

Abstract
The crescent-shaped BAR (Bin/Amphiphysin/Rvs-homology) domain dimer is a versatile protein module that senses and generates positive membrane curvature. The BAR domain dimer of human endophilin-A1, solved at 3.1 Å, has a unique structure consisting of a pair of helix–loop appendages sprouting out from the crescent. The appendage's short helices form a hydrophobic ridge, which runs across the concave surface at its center. Examining liposome binding and tubulation in vitro using purified BAR domain and its mutants indicated that the ridge penetrates into the membrane bilayer and enhances liposome tubulation. BAR domain-expressing cells exhibited marked plasma membrane tubulation in vivo. Furthermore, a swinging-arm mutant lost liposome tubulation activity yet retaining liposome binding. These data suggested that the rigid crescent dimer shape is crucial for the tubulation. We here propose that the BAR domain drives membrane curvature by coordinate action of the crescent's scaffold mechanism and the ridge's membrane insertion in addition to membrane binding via amino-terminal amphipathic helix.
Keywords: BAR domain, endophilin, liposome, membrane curvature, membrane insertion
Introduction
Membrane dynamics in a cell, such as membrane budding, tubulation, fission and fusion, is associated with changes in membrane curvature. The crystal structure of amphiphysin BAR (Bin/Amphiphysin/Rvs-homology) domain revealed an unexpected structural identity with arfaptin2, a binding protein to Arf and Rac small GTPases (Tarricone et al, 2001), and provided a common structural base for the sensing and the formation of positive curvature membrane by BAR-family proteins (Peter et al, 2004).
Endophilins are cytoplasmic proteins containing an N-terminal BAR domain and a C-terminal SH3 domain, and are involved in membrane dynamics (Schuske et al, 2003; Galli and Haucke, 2004; Wenk and De Camilli, 2004). There are five endophilin genes in the mammalian genomes, endophilin A1–3 and B1–2. Both A and B types are highly conserved from nematode to human. The most extensively studied one is endophilin-A1, a brain specific protein involved in clathrin-mediated synaptic vesicle endocytosis (Ringstad et al, 1997, 2001). Via SH3 domain, endophilins bind to the GTPase dynamin, a membrane scissor, and the polyphosphoinositide phosphatase synaptojanin, a clathrin-uncoater (Ringstad et al, 1997; de Heuvel et al, 1997; Verstreken et al, 2003). The BAR domain of endophilins is classified into the N-BAR subgroup characterized by a short amphipathic helical sequence preceding the consensus BAR-domain sequence (Peter et al, 2004). The N-BAR domain of endophilin-A1 binds to liposomes and induces the tubulation in vitro, requiring the short amphipathic helical sequence (Farsad et al, 2001).
The crescent-shaped BAR dimer structure implies a simple model to drive membrane curvature: the dimer may impress its positively charged concave surface on the negatively charged membrane to form a high-curvature membrane domain (Gallop and McMahon, 2005; McMahon and Gallop, 2005). This curvature-impressing or scaffold mechanism for membrane deformation is based on an assumption that the dimer behaves as a rigid body on the membrane (Zimmerberg and Kozlov, 2006). Although the essential requirement of positively charged residues on the concave surface has been suggested (McMahon and Mills, 2004; Peter et al, 2004), there have been no experimental supports for the scaffold mechanism. Here, we show the requirement of the molecular rigidity of the BAR dimer for membrane curvature on the basis of structure-oriented mutational analysis.
By determining the structure of endophilin-A1 BAR domain, we found a distinction from those of the known BAR domains: a helix–loop appendage of 30 amino acids stretch is inserted into the helix I of the canonical BAR domain. A pair of the helices of the appendages forms a hydrophobic ridge, which runs across the center of the concave surface of the dimer. We analyzed the function of this ridge as well as the previously proposed structure, the N-terminal amphipathic helix and the crescent main body, for membrane deformation (Peter et al, 2004). N-terminal amphipathic helix is essential for membrane binding. The crescent main body of the BAR dimer is required for impressing its intrinsic curvature to the membrane. The ridge contributes to deform the membrane presumably by penetrating into the membrane. Our results illustrate how these three components coordinate to induce membrane deformation.
