin, 1 mM Na3VO4, 1 mM NaF). Proteins were denatured by addition of 5 × sample buffer (312.5 mM Tris–HCl, pH 6.8, 5% SDS, 10 mM EDTA, 0.5 M DTT, 0.25% bromophenol blue, 50% glycerol) and subjected to SDS–PAGE followed by blotting onto nitrocellulose membrane. Membranes were blocked with 5% BSA solution and probed with phosphospecific eIF4B Ser422 antibodies (Raught et al, 2004). For loading control, membranes were stripped in acidic buffer (0.2 M glycine, 0.5 M NaCl, pH 2.8) and reprobed using anti-Flag (M2) antibody. Experiments were repeated at least three times. Data were quantified using NIH Image software (unless stated otherwise) and standard deviations ranged between 4 and 21%. Representative results are shown.
In vitro kinase assay
HeLa cells were transfected with HA-tagged wt RSK1 and S6K1 or kinase-dead RSK1 using Fugene 6 according to the manufacturer's instructions (Roche Diagnostics, Indianapolis, IN). At 24 h following transfection, cells were serum starved for 16–18 h, then stimulated with serum or insulin in the presence or absence of 10 μM U0126 or 20 nM rapamycin and lysed in cell lysis buffer (CLB: 10 mM K3PO4, 1 mM EDTA, 5 mM EGTA, 10 mM MgCl2, 50 mM β-glycerophosphate, 0.5% NP-40, 0.1% Brij 35, 0.1% deoxycholic acid, 1 mM sodium orthovanadate (Na3VO4), 1 mM phenylmethylsulfonyl fluoride, 5 μg/ml of leupeptin, 10 μg/ml of pepstatin). Lysates were incubated with anti-HA antibodies for 2 h and then with protein A-Sepharose for an additional 1 h at 4°C. Beads were washed three times in CLB and once in kinase buffer (25 mM Tris–HCl, pH 7.4, 10 mM MgCl2, 5 mM β-glycerophosphate). Kinase assays were performed with recombinant eIF4B (purified as in Pause and Sonenberg, 1992) as a substrate (2 μg per assay) and were completed in the linear range of substrate phosphorylation. Reaction products were subjected to SDS–PAGE, and 32P incorporation was quantified using a Bio-Rad PhosphorImager.
RNAi against RSK1 and RSK2
For the siRNA studies, 21 nt complementary RNA with symmetrical 2 nt overhangs was obtained from Qiagen (Valencia, CA). The DNA sequences against which double-stranded RNAs for RSK1 and RSK2 were created were CCCAACATCATCACTCTGAAA and AGCGCTGAGAATGGACAGCAA, respectively, and the mock sequence was TATTCTCCGAACGTGTCACGT. HeLa S3 cells were transfected using Oligofectamine and 0.25–0.5 μg siRNA per 35 mm dish according to the manufacturer's instructions (GIBCO-BRL, Grand Island, NY). Transfection efficiency was determined to be greater than 95% using a fluorescently labeled mock siRNA. At 24 h after transfection, cells were serum starved for 16–18 h, stimulated with either serum or insulin, and then harvested in CLB. The lysates were centrifuged for 5 min at 4°C, adjusted for protein concentration using Bradford assay, and processed for immunoblotting.
Bicistronic luciferase assay
For luciferase reporter experiments, HEK293E cells were transfected with pRL-HCV-FL reporter plasmid and the indicated siRNAs. At 48 h post-transfection, cells were harvested and the luciferase activity was measured using Dual-Luciferase Reporter Assay System (Promega) and Turner Designs TD-20/20 luminometer according to the manufacturers' instructions.
Acknowledgments
We thank D Alessi for PDK1 null and PIF pocket KI ES cells, N Methot and H Imataka for eIF4B and eIF4G antisera, M Altmann for eIF3a antibody, A-C Gingras for eIF4B point mutant constructs, and C Lister for invaluable technical assistance. This work was supported by grants from the Canadian Institute of Health Research (CIHR) and the Howard Hughes Medical Institute (HHMI) to NS; NIH grants #R37 CA46595 and #RO1 GM5140 to JB; INSERM Avenir program (R01131KS) and Association Française contre les Myopathies (9971) grants to MP; and NIH grant GM22135 from the USPHS to JH. JT is supported by NIGMS. PR is supported by International Human Frontier Science Program Organization (HFSPO). NS is a CIHR Distinguished Scientist and an HHMI International scholar.
