

Abstract
In vegetative cells, most recombination intermediates are metabolized without an association with a crossover (CO). The avoidance of COs allows for repair and prevents genomic rearrangements, potentially deleterious if the sequences involved are at ectopic locations. We have designed a system that permits to screen spontaneous intragenic recombination events in Saccharomyces cerevisiae and to investigate the CO outcome in different genetic contexts. We have analyzed the CO outcome in the absence of the Srs2 and Sgs1 helicases, DNA damage checkpoint proteins as well as in a mutant proliferating cell nuclear antigen (PCNA) and found that they all contribute to genome stability. Remarkably high effects on COs are mediated by srs2Δ, mrc1Δ and a pol30-RR mutation in PCNA. Our results support the view that Mrc1 plays a specific role in DNA replication, promoting the Srs2 recruitment to PCNA independently of checkpoint signaling. Srs2 would prevent formation of double Holliday junctions (dHJs) and thus CO formation. Sgs1 also negatively regulates CO formation but through a different process that resolves dHJs to yield non-CO products.
Keywords: checkpoint, crossover, helicase, mitotic recombination, PCNA
Introduction
When the replication machinery encounters a nick in DNA, the fork collapses and creates a double-strand break (DSB) (Kuzminov, 2001). Repair of this break is essential for cell survival and requires homologous recombination (HR). However, a large amount of replicative damage, like local single-stranded regions or gaps, does not result in the interruption of DNA integrity (Fabre et al, 2002). The observation that yeast cells are alive in the absence of the key recombination genes indicates that, unlike in meiosis, HR is not an essential process in vegetative yeast cells (for review, see Pâques and Haber, 1999). Additionally, while cell viability does not require HR genes following UV irradiation, recombination is strongly stimulated. This result underpins the idea that lesions other than DSBs are potent instigators of recombinational repair that may trigger genome instability.
Previous studies in Saccharomyces cerevisiae have established that in the absence of several combinations of genes, intermediates are formed that are toxic if the early steps of HR are functional. The sgs1 srs2, sgs1 mus81 (Gangloff et al, 2000; Fabre et al, 2002) and srs2 rad54 (Heude et al, 1995; Schild, 1995) double deletions belong to this category, and we have postulated that the corresponding gene products are involved at various stages of the recombination process (Fabre et al, 2002). Recombination structures formed spontaneously during normal growth are potential hazards to the cell, especially when they involve sequences present on either the same or a nonhomologous chromosome (Elliott and Jasin, 2002). It has been observed that wild-type (WT) cells generate deleterious rearrangements infrequently; therefore, mitotic cells have evolved efficient systems that limit the association of gene conversion intermediates with a crossover (CO) (Petes et al, 1991). Our previous results had led us to hypothesize that the Mus81/Mms4 complex, a structure-specific endonuclease (Bastin-Shanower et al, 2003), and Sgs1/Top3, a helicase (Lu et al, 1996) that associates with a type-IA topoisomerase (Kim and Wang, 1992; Gangloff et al, 1994), define alternative pathways for processing recombination intermediates (Fabre et al, 2002). Based on its substrate and cleavage specificities, we proposed that Mus81 is active during mitotic recombination in a synthesis-dependent strand annealing (SDSA) pathway (Formosa and Alberts, 1986) in which it efficiently cleaves 3′ protruding ends that result from over-replicating the donor template. On the other hand, Sgs1 and Top3 could operate in a dissolution pathway processing double Holliday junction intermediates (dHJ), as supported by in vitro data with the Blm and Top3α proteins, human orthologues of Sgs1 and Top3, respectively (Wu and Hickson, 2003).
The Sgs1 and Srs2 helicases as well as the Top3 topoisomerase have been described as negative regulators of CO formation in a study in which the recombination event was initiated by a single targeted DSB during vegetative growth (Ira et al, 2003). Sgs1 also downregulates COs during meiosis (Rockmill et al, 2003), where most of the events are initiated with a DSB in the promoter regions of the genes (Baudat and Nicolas, 1997). However, very little is known about the mechanisms and the genes that control the outcome of a spontaneously occurring recombination intermediate.
In our present study, we have designed and used a genetic screen based on an ectopic assay in haploid yeast cells that allows conversion events associated or not with a CO to be differentiated. We used this system to determine the effects of mutations in Mus81, Srs2, Top3 and Sgs1 on the outcome of recombination intermediates formed spontaneously in the course of normal growth. We also address the role of DNA damage checkpoint genes in the resolution of recombination intermediates, as it was shown that their absence causes chromosome loss in diploid cells (Klein, 2001) and a resolution bias in favor of COs between a plasmid and the chromosome (Haghnazari and Heyer, 2004). Finally, because the srs2 mutation increases the bias and because Srs2 was shown to bind preferentially sumoylated proliferating cell nuclear antigen (PCNA), we questioned the role of PCNA in CO control.
Here we show that cells have evolved many strategies to prevent the unnecessary and potential harmful formation of COs during mitotic growth. Sgs1 regulates negatively spontaneous COs probably by merging dHJs into a hemi-catenated structure that requires Top3 to be efficiently resolved as a non-CO. Srs2 favors an SDSA-type of repair at the expense of dHJ formation by acting at two steps, one early by dismantling the Rad51 nucleofilament, and one later in the recombination process. We show that PCNA plays an important role, which is likely the recruitment of Srs2. DNA damage checkpoints all contribute to maintain low levels of COs during mitotic growth, both by stabilizing replication forks and by modulating the resolution process. Finally, our results clearly indicate that Mrc1 has an exclusive function during DNA replication that is independent of checkpoint signaling. This role would consist in controlling the Srs2 activity and therefore promoting negative regulation of COs.
Results
Spontaneous CO recovery assay
We have constructed an assay that allows the intragenic recombination rate of Arg+ formation to be determined and to calculate the frequency of gene conversion associated with a CO among surviving haploid yeast cells. The system is based on two arg4 alleles each carrying a different mutation separated by 1 kb. One allele is located at its endogenous locus on chromosome VIII and the other between a WT and a mutated allele of URA3 on chromosome V, in the same orientation with respect to the centromere (Figure 1A). A recombination event between these ectopically located arg4 alleles can generate a functional copy of the ARG4 gene by gene conversion of a maximum tract length of 1.5 kb associated or not with a CO. A CO leads to a reciprocal translocation that separates the duplicated URA3 and ura3-1 alleles to individual chromosomes. It is therefore possible to directly infer CO events in a secondary screen based on replica plating onto a medium containing a drug (5-fluoroorotic acid (5-FOA)) that kills Ura+ cells (Figure 1B) (Boeke et al, 1984). Indeed, Arg+ colonies resulting from a recombination event associated with a CO will not form many papillations when replica-plated onto 5-FOA, whereas those resulting from a simple gene conversion event that retains the URA3 direct repeats will. To ascertain that events detected in our genetic screen are the outcome of actual COs, we analyzed the DNA of putative CO colonies isolated in our primary screen by pulsed-field gel electrophoresis (PFGE) and probed them for reciprocal translocations (Figure 1C). For every genotype tested, we found a near-perfect correlation between our genetic approach to estimate CO frequencies and the molecular analysis.
Figure 1.