Results
Endophilin-A1 BAR domain has a unique appendage
The structure of the BAR domain of human endophilin-A1 (amino acid 1–247, hereafter EndA1-BAR) was solved at 3.1 Å resolution by a multi-wavelength anomalous dispersion method. The structure of EndA1-BAR dimer is similar to that of amphiphysin (Peter et al, 2004) and arfaptin2 (Tarricone et al, 2001): a crescent-shaped dimer composed of a 6-helix bundle core and two 3-helix bundle arms extended from the core (Figure 1A). The whole structure of EndA1-BAR dimer can be precisely superimposed on that of amphiphysin and arfaptin (Figure 1B). All three structures show nearly identical dimer shapes. Notably, the present EndA1-BAR structure from a tetragonal crystal packing is almost completely the same as an independent crystal structure from an orthogonal crystal packing (Supplementary Figure 1; and Weissenhorn, 2005). The RMS deviations are 0.63, 0.86 and 0.80 Å for Cα atoms in monomers A, B and dimer, respectively. The structural identity indicates that the crescent shape is stably present in solution. Consistent with previous results (Habermann, 2004; Peter et al, 2004), structure-based sequence alignment reveals that these three proteins are poorly conserved in amino-acid sequence including the residues possibly important for the crescent-shape formation (Supplementary Figure 2).
Figure 1.
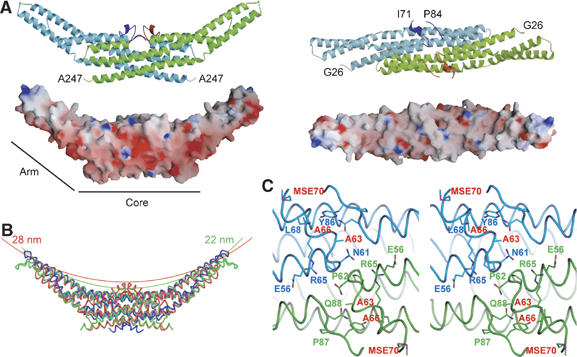
Structure of human endophilin-A1 BAR domain dimer. (A) Ribbon representation (a green monomer with a red appendage and a pale-blue monomer with a blue appendage) and surface electrostatic potential (red, –15 kTe−1; blue, 15 kTe−1) of the dimer viewed from the side (left) and from the top (right). The numbered amino-acid residues are the first and the last ones in consecutive polypeptide segments determined in this model. (B) Comparison of three BAR domain structures in trace representation. Red, endophilin-A1 (PDB ID: 1X03); green, amphiphysin (1URU); blue, arfaptin2 (1I4D). The red and green arcs with indicated diameters represent curved membranes fit the concave surface of endophilin-A1 and amphiphysin, respectively. (C) Stereo view of the appendages. Side-chains of the residues forming the hydrophobic ridge and those of interacting with residues of the main body are shown.
We find a unique structure of the EndA1-BAR, an appendage-like structure protruded from the center of the dimer (Figure 1A). The sequence alignments of the BAR-family proteins indicated that this appendage appears unique to the endophilin-family proteins including nadrin (Habermann, 2004; Peter et al, 2004) and the candidates from yeasts (Supplementary Figure 2). The appendage (Q59–Q88) has an N-terminal short helix and a loop of which electron density is mostly missing (N72–G85). The pair of helices appears to stay on the main body and forms a ridge across the center of the concave dimer surface. The helix displays, on its top surface, a series of hydrophobic residues (P62, A63, A66 and M70) aligned 60° against the longitudinal axis of the dimer (Figure 1C). Other than the conserved hydrophobic amino acids of the ridge, the appendage sequences show clear distinction between endophilin-A and endophilin-B (Supplementary Figure 2). The B type endophilins show cytoplasmic localization, presumably being involved in intracellular membrane dynamics (Farsad et al, 2001; Modregger et al, 2003; Karbowski et al, 2004). Analyses of chimeric mutations in the appendage between EndA1-BAR and EndB1-BAR suggest that BAR domain may contribute to defining where to target, plasma membrane or intracellular organ membrane (Supplementary Figure 3).