References
- Alessi DR, Andjelkovic M, Caudwell B, Cron P, Morrice N, Cohen P, Hemmings BA (1996) Mechanism of activation of protein kinase B by insulin and IGF-1. EMBO J 15: 6541–6551 [PMC free article] [PubMed] [Google Scholar]
- Alessi DR, Kozlowski MT, Weng QP, Morrice N, Avruch J (1998) 3-Phosphoinositide-dependent protein kinase 1 (PDK1) phosphorylates and activates the p70 S6 kinase in vivo and in vitro. Curr Biol 8: 69–81 [DOI] [PubMed] [Google Scholar]
- Angenstein F, Greenough WT, Weiler IJ (1998) Metabotropic glutamate receptor-initiated translocation of protein kinase p90rsk to polyribosomes: a possible factor regulating synaptic protein synthesis. Proc Natl Acad Sci USA 95: 15078–15083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avruch J, Lin Y, Long X, Murthy S, Ortiz-Vega S (2005) Recent advances in the regulation of the TOR pathway by insulin and nutrients. Curr Opin Clin Nutr Metab Care 8: 67–72 [DOI] [PubMed] [Google Scholar]
- Balendran A, Currie R, Armstrong CG, Avruch J, Alessi DR (1999) Evidence that 3-phosphoinositide-dependent protein kinase-1 mediates phosphorylation of p70 S6 kinase in vivo at Thr-412 as well as Thr-252. J Biol Chem 274: 37400–37406 [DOI] [PubMed] [Google Scholar]
- Biondi RM, Kieloch A, Currie RA, Deak M, Alessi DR (2001) The PIF-binding pocket in PDK1 is essential for activation of S6K and SGK, but not PKB. EMBO J 20: 4380–4390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blenis J (1993) Signal transduction via the MAP kinases: proceed at your own RSK. Proc Natl Acad Sci USA 90: 5889–5892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown-Luedi ML, Meyer LJ, Milburn SC, Yau PM, Corbett S, Hershey JW (1982) Protein synthesis initiation factors from human HeLa cells and rabbit reticulocytes are similar: comparison of protein structure, activities, and immunochemical properties. Biochemistry 21: 4202–4206 [DOI] [PubMed] [Google Scholar]
- Burnett PE, Barrow RK, Cohen NA, Snyder SH, Sabatini DM (1998) RAFT1 phosphorylation of the translational regulators p70 S6 kinase and 4E-BP1. Proc Natl Acad Sci USA 95: 1432–1437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen MS, Zhang C, Shokat KM, Taunton J (2005) Structural bioinformatics-based design of selective, irreversible kinase inhibitors. Science 308: 1318–1321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins BJ, Deak M, Arthur JS, Armit LJ, Alessi DR (2003) In vivo role of the PIF-binding docking site of PDK1 defined by knock-in mutation. EMBO J 22: 4202–4211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dasgupta B, Yi Y, Chen DY, Weber JD, Gutmann DH (2005) Proteomic analysis reveals hyperactivation of the mammalian target of rapamycin pathway in neurofibromatosis 1-associated human and mouse brain tumors. Cancer Res 65: 2755–2760 [DOI] [PubMed] [Google Scholar]
- Dmitriev SE, Terenin IM, Dunaevsky YE, Merrick WC, Shatsky IN (2003) Assembly of 48S translation initiation complexes from purified components with mRNAs that have some base pairing within their 5′ untranslated regions. Mol Cell Biol 23: 8925–8933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duncia JV, Santella JB III, Higley CA, Pitts WJ, Wityak J, Frietze WE, Rankin FW, Sun JH, Earl RA, Tabaka AC, Teleha CA, Blom KF, Favata MF, Manos EJ, Daulerio AJ, Stradley DA, Horiuchi K, Copeland RA, Scherle PA, Trzaskos JM, Magolda RL, Trainor GL, Wexler RR, Hobbs FW, Olson RE (1998) MEK inhibitors: the chemistry and biological activity of U0126, its analogs, and cyclization products. Bioorg Med Chem Lett 8: 2839–2844 [DOI] [PubMed] [Google Scholar]
- Eldar-Finkelman H, Seger R, Vandenheede JR, Krebs EG (1995) Inactivation of glycogen synthase kinase-3 by epidermal growth factor is mediated by mitogen-activated protein kinase/p90 ribosomal protein S6 kinase signaling pathway in NIH/3T3 cells. J Biol Chem 270: 987–990 [DOI] [PubMed] [Google Scholar]
- Etchison D, Milburn SC, Edery I, Sonenberg N, Hershey JW (1982) Inhibition of HeLa cell protein synthesis following poliovirus infection correlates with the proteolysis of a 220 000-dalton polypeptide associated with eucaryotic initiation factor 3 and a cap binding protein complex. J Biol Chem 257: 14806–14810 [PubMed] [Google Scholar]