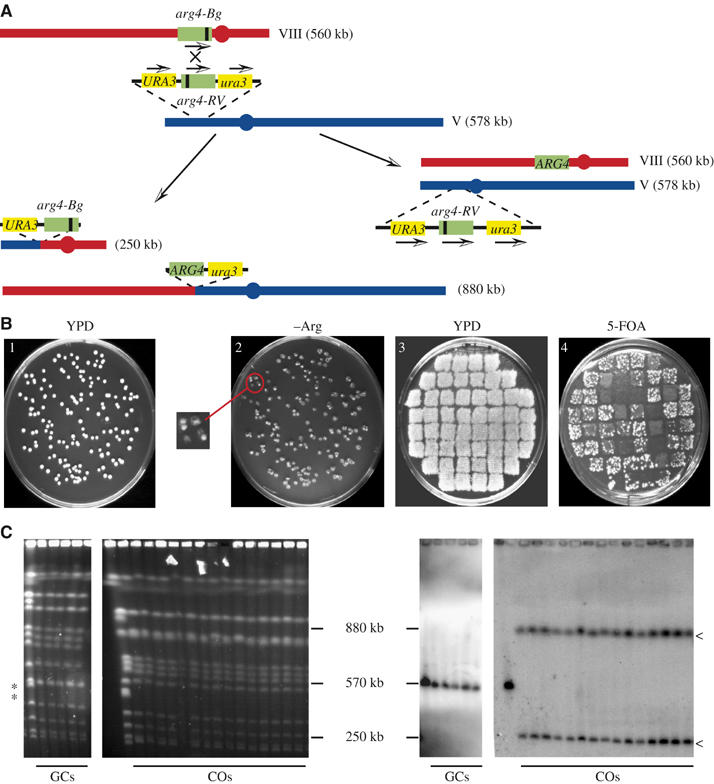
Description of the assay. (A) arg4 heteroalleles are located at its endogenous locus on chromosome VIII and between duplicated alleles of URA3 on chromosome V, in the same orientation with respect to the centromere. A recombination event between these ectopically located arg4 alleles can generate a functional copy of the ARG4 gene by gene conversion associated (left arrow) or not (right arrow) with a CO. (B) Determination of conversion events associated with a CO event. (1) Cells are plated on rich medium, (2) replicated onto arginine-free synthetic medium (a magnified region of the plate shows individual recombinants forming a papillae on a lawn of ghost cells), (3) individual recombinants are patched on rich medium (4) before being replica-plated onto a medium containing 5-FOA. (C) DNA from colonies yielding either papillae (putative GCs) or no papillae (putative COs) was prepared and subjected to clamped homogenous electrical field (CHEF) electrophoresis. Ethidium bromide staining (left panel) or URA3-hybridized Hybond N+ transferred DNA (right panel) confirms the results of the genetic screen. Asterisks (*) identify the mobility of chromosomes VIII (560 kb) and V (578 kb), whereas arrowheads (<) point at the reciprocally translocated products at 880 and 250 kb.
Respective roles of Mus81 and Sgs1 in spontaneous intragenic recombination
Based on our genetic analysis (Fabre et al, 2002) and on the respective known biochemical activities for these proteins (Bennett et al, 1999; Kaliraman et al, 2001), it was proposed that Sgs1 acts on dHJ intermediates, whereas Mus81 cleaves 3′ protruding sequences that can arise during SDSA (de los Santos et al, 2001). The increase in spontaneous gene conversion observed in the absence of SGS1 underpins a replicative function for Sgs1 (Gangloff et al, 1994). However, it is still unclear whether Mus81 also acts upstream of HR in promoting replication damage to be processed by recombination. We addressed the involvement of Mus81 in a similar early function by measuring conversion rates of a mus81Δ deletion mutant. In agreement with previous studies in yeast (Interthal and Heyer, 2000), no significant increase was detected in the absence of Mus81 (Figure 2), suggesting that the mus81Δ deletion mutant does not generate recombinogenic damage during unperturbed replication. With respect to COs, previous studies focusing on induced mitotic DSB repair and meiosis have indicated that Sgs1 negatively regulates COs (Ira et al, 2003; Rockmill et al, 2003) whereas Mus81 participates in meiotic recombination (de los Santos et al, 2003) but does not affect mitotic DSB-induced COs. However, their role in spontaneous recombination is unknown. Therefore, we measured uninduced CO formation using the ectopic assay described above. In WT cells, we found that 11.2% of the conversion events yielding arginine prototrophs (Arg+) are associated with a CO (Figure 2), confirming that CO resolution in vegetative cells is not a prominent event. mus81Δ mutants yield a modest but significant increase in COs (1.5-fold, P<0.01). The absence of Sgs1 generates a 2.5-fold increase in the spontaneous CO frequency, an elevation very similar to that recorded during DSB-induced repair (Ira et al, 2003). This result indicates that Sgs1 indeed plays a role in recombination as a negative regulator of CO during both spontaneous and DSB-induced events.
Figure 2.
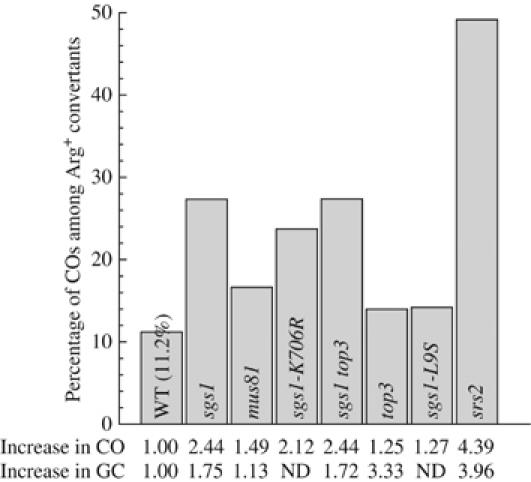
CO bias observed in single mutants. The percentage of CO was determined on a minimum of three individual segregants for each genotype. Homogeneity among each genotype was ascertained first through an ɛ test, and the mean value corresponding to the number of COs divided by the total number of convertants is represented (no s.d.). When percentages were compared to one another, we used the same ɛ test to determine the significance (see Materials and methods). Underneath the graph are the fold increase compared to WT of both COs and total conversion rates (COs and non-COs). The total conversion rate is 6.04 10−7 for our WT control.
The helicase activity of Sgs1 is required for mitotic CO control
We found that Sgs1 negatively controls gene conversion associated with CO. It was shown previously that the helicase activity of Sgs1 is essential for mitotic functions, like MMS and HU sensitivity (Frei and Gasser, 2000; Miyajima et al, 2000; Mullen et al, 2000; Onoda et al, 2000), control of spontaneous intragenic recombination (Ui et al, 2001) and suppression of the growth defect of top3Δ null mutants (S Gangloff, unpublished results). However, it is not known whether the helicase activity of Sgs1 during vegetative growth has a role in the control of CO outcome. We found that, like sgs1Δ, the helicase-dead sgs1-K706R mutant still results in synthetic lethality with mus81Δ (data not shown), indicating that helicase activity is essential in the absence of Mus81 to prevent the formation of toxic structures. Using our molecular genetic screen, we found that the helicase activity of Sgs1 is also essential for the negative control of CO formation (Figure 2), as the helicase-dead allele behaves like the deletion mutant.
Absence of Top3 does not influence the spontaneous CO bias
Sgs1 interacts genetically and physically with the Top3 topoisomerase (Gangloff et al, 1994). The physical interaction with Top3 has been mapped to the amino-terminus of Sgs1 (Bennett et al, 2000) and is required both for the resistance to MMS and the suppression of the hyper-recombination observed in sgs1Δ disruptants (Mullen et al, 2000; Ui et al, 2001). Top3 mutants are extremely sick (Wallis et al, 1989), and unlike Sgs1, Top3 is essential in meiosis (Gangloff et al, 1999). Such a separation of function has also been reported for some mitotic processes (Onodera et al, 2002). Because Top3 is implicated in the resolution of meiotic recombination structures (Gangloff et al, 1999), it is important to determine whether the negative control of Sgs1 on spontaneous mitotic CO depends on Top3. To investigate this question, we first measured the CO frequency in sgs1Δ top3Δ mutants and found that it is identical to that of sgs1Δ alone (Figure 2). We next assayed the CO frequency in a strain deleted for TOP3 alone and did not find an increase of CO although we found a strong increment in the rate of spontaneous intragenic recombination (Figure 2). This result indicates that the absence of Top3 stimulates recombination initiation, but does not perturb the CO frequency. Therefore, whereas DSB-induced COs in the absence of Top3 are elevated to the sgs1Δ level (Ira et al, 2003), no effect was found in spontaneously occurring COs. This result is further supported by the observation that sgs1-L9S mutants, which have lost the ability to interact with Top3 (Duno et al, 2000), behave similarly to WT and top3Δ mutants with respect to COs (Figure 2).
Srs2 may operate at different levels
A key role of the Srs2 helicase consists in downregulating HR by dismantling the Rad51 nucleofilament (Krejci et al, 2003; Veaute et al, 2003). Consistent with this role, absence of Srs2 elevates the spontaneous levels of intragenic recombination four-fold in our assay (Figure 2). In this experimental setup, viability is not affected in a detectable way. This situation is quite different from that where recombination was artificially initiated with a DSB in mostly every cell. In this latter case, the srs2Δ haploid cells, containing an ectopic copy of the target sequence, lose viability, probably because of poor repair (about 30% as measured by densitometry on Southern blots) as well as failure to recover from checkpoint arrest (Vaze et al, 2002; Aylon et al, 2003; Ira et al, 2003). When we measured the percentage of CO among spontaneous Arg+ convertants, we found that up to half the conversion events were associated with a CO (Figure 2), a much more dramatic increase than that observed following a DSB in the ectopic copy (Ira and Haber, 2002). If Srs2 were only active on preventing substrates from being channeled into the recombination pathway by removing Rad51 from single-stranded DNA before strand invasion, we would not expect to observe a biased distribution of COs among the intermediates that escaped this early control. Therefore, we postulate that in the absence of Srs2, either a different intermediate is created that is processed to mainly generate COs or that Srs2 can also operate at another stage in the recombination process.
DNA damage checkpoint genes affect CO resolution to various extents
Earlier work using the yeast Schizosaccharomyces pombe had suggested that the BRCT domain-containing Crb2 checkpoint protein has a function in the later steps of HR that require Rqh1, the S. pombe orthologue of Sgs1 (Caspari et al, 2002). In humans, BRCA1 also contains two BRCT domains at its C-terminus (Scully et al, 1997) and was shown to interact with the BLM DNA helicase, one of several human orthologues of Sgs1 (Wang et al, 2000). As it was shown that BRCA1 binds directly to branched structures and four-way junctions (Paull et al, 2001), it was suggested that BRCA1 and Crb2 could promote the processing of recombination intermediates (Caspari et al, 2002). To test whether Rad9, the Crb2 orthologue in S. cerevisiae (Saka et al, 1997; Willson et al, 1997), influences the CO outcome, we analyzed the effect of a RAD9 deletion in our ectopic assay. We found that the absence of Rad9 elevates the spontaneous rates of ectopic conversion, by a factor of 1.45 (P<0.01) (Figure 3). In addition, resolution associated with a CO is found among 34% of the convertants, indicating an involvement of Rad9 in the resolution process (Figure 3).
Figure 3.
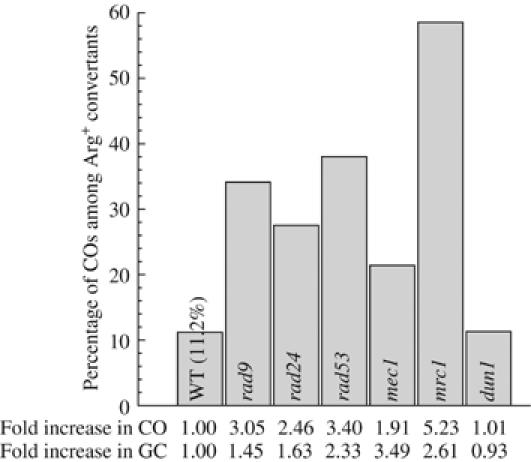
CO bias observed in single checkpoint mutants. Experiments were carried out and compared as in Figure 2.
As a negative effect of mec1, rad53 and dun1 mutants on CO control has been reported previously using a plasmid transformation assay (Haghnazari and Heyer, 2004), we wanted to determine whether the function of Rad9 in modulating resolution is linked to its checkpoint function. Therefore, to reach a comprehensive picture of the process, we explored the consequences of deletions in representative members of the DNA damage sensors (RAD24 and MEC1), adaptors (RAD9 and MRC1) and FHA domain-containing effector kinases (RAD53 and DUN1) (reviewed by Melo and Toczyski, 2002; Nyberg et al, 2002) for their effect on spontaneous recombination and CO. The results summarized in Figure 3 indicate that, except for the Dun1 downstream kinase required for DNA damage-induced transcription (Zhao et al, 2001), loss of any of the other DNA damage checkpoint proteins analyzed leads both to an increase in the spontaneous conversion rate (P<0.05) and an increase in the proportion of associated COs (P<0.01). Surprisingly, we found that the absence of Mec1 leads to the highest level of recombinogenic substrates (a 3.5-fold increase in convertants) whereas association with a CO is the lowest (two-fold increase; Figure 3). However, the absence of the downstream Rad53 effector PI3-kinase, to which most of the signaling converges (reviewed by Melo and Toczyski, 2002; Nyberg et al, 2002), exhibits the strongest effect of all the DNA damage checkpoint mutants with respect to CO control, with the noticeable exception of mrc1Δ. This result suggests that the upstream DNA damage checkpoints transduce a signal to Rad53, which in turn modifies proteins involved in the recombination process. The convergence of this signaling to Rad53 prevents spontaneous recombination intermediates to be resolved in association with a CO.
The checkpoint function of Mrc1 does not play a major role in CO control
The absence of Mrc1 leads to the highest level of spontaneous CO among the Arg+ convertants (58%) (Figure 4A). Mrc1 and Tof1 form a complex present at the replication fork (Katou et al, 2003), but separable functions have been documented recently for replication restart following hydroxyurea treatment (Calzada et al, 2005; Tourrière et al, 2005). Therefore, we determined whether Tof1 and Mrc1 play a different role with respect to CO. Using our assay, we have established that absence of Tof1 only leads to 20% of mitotic COs (Figure 4A). We next investigated if the increase in COs observed in mrc1Δ mutants is linked to its activation by Mec1 during checkpoint response. To address this question, we took advantage of the mrc1AQ allele, in which all the putative SQ or TQ phosphorylation sites for the Mec1 and Tel1 PI3-kinases have been mutated to AQ (Osborn and Elledge, 2003). In this mutant, we found a 2.5-fold increase in COs (25%). Thus, mrc1AQ behaves like a checkpoint mutant and not like the deletion (see Figure 3). Therefore, Mrc1 regulation of COs has both phosphorylation-dependent and phosphorylation-independent components.
Figure 4.
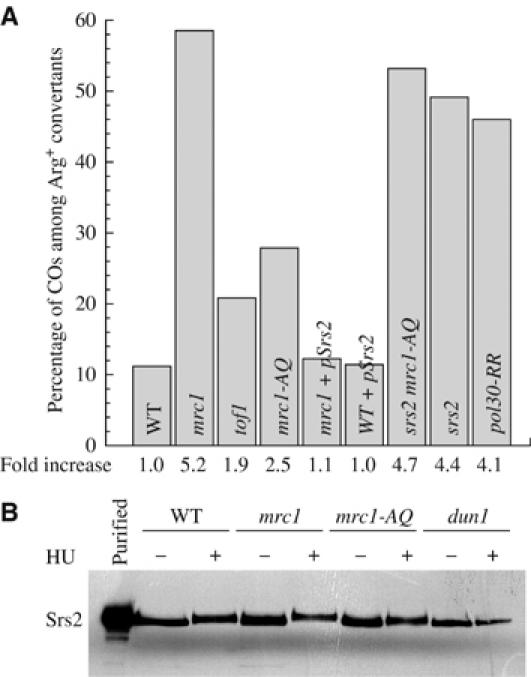
CO control by Mrc1 and Srs2. (A) CO bias in various backgrounds was determined and analyzed as in Figure 2. (B) Phosphorylation status of Srs2 in various backgrounds in the presence or absence of 0.2 M of HU. A 5 ng portion of purified Srs2 was loaded in the control lane.