- Ferraiuolo MA, Basak S, Dostie J, Murray EL, Schoenberg DR, Sonenberg N (2005) A role for the eIF4E-binding protein 4E-T in P-body formation and mRNA decay. J Cell Biol 170: 913–924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frodin M, Gammeltoft S (1999) Role and regulation of 90 kDa ribosomal S6 kinase (RSK) in signal transduction. Mol Cell Endocrinol 151: 65–77 [DOI] [PubMed] [Google Scholar]
- Frodin M, Jensen CJ, Merienne K, Gammeltoft S (2000) A phosphoserine-regulated docking site in the protein kinase RSK2 that recruits and activates PDK1. EMBO J 19: 2924–2934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gingras AC, Raught B, Sonenberg N (1999) eIF4 initiation factors: effectors of mRNA recruitment to ribosomes and regulators of translation. Annu Rev Biochem 68: 913–963 [DOI] [PubMed] [Google Scholar]
- Hershey JW, Merrick WC (2000) Pathway and mechanism of initiation of protein synthesis. In Translational Control of Gene Expression, Sonenberg N, Hershey JW, Mathews MB (eds) pp 33–89. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press [Google Scholar]
- Holland EC, Sonenberg N, Pandolfi PP, Thomas G (2004) Signaling control of mRNA translation in cancer pathogenesis. Oncogene 23: 3138–3144 [DOI] [PubMed] [Google Scholar]
- Holz MK, Ballif BA, Gygi SP, Blenis J (2005) mTOR and S6K1 mediate assembly of the translation preinitiation complex through dynamic protein interchange and ordered phosphorylation events. Cell 123: 569–580 [DOI] [PubMed] [Google Scholar]
- Jensen CJ, Buch MB, Krag TO, Hemmings BA, Gammeltoft S, Frodin M (1999) 90-kDa ribosomal S6 kinase is phosphorylated and activated by 3-phosphoinositide-dependent protein kinase-1. J Biol Chem 274: 27168–27176 [DOI] [PubMed] [Google Scholar]
- Jones SM, Kazlauskas A (2001) Growth-factor-dependent mitogenesis requires two distinct phases of signalling. Nat Cell Biol 3: 165–172 [DOI] [PubMed] [Google Scholar]
- Kruger M, Beger C, Welch PJ, Barber JR, Manns MP, Wong-Staal F (2001) Involvement of proteasome alpha-subunit PSMA7 in hepatitis C virus internal ribosome entry site-mediated translation. Mol Cell Biol 21: 8357–8364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawson TG, Lee KA, Maimone MM, Abramson RD, Dever TE, Merrick WC, Thach RE (1989) Dissociation of double-stranded polynucleotide helical structures by eukaryotic initiation factors, as revealed by a novel assay. Biochemistry 28: 4729–4734 [DOI] [PubMed] [Google Scholar]
- Leighton IA, Dalby KN, Caudwell FB, Cohen PT, Cohen P (1995) Comparison of the specificities of p70 S6 kinase and MAPKAP kinase-1 identifies a relatively specific substrate for p70 S6 kinase: the N-terminal kinase domain of MAPKAP kinase-1 is essential for peptide phosphorylation. FEBS Lett 375: 289–293 [DOI] [PubMed] [Google Scholar]
- Ma L, Chen Z, Erdjument-Bromage H, Tempst P, Pandolfi PP (2005) Phosphorylation and functional inactivation of TSC2 by Erk implications for tuberous sclerosis and cancer pathogenesis. Cell 121: 179–193 [DOI] [PubMed] [Google Scholar]
- Mengod G, Trachsel H (1985) Eukaryotic protein synthesis initiation factor eIF-3: determination of concentration and association with ribosomes in rabbit reticulocyte and HeLa cell lysates. Biochim Biophys Acta 825: 169–174 [DOI] [PubMed] [Google Scholar]
- Methot N, Song MS, Sonenberg N (1996) A region rich in aspartic acid, arginine, tyrosine, and glycine (DRYG) mediates eukaryotic initiation factor 4B (eIF4B) self-association and interaction with eIF3. Mol Cell Biol 16: 5328–5334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirza AM, Gysin S, Malek N, Nakayama K, Roberts JM, McMahon M (2004) Cooperative regulation of the cell division cycle by the protein kinases RAF and AKT. Mol Cell Biol 24: 10868–10881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyazaki T, Liu ZJ, Kawahara A, Minami Y, Yamada K, Tsujimoto Y, Barsoumian EL, Permutter RM, Taniguchi T (1995) Three distinct IL-2 signaling pathways mediated by bcl-2, c-myc, and lck cooperate in hematopoietic cell proliferation. Cell 81: 223–231 [DOI] [PubMed] [Google Scholar]
- Mora A, Komander D, van Aalten DM, Alessi DR (2004) PDK1, the master regulator of AGC kinase signal transduction. Semin Cell Dev Biol 15: 161–170 [DOI] [PubMed] [Google Scholar]