Mrc1 does not downregulate spontaneous COs by preventing Srs2 phosphorylation
We found that the absence of either Mrc1 or Srs2 leads to a similar increase in COs (Figure 4A), suggesting that Mrc1 and Srs2 could function in the same pathway of CO suppression. Unfortunately, we could not carry out the epistasis analysis, as the mrc1Δ srs2Δ double mutant is not viable (Ooi et al, 2003), a phenotype explained previously as the result of elevated HR initiation in the absence of Mrc1 leading to toxic recombination intermediates formed in the absence of Srs2 (Xu et al, 2004). Interestingly, the mrc1AQ srs2 mutant is fully viable, confirming that Mrc1 phosphorylation is dispensable for cell viability in the absence of Srs2 (Xu et al, 2004). However, it yields a CO bias that is indistinguishable from that observed in srs2 mutants (Figure 4A), suggesting that the Mrc1 checkpoint function operates in the same pathway as Srs2 for limiting COs. Srs2 was shown to be phosphorylated in a Dun1-dependent way during replicative stress, like during growth on hydroxyurea (Liberi et al, 2000). We show here that its phosphorylation status is altered neither in the mrc1Δ nor in the mrc1AQ background (Figure 4B), supporting the idea that a phosphorylation-independent component of Mrc1 plays a major role in CO control. To determine whether the phosphorylation-independent activity of Mrc1 in CO suppression could be related to its ability to help put Srs2 on its cognate substrate, we increased the gene dosage of the Srs2 helicase by introducing a multicopy plasmid containing the SRS2 gene under the control of its own promoter in both an mrc1Δ and a WT strain. We found that excess Srs2 can overcome the CO bias in the absence of Mrc1 but has no effect in its presence (Figure 4A). This result indicates that a physiological amount of Srs2 cannot correctly regulate COs when Mrc1 is absent, a phenotype that can be compensated by additional Srs2 activity. Moreover, it also indicates that the Srs2 CO suppression is maximal when physiological amounts of Mrc1 are present in the cell.
PCNA links Mrc1 to Srs2
One way to explain our results involves a direct interaction between Srs2 and Mrc1. To investigate this possibility, we have performed both two-hybrid and co-immunoprecipitation experiments to uncover a putative physical interaction between these proteins. None of our experiments allowed us to reach this conclusion, even in situations where all the cells were synchronized in S phase by α-factor arrest and released into hydroxyurea-containing medium (data not shown). Another possibility is that the interaction is indirect. It has been shown recently that Srs2 could be recruited to the replication fork through an interaction with a sumoylated form of PCNA (Papouli et al, 2005; Pfander et al, 2005). We tested this idea by measuring the CO bias in the pol30-RR mutant encoding a PCNA that can neither be ubiquitinated on lysine 164 nor sumoylated on lysines 127 and 164 (Hoege et al, 2002). As shown in Figure 4A, we found that this mutant behaves like srs2 mutants with respect to CO control, strongly suggesting that sumoylated PCNA is the link between Mrc1 and Srs2.
Discussion
Several reports had suggested that Mus81 could be involved in generating a recombinogenic substrate at the replication fork under normal growth conditions (Kaliraman et al, 2001; Doe et al, 2002). Here, we show that mus81 mutants do not affect the rate of spontaneous gene conversion in our assay, which indicates that Mus81 is not creating recombinogenic substrates in the absence of exogenous damage. With respect to the processing of recombination intermediates, we found that the absence of Mus81 leads to a slight but significant (P<0.01) increase in the percentage of COs. One way to explain this result is consistent with the model in which Mus81 cuts the 3′ protruding flap resulting from the rejection of the elongated invading strand during SDSA. Failure to process this flap structure allows the single strand to re-invade the donor duplex, offering a new chance to engage into a dHJ that can be potentially resolved associated with a CO (Fabre et al, 2002). The observation that a mus81 mutation does not decrease the occurrence of spontaneous COs further strengthens the idea that Mus81 is not a key player in dHJ resolution during mitosis in S. cerevisiae.
The helicase activity of Sgs1 acts as a negative regulator of spontaneous COs, a result consistent with that reported for DSB-induced lesions, both mitotically and meiotically (Ira et al, 2003; Rockmill et al, 2003). The observation that absence of Top3 stimulates recombination greatly at the level of initiation but has no effect on the resolution bias can be explained in light of the results described for the human counterparts of Top3 and Sgs1 (Wu and Hickson, 2003). As the sgs1Δ top3Δ double mutants behave like sgs1Δ mutants or sgs1-K706R helicase-defective mutants (Figure 2), we have to assume that it is the helicase function of Sgs1 that is important for the negative control. As was shown in vitro for BLM, Sgs1 could merge a dHJ and create a stable hemi-catenated intermediate that can be subsequently resolved by dissolution through the specific single-strand decatenating activity of Top3 (Wang, 2002). Hence, like during DNA replication termination (Gangloff et al, 1994), such an intermediate would not be formed in the absence of Sgs1 and therefore Top3 would not be required. In the absence of Top3, however, Sgs1 would still promote the formation of a stable hemi-catenated structure that cannot easily branch migrate back and yield a CO. Indeed, as no other activity is capable of efficiently processing this intermediate, we have to assume that it must somehow undergo a recombinogenic strand break during anaphase to liberate the connected molecules. This hypothesis is supported by the observation that sgs1-L9S mutants that abrogate the interaction with Top3 (Duno et al, 2000) exhibit, like top3Δ mutants, no effect on COs (Figure 2). Furthermore, the deletion of Rad1, which is capable of cleaving single-stranded DNA, reduces the plating efficiency of top3Δ mutants even further, suggesting that a RAD1-dependent function is involved in the processing of intertwined DNA that persists in the absence of Top3 activity (Bailis et al, 1992).
Both Sgs1 and Srs2 helicases regulate spontaneous mitotic COs, as was previously concluded from DSB-induced experiments (Ira et al, 2003). However, our results clearly indicate that the absence of Srs2 leads to a more dramatic effect on COs than that of Sgs1 when conversion is triggered during unperturbed growth conditions. There are at least two non-exclusive ways of explaining why the absence of Srs2 would increase COs—either Srs2 specifically acts on structures that are otherwise predestined to be resolved mainly as COs, or Srs2 is capable of assisting strand displacement of the newly synthesized DNA strand (Model in Figure 5). In the first case, one has to hypothesize that failure to load Srs2 at the site of damage allows recombination proteins to stay bound to DNA favoring either the invasion by the single-stranded gap or invasion by both ends of the broken DNA, therefore increasing the frequency of dHJs formation. Alternatively, it may permit resolution proteins to process an intermediate structure in a manner that leads to a CO. Such a DNA cleaning function for Srs2 is in agreement with biochemical studies (Krejci et al, 2003; Veaute et al, 2003). In the second case, Srs2 acts at a later stage of the process, when the invading strand becomes elongated. At this point, the helicase activity of Srs2 could melt the hydrogen bonds that link together the template strand and the newly synthesized strand, leading to SDSA. In this case, a possibility is that a DNA polymerase extends the invading strand whereas the Srs2 helicase tracks behind. If the helicase progresses faster than the polymerase, there will be a point at which both machineries will collide resulting eventually in the rejection of the invading strand. In this scenario, the steps involved in the concerted capture of the second end of the break are no longer required, therefore favoring repair by SDSA. One prediction of this model is that conversion tracts should be longer in the absence of Srs2. In support of our view, several reports have pointed out that longer mitotic conversion tracts are associated with elevated levels of CO (reviewed by Prado and Aguilera, 2003; Aylon and Kupiec, 2004), the hallmark of srs2 mutants in our assay.
Figure 5.
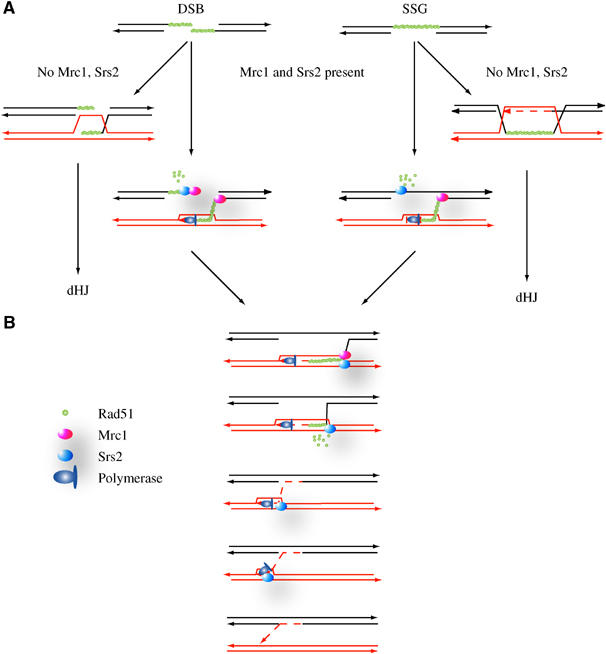
Model for CO inhibition by Srs2 and Mrc1. The initiating lesions are either DSBs (left) or single-strand gap (right) that can arise spontaneously in the course of DNA replication. (A) Early steps: When a DSB is created, the ends of the break are resected and covered with Rad51 before initiating strand invasion. In the presence of Mrc1 and Srs2, dismantling of the nucleofilaments may either prevent one of the ends of a break to become invasive or prevent the second end to be captured and therefore form a dHJ. In the case of a single-strand gap (SSG), Srs2 could prevent the formation of a dHJ by removing Rad51 from the gapped single-stranded DNA, therefore favoring a one-ended event. (B) Later steps: The invading strand pairs with its homologue and establishes a D-loop that becomes stabilized through reverse branch migration. This process brings Mrc1 present at the stalled fork in close contact with the donor DNA and may offer an entryway for Srs2 to the copied strand (3′ to 5′). The helicase activity of Srs2 will melt the newly formed duplex DNA, therefore rejecting the invading strand. Additionally, if the tracking speed of Srs2 is faster than that of the polymerase, the two machineries will collide and result in the complete rejection of the invading strand. In this model, the late steps do not depend on the type of initiating lesions (only SSG is shown), except that in the case of a DSB, a second event of DNA synthesis is necessary to seal the single-strand break present on the recipient molecule.
When the dissolution pathway is impaired in sgs1 mutants, we observe a doubling (25%) of the percentage of COs. This observation suggests that dHJs are the outcome of about 25% of the recombination intermediates and that 75% of the conversion events are normally processed through the SDSA pathway. Such a bias has beneficial physiological consequences in mitosis, because it allows repair to take place, most often without a potentially harmful reciprocal translocation. If this view is true, the fact that srs2 mutants bring the percentage of COs up to 50% suggests firstly that the dHJs that are not dissolved through the Sgs1 pathway are bound to be resolved mainly in association with a CO and secondly that absence of Srs2 function leads to the near exclusive formation of dHJs at the expense of SDSA.
We have accumulated evidence indicating that two DNA helicases involved in recombinational repair control the mitotic CO outcome through individual pathways. Sgs1 is instrumental in the dissolution process of dHJs, whereas Srs2 prevents their formation therefore promoting SDSA. Previous studies had revealed that both Sgs1 (Frei and Gasser, 2000) and Srs2 (Liberi et al, 2000) participate in the checkpoint response. However, little is known about the involvement of DNA damage signaling in the processing of spontaneous recombination intermediates. With the exception of dun1, we found that the absence of a member of any category of DNA damage checkpoint tested results both in elevated levels of conversion and an increased proportion of COs (Figure 3). Although their hyper-recombinogenic phenotype can be readily explained in light of their replication fork stabilization role (Lopes et al, 2001; Sogo et al, 2002), it is more difficult to ascribe a function for their resolution bias. At this point, we can only propose that checkpoint proteins inhibit resolution associated with a CO either directly or by phosphorylating yet unknown targets. In support of the second view, phosphorylation of several proteins involved in DNA recombination is dependent on checkpoint proteins (Bashkirov et al, 2000; Liberi et al, 2000; Bartrand et al, 2004). With respect to dun1, we found no effect on either conversion rates or COs. Interestingly, phosphorylation of Srs2 following DNA damage or fork stalling was shown to be dependent on a functional Dun1 protein (Liberi et al, 2000). Therefore, Srs2 phosphorylation is probably not involved in the processing of recombination intermediates, although we cannot formally rule out the possibility that under stress, phosphorylation of Srs2 may be needed. Interestingly, when the effects of dun1 mutants were examined with a plasmid gap repair assay, an increase in COs was found (Haghnazari and Heyer, 2004). This observation suggests that the proteins involved in the control of COs depend both on the cell cycle and on the nature of the initiating lesion. Additional support for this idea comes from the mrc1 deletion mutant that we have tested using a similar system. We found no effect of Mrc1 on COs in the transformation assay (data not shown), whereas it exhibits the strongest bias in our spontaneous system. As Mrc1 is present at replication forks (Katou et al, 2003), we believe that Mrc1 processes exclusively replicative damage.
The role of Mrc1 in CO control cannot be entirely explained by the checkpoint function of the protein: firstly, because absence of Mrc1 triggers a much higher stimulation of the COs than that of Rad53; secondly, because mrc1AQ, which is present at the fork but unable to mediate the checkpoint signaling, behaves mostly like the other checkpoint mutants rather than the deletion mutant; thirdly, Mrc1 belongs to a complex present at the replication fork together with Tof1 and Csm3 (Ito et al, 2001; Katou et al, 2003). However, removing Tof1 from the cells has an effect similar to that of mrc1AQ mutants and does not lead to the extreme increase in COs observed in the absence of Mrc1. Such an uncoupling of Tof1 and Mrc1 has recently been observed by several laboratories for induced replication fork pausing (Calzada et al, 2005; Szyjka et al, 2005; Tourrière et al, 2005). These results clearly indicate that Mrc1 exercises a function at the fork that is not shared with Tof1 or any checkpoint protein tested but depends on its presence and not on its capability of being phosphorylated.
Because mrc1Δ mutants behave like Srs2 mutants with respect to COs and because mrc1AQ does not exhibit the extreme hyper-CO phenotype of the deletion, we suggest that Mrc1 present at the replication fork is involved in the recruitment of Srs2 to PCNA, promoting a mainly CO-free processing of the recombination intermediate. In support of this model is the fact that overexpression of Srs2 can partially compensate for the hyper-CO phenotype observed in the absence of Mrc1, whereas it has no effect in cells where Mrc1 is present. This function of Srs2 does not depend on its phosphorylation status, as the absence of Dun1, which triggers the loss of Srs2 phosphorylation following HU, MMS or UV treatment, has no effect on CO control (Liberi et al, 2000; our study). Additionally, in mrc1Δ or mrc1AQ mutants, Srs2 phosphorylation is indistinguishable from that observed in WT cells, whereas COs are differentially regulated.
We found no evidence for an interaction between Mrc1 and Srs2, raising the question of the recruitment of Srs2 to the replication fork. In vitro and in vivo studies on repair following genotoxic treatment showed that sumoylated forms of PCNA bind Srs2 (Papouli et al, 2005; Pfander et al, 2005). We therefore asked if mutations in PCNA that prevent post-translational modifications increase the frequency of conversion-associated COs. Indeed, when lysine 164 can neither be ubiquitinated nor sumoylated and lysine 127 cannot be sumoylated, the CO association went up to 50%, a value similar to that observed in mrc1Δ and in srs2Δ cells. This result strongly suggests that a modified PCNA recruits Srs2. Because the rad18 mutation that prevents PCNA ubiquitination has no effect on the CO outcome (data not shown), we infer that it is the sumoylated and not the ubiquitinated form of PCNA that mediates Srs2 recruitment, a question that is under further investigation. Thus, for both inhibition of recombinational repair (Papouli et al, 2005; Pfander et al, 2005) and inhibition of COs, Srs2 appears to be recruited by a similar process. Such a recruitment scheme for Srs2 through specific protein–protein interaction could be used to achieve fine-tuned regulation of biological processes in cells under various conditions of stress.