- Naegele S, Morley SJ (2004) Molecular cross-talk between MEK1/2 and mTOR signaling during recovery of 293 cells from hypertonic stress. J Biol Chem 279: 46023–46034 [DOI] [PubMed] [Google Scholar]
- Naranda T, Strong WB, Menaya J, Fabbri BJ, Hershey JW (1994) Two structural domains of initiation factor eIF-4B are involved in binding to RNA. J Biol Chem 269: 14465–14472 [PubMed] [Google Scholar]
- Obata T, Yaffe MB, Leparc GG, Piro ET, Maegawa H, Kashiwagi A, Kikkawa R, Cantley LC (2000) Peptide and protein library screening defines optimal substrate motifs for AKT/PKB. J Biol Chem 275: 36108–36115 [DOI] [PubMed] [Google Scholar]
- Park IH, Bachmann R, Shirazi H, Chen J (2002) Regulation of ribosomal S6 kinase 2 by mammalian target of rapamycin. J Biol Chem 277: 31423–31429 [DOI] [PubMed] [Google Scholar]
- Pause A, Sonenberg N (1992) Mutational analysis of a DEAD box RNA helicase: the mammalian translation initiation factor eIF-4A. EMBO J 11: 2643–2654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pende M, Um SH, Mieulet V, Sticker M, Goss VL, Mestan J, Mueller M, Fumagalli S, Kozma SC, Thomas G (2004) S6K1(−/−/S6K2(−/−) mice exhibit perinatal lethality and rapamycin-sensitive 5′-terminal oligopyrimidine mRNA translation and reveal a mitogen-activated protein kinase-dependent S6 kinase pathway. Mol Cell Biol 24: 3112–3124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajasekhar VK, Viale A, Socci ND, Wiedmann M, Hu X, Holland EC (2003) Oncogenic Ras and Akt signaling contribute to glioblastoma formation by differential recruitment of existing mRNAs to polysomes. Mol Cell 12: 889–901 [DOI] [PubMed] [Google Scholar]
- Raught B, Gingras A-C, Sonenberg N (2000) Regulation of ribosomal recruitment in eukaryotes. In Translational Control of Gene Expression, Sonenberg N, Hershey JW, Mathews MB (eds) pp 245–295. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press [Google Scholar]
- Raught B, Peiretti F, Gingras AC, Livingstone M, Shahbazian D, Mayeur GL, Polakiewicz RD, Sonenberg N, Hershey JW (2004) Phosphorylation of eucaryotic translation initiation factor 4B Ser422 is modulated by S6 kinases. EMBO J 23: 1761–1769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roux PP, Ballif BA, Anjum R, Gygi SP, Blenis J (2004) Tumor-promoting phorbol esters and activated Ras inactivate the tuberous sclerosis tumor suppressor complex via p90 ribosomal S6 kinase. Proc Natl Acad Sci USA 101: 13489–13494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roux PP, Blenis J (2004) ERK and p38 MAPK-activated protein kinases: a family of protein kinases with diverse biological functions. Microbiol Mol Biol Rev 68: 320–344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozen F, Edery I, Meerovitch K, Dever TE, Merrick WC, Sonenberg N (1990) Bidirectional RNA helicase activity of eucaryotic translation initiation factors 4A and 4F. Mol Cell Biol 10: 1134–1144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shima H, Pende M, Chen Y, Fumagalli S, Thomas G, Kozma SC (1998) Disruption of the p70(s6k)/p85(s6k) gene reveals a small mouse phenotype and a new functional S6 kinase. EMBO J 17: 6649–6659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torres MA, Eldar-Finkelman H, Krebs EG, Moon RT (1999) Regulation of ribosomal S6 protein kinase-p90(rsk), glycogen synthase kinase 3, and beta-catenin in early Xenopus development. Mol Cell Biol 19: 1427–1437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volarevic S, Thomas G (2001) Role of S6 phosphorylation and S6 kinase in cell growth. Prog Nucleic Acid Res Mol Biol 65: 101–127 [DOI] [PubMed] [Google Scholar]
- Vornlocher HP, Hanachi P, Ribeiro S, Hershey JW (1999) A 110-kilodalton subunit of translation initiation factor eIF3 and an associated 135-kilodalton protein are encoded by the Saccharomyces cerevisiae TIF32 and TIF31 genes. J Biol Chem 274: 16802–16812 [DOI] [PubMed] [Google Scholar]
- Wang X, Li W, Williams M, Terada N, Alessi DR, Proud CG (2001) Regulation of elongation factor 2 kinase by p90(RSK1) and p70 S6 kinase. EMBO J 20: 4370–4379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams MR, Arthur JS, Balendran A, van der Kaay J, Poli V, Cohen P, Alessi DR (2000) The role of 3-phosphoinositide-dependent protein kinase 1 in activating AGC kinases defined in embryonic stem cells. Curr Biol 10: 439–448 [DOI] [PubMed] [Google Scholar]