Materials and methods
Yeast strains, plasmids and media
All media were prepared as previously described (Sherman and Hicks, 1991). The strains used in this study (Table I) are isogenic derivatives of D325-7D (MATa ade2-1 arg4ΔBglII his3-11,15 leu2-3,112 trp1-1 URA3∷arg4ΔRV∷ura3-1) obtained after mating, sporulation and dissection. The sgs1-K706R mutation derives from J726 (R Rothstein), and is integrated into the chromosome at its native locus. Plasmid pNM20, derived from YIp5, was digested with NcoI and used to direct the integration of arg4ΔEcoRV to the endogenous ura3-1 locus to generate the ectopic assay (see Table I). pNM20 contains the URA3 gene in the same orientation as the arg4ΔEcoRV allele. After integration, the URA3 and ura3-1 alleles flank the arg4ΔEcoRV allele, which is in the same orientation with respect to the centromere as the endogenous arg4ΔBglII (Figure 1). To construct the sgs1-L9S allele, we used ‘QuickChange site-directed mutagenesis kit' (Stratagene). Plasmid pSG003 (pUC18-SGS1) was used as a template and oligonucleotides sgs1-L9S-C (CCA TTT GTG CTC CCT TCT TCA GTT ATG TGA CGG C) and sgs1-L9S-W (GCC GTC ACA TAA CTG AAG AAG GGA GCA CAA ATG G) were used for PCR. The pUC18-sgs1-L9S plasmid was obtained and an XhoI–AgeI fragment containing the mutation was swapped into pRS414-SGS1 to form pSG085 (pRS414-sgs1-L9S). Overexpression of SRS2 was achieved by transforming pSG113 into D375-1D. pSG113 derives from YEp13 in which an SphI–HindIII fragment carrying SRS2 under the control of its native promoter was cloned into the corresponding sites of the vector.
Table 1.
Strain list
| Strain name | Relevant genotype |
|---|---|
| D325-7D | α WT (arg4ΔBglII, URA3∷arg4ΔRV∷ura3-1) |
| D325-2C | α sgs1Δ∷LEU2 |
| D325-3D | α mus81Δ∷TRP1 |
| D330-10D | a top3Δ∷LEU2 |
| D330-1A | a top3Δ∷LEU2 sgs1Δ∷TRP1 |
| D377-5C | α srs2Δ∷LEU2 |
| D338-2A | α sgs1-K706R |
| D447-4C | a rad9Δ∷HIS3 |
| D369-2A | α rad24Δ∷TRP1 |
| D374-5A | α mec1Δ∷TRP1 sml1Δ∷HIS3 |
| D380-5C | α rad53Δ∷HIS3 sml1-1 |
| D375-1D | a mrc1Δ∷KanMX |
| D422-6B | a dun1Δ∷LEU2 |
| D449-2B | a mrc1AQ∷his5+MYC13 |
| D455-14C | a tof1Δ∷TRP1 |
| D372-2B | α sgs1Δ∷LEU2 rad9Δ∷HIS3 |
| D373-1A | a sgs1Δ∷LEU2 rad24Δ∷TRP1 |
| D381-9C | α sgs1Δ∷LEU2 mec1Δ∷TRP1 sml1Δ∷HIS3 |
| D383-9C | α sgs1Δ∷LEU2 rad53Δ∷HIS3 sml1-1 |
| D378-10C | a srs2Δ∷LEU2 rad9Δ∷HIS3 |
| D411-1C | a srs2Δ∷LEU2 rad24Δ∷TRP1 |
| D393-6B | α srs2Δ∷LEU2 mec1Δ∷TRP1 sml1Δ∷HIS3 |
| D382-6D | α srs2Δ∷LEU2 rad53Δ∷HIS3 sml1-1 |
| D412-4A | α srs2Δ∷LEU2 mrc1AQ∷his5+MYC13 |
| D429-18D | α sgs1Δ∷HIS3 dun1Δ∷LEU2 |
| D430-7D | a srs2Δ∷HIS3 dun1Δ∷LEU2 |
| D488-1D | a pol30-RR |
Recombination rates determination
Spontaneous rates of recombination were measured by fluctuation analysis using the algorithm developed in the Robertson laboratory (Spell and Jinks-Robertson, 2004). The median was determined from at least seven independent cultures, whereas the experiment was repeated at least three times. Cells in the stationary phase were washed in 0.9% NaCl and plated at appropriate dilutions on YPD medium for survival and on medium lacking arginine for recombination.
Selection of COs
Cells were plated on YPD medium and grown for 3 days at 30°C. Colonies were replica-plated on complete medium lacking arginine to select for recombinants. After 3 days at 30°C, papillae appear among colonies. Each papilla corresponds to an independent recombination event that was patched on synthetic medium lacking arginine. To determine if the conversion events were associated with a CO, patched convertants were replica-plated first onto YPD and later onto 5-FOA-containing plates (Boeke et al, 1984). Confluent growth on 5-FOA reflects a high probability of the URA3 allele loss event, such as that associated with a direct repeat recombination event. This conversion event is unlikely to result from a CO that would yield occasional papillae at a frequency of three orders of magnitude lower. Convertants that yielded no papillae on 5-FOA were picked and grown again on YPD plates as larger patches. They were replica-plated a second time onto 5-FOA to discard the convertants that would not have produced papillae in the first screen. Extensive PFG analysis showed that if more than five individual papillae were growing, they are always the result of a conversion event not associated with a CO.
PFGE and Southern blot
Yeast cells were embedded in low-melting point agarose plugs and yeast chromosomes were separated by PFGE. All steps were carried out as described by the manufacturer (Bio-Rad). Chromosome transfer onto a nitrocellulose membrane was achieved in 0.4 M NaOH and 1.5 M NaCl using a ‘Vacuum blotter' from Apligene. The membrane was hybridized to DNA probes made with ‘Rediprime II Random Prime labeling system' from Amersham Biosciences and revealed on a Storm PhosphorImager.
Protein separation and immunodetection
Cultures were grown in rich medium to reach a density of 0.7 at OD 600 nm either in the absence or presence of 0.2 M hydroxyurea. Proteins were extracted with TCA and treated as described previously (Liberi et al, 2000). Goat anti-Srs2 polyclonal antibodies (Santa Cruz Biotechnology) diluted at 1:500 were used as a primary antibody. Horseradish peroxidase-conjugated anti-goat immunoglobulin G was used as the secondary antibody at a dilution of 1:5000 (Santa Cruz Biotechnology).
Statistical analyses
Recombination rates were compared using the Student–Fisher test (α=0.05). The percentage of COs determined among at least three independent segregants was first shown to be homogeneous for each genotype (ɛ test to compare observed percentages; α=0.05). We next calculated the CO percentage for each genotype by adding all the CO events that we divided by the total number of convertants analyzed (no SD in the graphs). We next used the ɛ test to compare the genotypes to one another. If ɛ is greater than 1.96, the percentages were considered different with a confidence of 95%.
Acknowledgments
We thank Steve Elledge, Marco Foiani, Philippe Pasero, Martine Heude and Rodney Rothstein for plasmids or strains and Sue Jinks Robertson for her rate analysis software. We also thank Stéphane Marcand, Julianne Smith and all the members of our laboratory for the critical reading of the manuscript. This study was supported by a grant of the European Community (LSHG-CT-2003-503303), a grant (to SG) and a fellowship of ARC (to TR), the CNRS and the CEA.
References
- Aylon Y, Kupiec M (2004) New insights into the mechanism of homologous recombination in yeast. Mutat Res 566: 231–248 [DOI] [PubMed] [Google Scholar]
- Aylon Y, Liefshitz B, Bitan-Banin G, Kupiec M (2003) Molecular dissection of mitotic recombination in the yeast Saccharomyces cerevisiae. Mol Cell Biol 23: 1403–1417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailis AM, Arthur L, Rothstein R (1992) Genome rearrangement in top3 mutants of Saccharomyces cerevisiae requires a functional RAD1 excision repair gene. Mol Cell Biol 12: 4988–4993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartrand AJ, Iyasu D, Brush GS (2004) DNA stimulates Mec1-mediated phosphorylation of replication protein A. J Biol Chem 279: 26762–26767 [DOI] [PubMed] [Google Scholar]
- Bashkirov VI, King JS, Bashkirova EV, Schmuckli-Maurer J, Heyer WD (2000) DNA repair protein Rad55 is a terminal substrate of the DNA damage checkpoints. Mol Cell Biol 20: 4393–4404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bastin-Shanower SA, Fricke WM, Mullen JR, Brill SJ (2003) The mechanism of Mus81–Mms4 cleavage site selection distinguishes it from the homologous endonuclease Rad1–Rad10. Mol Cell Biol 23: 3487–3496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baudat F, Nicolas A (1997) Clustering of meiotic double-strand breaks on yeast chromosome III. Proc Natl Acad Sci USA 94: 5213–5218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett RJ, Keck JL, Wang JC (1999) Binding specificity determines polarity of DNA unwinding by the Sgs1 protein of S. cerevisiae. J Mol Biol 289: 235–248 [DOI] [PubMed] [Google Scholar]
- Bennett RJ, Noirot-Gros MF, Wang JC (2000) Interaction between yeast Sgs1 helicase and DNA topoisomerase III. J Biol Chem 275: 26898–26905 [DOI] [PubMed] [Google Scholar]
- Boeke J, Lacroute F, Fink G (1984) A positive selection for mutants lacking orotidine-5′-phosphate decarboxylase activity in yeast: 5-fluoro-orotic acid resistance. Mol Gen Genet 197: 342–346 [DOI] [PubMed] [Google Scholar]
- Calzada A, Hodgson B, Kanemaki M, Bueno A, Labib K (2005) Molecular anatomy and regulation of a stable replisome at a paused eukaryotic DNA replication fork. Genes Dev 19: 1905–1919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caspari T, Murray JM, Carr AM (2002) Cdc2-cyclin B kinase activity links Crb2 and Rqh1-topoisomerase III. Genes Dev 16: 1195–1208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de los Santos T, Hunter N, Lee C, Larkin B, Loidl J, Hollingsworth NM (2003) The Mus81/Mms4 endonuclease acts independently of double-holliday junction resolution to promote a distinct subset of crossovers during meiosis in budding yeast. Genetics 164: 81–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de los Santos T, Loidl J, Larkin B, Hollingsworth NM (2001) A role for MMS4 in the processing of recombination intermediates during meiosis in Saccharomyces cerevisiae. Genetics 159: 1511–1525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doe CL, Ahn JS, Dixon J, Whitby MC (2002) Mus81–Eme1 and Rqh1 involvement in processing stalled and collapsed replication forks. J Biol Chem 277: 32753–32759 [DOI] [PubMed] [Google Scholar]
- Duno M, Thomsen B, Westergaard O, Krejci L, Bendixen C (2000) Genetic analysis of the Saccharomyces cerevisiae Sgs1 helicase defines an essential function for the Sgs1–Top3 complex in the absence of SRS2 or TOP1. Mol Gen Genet 264: 89–97 [DOI] [PubMed] [Google Scholar]
- Elliott B, Jasin M (2002) Double-strand breaks and translocations in cancer. Cell Mol Life Sci 59: 373–385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabre F, Chan A, Heyer WD, Gangloff S (2002) Alternate pathways involving Sgs1/Top3, Mus81/ Mms4, and Srs2 prevent formation of toxic recombination intermediates from single-stranded gaps created by DNA replication. Proc Natl Acad Sci USA 99: 16887–16892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Formosa T, Alberts BM (1986) DNA synthesis dependent on genetic recombination: characterization of a reaction catalyzed by purified bacteriophage T4 proteins. Cell 47: 793–806 [DOI] [PubMed] [Google Scholar]
- Frei C, Gasser SM (2000) The yeast Sgs1p helicase acts upstream of Rad53p in the DNA replication checkpoint and colocalizes with Rad53p in S-phase-specific foci. Genes Dev 14: 81–96 [PMC free article] [PubMed] [Google Scholar]
- Gangloff S, de Massy B, Arthur L, Rothstein R, Fabre F (1999) The essential role of yeast topoisomerase III in meiosis depends on recombination. EMBO J 18: 1701–1711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gangloff S, McDonald JP, Bendixen C, Arthur L, Rothstein R (1994) The yeast type I topoisomerase Top3 interacts with Sgs1, a DNA helicase homolog: a potential eukaryotic reverse gyrase. Mol Cell Biol 14: 8391–8398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gangloff S, Soustelle C, Fabre F (2000) Homologous recombination is responsible for cell death in the absence of the Sgs1 and Srs2 helicases. Nat Genet 25: 192–194 [DOI] [PubMed] [Google Scholar]
- Haghnazari E, Heyer WD (2004) The DNA damage checkpoint pathways exert multiple controls on the efficiency and outcome of the repair of a double-stranded DNA gap. Nucleic Acids Res 32: 4257–4268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heude M, Chanet R, Fabre F (1995) Regulation of the Saccharomyces cerevisiae Srs2 helicase during the mitotic cell cycle, meiosis and after irradiation. Mol Gen Genet 248: 59–68 [DOI] [PubMed] [Google Scholar]
- Hoege C, Pfander B, Moldovan GL, Pyrowolakis G, Jentsch S (2002) RAD6-dependent DNA repair is linked to modification of PCNA by ubiquitin and SUMO. Nature 419: 135–141 [DOI] [PubMed] [Google Scholar]
- Interthal H, Heyer WD (2000) MUS81 encodes a novel helix–hairpin–helix protein involved in the response to UV- and methylation-induced DNA damage in Saccharomyces cerevisiae. Mol Gen Genet 263: 812–827 [DOI] [PubMed] [Google Scholar]
- Ira G, Haber JE (2002) Characterization of RAD51-independent break-induced replication that acts preferentially with short homologous sequences. Mol Cell Biol 22: 6384–6392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ira G, Malkova A, Liberi G, Foiani M, Haber JE (2003) Srs2 and Sgs1–Top3 suppress crossovers during double-strand break repair in yeast. Cell 115: 401–411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito T, Chiba T, Ozawa R, Yoshida M, Hattori M, Sakaki Y (2001) A comprehensive two-hybrid analysis to explore the yeast protein interactome. Proc Natl Acad Sci USA 98: 4569–4574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaliraman V, Mullen JR, Fricke WM, Bastin-Shanower SA, Brill SJ (2001) Functional overlap between Sgs1–Top3 and the Mms4–Mus81 endonuclease. Genes Dev 15: 2730–2740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katou Y, Kanoh Y, Bando M, Noguchi H, Tanaka H, Ashikari T, Sugimoto K, Shirahige K (2003) S-phase checkpoint proteins Tof1 and Mrc1 form a stable replication-pausing complex. Nature 424: 1078–1083 [DOI] [PubMed] [Google Scholar]
- Kim RA, Wang JC (1992) Identification of the yeast TOP3 gene product as a single strand-specific DNA topoisomerase. J Biol Chem 267: 17178–17185 [PubMed] [Google Scholar]
- Klein HL (2001) Spontaneous chromosome loss in Saccharomyces cerevisiae is suppressed by DNA damage checkpoint functions. Genetics 159: 1501–1509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krejci L, Van Komen S, Li Y, Villemain J, Reddy MS, Klein H, Ellenberger T, Sung P (2003) DNA helicase Srs2 disrupts the Rad51 presynaptic filament. Nature 423: 305–309 [DOI] [PubMed] [Google Scholar]
- Kuzminov A (2001) Single-strand interruptions in replicating chromosomes cause double-strand breaks. Proc Natl Acad Sci USA 98: 8241–8246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liberi G, Chiolo I, Pellicioli A, Lopes M, Plevani P, Muzi-Falconi M, Foiani M (2000) Srs2 DNA helicase is involved in checkpoint response and its regulation requires a functional Mec1-dependent pathway and Cdk1 activity. EMBO J 19: 5027–5038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopes M, Cotta-Ramusino C, Pellicioli A, Liberi G, Plevani P, Muzi-Falconi M, Newlon CS, Foiani M (2001) The DNA replication checkpoint response stabilizes stalled replication forks. Nature 412: 557–561 [DOI] [PubMed] [Google Scholar]
- Lu J, Mullen JR, Brill SJ, Kleff S, Romeo AM, Sternglanz R (1996) Human homologues of yeast helicase. Nature 383: 678–679 [DOI] [PubMed] [Google Scholar]
- Melo J, Toczyski D (2002) A unified view of the DNA-damage checkpoint. Curr Opin Cell Biol 14: 237–245 [DOI] [PubMed] [Google Scholar]
- Miyajima A, Seki M, Onoda F, Shiratori M, Odagiri N, Ohta K, Kikuchi Y, Ohno Y, Enomoto T (2000) Sgs1 helicase activity is required for mitotic but apparently not for meiotic functions. Mol Cell Biol 20: 6399–6409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullen JR, Kaliraman V, Brill SJ (2000) Bipartite structure of the SGS1 DNA helicase in Saccharomyces cerevisiae. Genetics 154: 1101–1114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nyberg KA, Michelson RJ, Putnam CW, Weinert TA (2002) Toward maintaining the genome: DNA damage and replication checkpoints. Annu Rev Genet 36: 617–656 [DOI] [PubMed] [Google Scholar]
- Onoda F, Seki M, Miyajima A, Enomoto T (2000) Elevation of sister chromatid exchange in Saccharomyces cerevisiae sgs1 disruptants and the relevance of the disruptants as a system to evaluate mutations in Bloom's syndrome gene. Mutat Res-DNA Repair 459: 203–209 [DOI] [PubMed] [Google Scholar]
- Onodera R, Seki M, Ui A, Satoh Y, Miyajima A, Onoda F, Enomoto T (2002) Functional and physical interaction between Sgs1 and Top3 and Sgs1-independent function of Top3 in DNA recombination repair. Genes Genet Syst 77: 11–21 [DOI] [PubMed] [Google Scholar]
- Ooi SL, Shoemaker DD, Boeke JD (2003) DNA helicase gene interaction network defined using synthetic lethality analyzed by microarray. Nat Genet 35: 277–286 [DOI] [PubMed] [Google Scholar]
- Osborn AJ, Elledge SJ (2003) Mrc1 is a replication fork component whose phosphorylation in response to DNA replication stress activates Rad53. Genes Dev 17: 1755–1767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papouli E, Chen S, Davies AA, Huttner D, Krejci L, Sung P, Ulrich HD (2005) Crosstalk between SUMO and ubiquitin on PCNA is mediated by recruitment of the helicase Srs2p. Mol Cell 19: 123–133 [DOI] [PubMed] [Google Scholar]
- Pâques F, Haber JE (1999) Multiple pathways of recombination induced by double-strand breaks in Saccharomyces cerevisiae. Microbiol Mol Biol Rev 63: 349–404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paull TT, Cortez D, Bowers B, Elledge SJ, Gellert M (2001) Direct DNA binding by Brca1. Proc Natl Acad Sci USA 98: 6086–6091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petes TD, Malone RE, Symington LS (1991) Recombination in yeast. In The Molecular & Cellular Biology of the Yeast Saccharomyces: Genome Dynamics, Protein Synthesis and Energetics.(1), Broach JR, Pringle JR, Jones EW (eds) pp 407–521. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press [Google Scholar]
- Pfander B, Moldovan G-L, Sacher M, Hoege C, Jentsch S (2005) SUMO-modified PCNA recruits Srs2 to prevent recombination during S phase. Nature 436: 428–433 [DOI] [PubMed] [Google Scholar]
- Prado F, Aguilera A (2003) Control of cross-over by single-strand DNA resection. Trends Genet 19: 428–431 [DOI] [PubMed] [Google Scholar]
- Rockmill B, Fung JC, Branda SS, Roeder GS (2003) The Sgs1 helicase regulates chromosome synapsis and meiotic crossing over. Curr Biol 13: 1954–1962 [DOI] [PubMed] [Google Scholar]
- Saka Y, Esashi F, Matsusaka T, Mochida S, Yanagida M (1997) Damage and replication checkpoint control in fission yeast is ensured by interactions of Crb2, a protein with BRCT motif, with Cut5 and Chk1. Genes Dev 11: 3387–3400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schild D (1995) Suppression of a new allele of the yeast RAD52 gene by overexpression of RAD51, mutations in srs2 and ccr4, or mating-type heterozygosity. Genetics 140: 115–127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scully R, Chen J, Ochs RL, Keegan K, Hoekstra M, Feunteun J, Livingston DM (1997) Dynamic changes of BRCA1 subnuclear location and phosphorylation state are initiated by DNA damage. Cell 90: 425–435 [DOI] [PubMed] [Google Scholar]
- Sherman F, Hicks J (1991) Micromanipulation and dissection of asci. Methods Enzymol 194: 21–37 [DOI] [PubMed] [Google Scholar]
- Sogo JM, Lopes M, Foiani M (2002) Fork reversal and ssDNA accumulation at stalled replication forks owing to checkpoint defects. Science 297: 599–602 [DOI] [PubMed] [Google Scholar]
- Spell RM, Jinks-Robertson S (2004) Determination of mitotic recombination rates by fluctuation analysis in Saccharomyces cerevisiae. Methods Mol Biol 262: 3–12 [DOI] [PubMed] [Google Scholar]
- Szyjka SJ, Viggiani CJ, Aparicio OM (2005) Mrc1 is required for normal progression of replication forks throughout chromatin in S. cerevisiae. Mol Cell 19: 691–697 [DOI] [PubMed] [Google Scholar]
- Tourrière H, Versini G, Cordón-Preciado V, Alabert C, Pasero P (2005) Mrc1 and Tof1 promote replication fork progression and recovery independently of Rad53. Mol Cell 19: 699–706 [DOI] [PubMed] [Google Scholar]
- Ui A, Satoh Y, Onoda F, Miyajima A, Seki M, Enomoto T (2001) The N-terminal region of Sgs1, which interacts with Top3, is required for complementation of MMS sensitivity and suppression of hyper-recombination in sgs1 disruptants. Mol Genet Genomics 265: 837–850 [DOI] [PubMed] [Google Scholar]
- Vaze MB, Pellicioli A, Lee SE, Ira G, Liberi G, Arbel-Eden A, Foiani M, Haber JE (2002) Recovery from checkpoint-mediated arrest after repair of a double-strand break requires Srs2 helicase. Mol Cell 10: 373–385 [DOI] [PubMed] [Google Scholar]
- Veaute X, Jeusset J, Soustelle C, Kowalczykowski SC, Le Cam E, Fabre F (2003) The Srs2 helicase prevents recombination by disrupting Rad51 nucleoprotein filaments. Nature 423: 309–312 [DOI] [PubMed] [Google Scholar]
- Wallis JW, Chrebet G, Brodsky G, Rolfe M, Rothstein R (1989) A hyper-recombination mutation in S. cerevisiae identifies a novel eukaryotic topoisomerase. Cell 58: 409–419 [DOI] [PubMed] [Google Scholar]
- Wang JC (2002) Cellular roles of DNA topoisomerases: a molecular perspective. Nat Rev Mol Cell Biol 3: 430–440 [DOI] [PubMed] [Google Scholar]
- Wang Y, Cortez D, Yazdi P, Neff N, Elledge SJ, Qin J (2000) BASC, a super complex of BRCA1-associated proteins involved in the recognition and repair of aberrant DNA structures. Genes Dev 14: 927–939 [PMC free article] [PubMed] [Google Scholar]
- Willson J, Wilson S, Warr N, Watts FZ (1997) Isolation and characterization of the Schizosaccharomyces pombe rhp9 gene: a gene required for the DNA damage checkpoint but not the replication checkpoint. Nucleic Acids Res 25: 2138–2146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu L, Hickson ID (2003) The Bloom's syndrome helicase suppresses crossing over during homologous recombination. Nature 426: 870–874 [DOI] [PubMed] [Google Scholar]
- Xu H, Boone C, Klein HL (2004) Mrc1 is required for sister chromatid cohesion to aid in recombination repair of spontaneous damage. Mol Cell Biol 24: 7082–7090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao X, Chabes A, Domkin V, Thelander L, Rothstein R (2001) The ribonucleotide reductase inhibitor Sml1 is a new target of the Mec1/Rad53 kinase cascade during growth and in response to DNA damage. EMBO J 20: 3544–3553 [DOI] [PMC free article] [PubMed] [Google Scholar]