The appendage's penetration enhances liposome tubulation
To investigate the functional significance of the hydrophobic ridge of the endophilin-specific appendage, we first examined the effects of point mutations in this region (red residues in Figure 1C) on the liposome binding and tubulation activities in vitro (Figures 2A and 3). Introduction of membrane-repulsive negative charge (A66D) lost the ability to form tubes from liposomes. Hydrophilic mutations (A63S/A66S (SS) and A63S/A66S/M70Q (SSQ)) reduced the number of tubes (<1/100) and induced three-time enlargement of the tube diameter. In contrast, a bulky hydrophobic residue (A66W) led to extensive vesiculation and less tubulation. All these mutations did not affect the liposome binding. These results suggest an important role for the hydrophobic ridge in the membrane curvature formation but not in the membrane binding.
Figure 2.
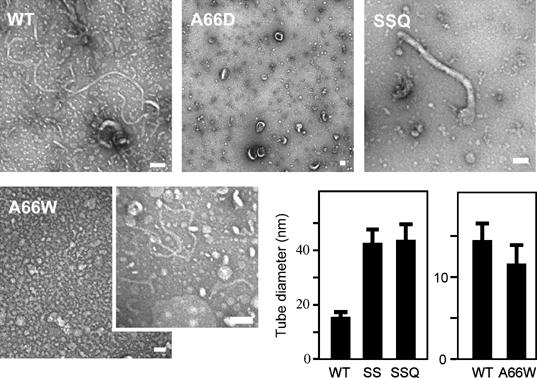
Liposome tubulation by endophilin-A1 BAR domains with mutations in the hydrophobic ridge. WT, 7 μM wild-type BAR domain incubated for 10 min; A66D, 28 μM, 10 min; SSQ, A63S/A66S/M70Q triple mutant, 28 μM, 10 min; A66W, 1.4 μM, 10 min (vesiculated, left panel) and 10 s (tubulated, right panel). Tubulation was not observed when incubated for longer than 1 min. Scale, 100 nm. The bar graphs show tubule diameter (mean and s.d.). SS, A63S/A66S double mutant, 28 μM, 10 min.
Figure 3.
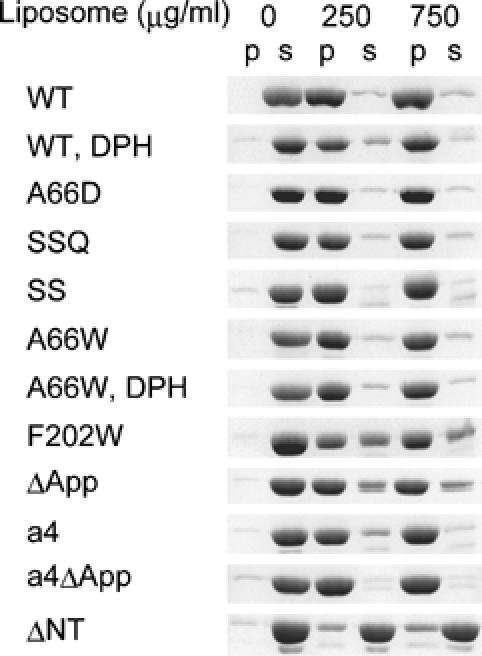
Liposome binding assays of endophilin-A1 BAR domain and its mutants. Protein (200 μg/ml) was co-sedimented with liposomes (0, 250 and 750 μg/ml). Proteins recovered from the pellet (p) and the supernatant (s) were analyzed by SDS–PAGE. The DPH-liposomes show similar binding capacity for the wild type (WT) and the A66W mutants. The liposome binding activity is slightly reduced in the F202W and the appendage-less mutants (ΔApp) and is almost lost in the helix 0 truncated mutant (ΔNT).
Although the ridge reduces the intrinsic curvature of the concave surface (red line in Figure 1B), it appears to promote the membrane curvature formation with conserved hydrophobicity. This raises the possibility that the ridge penetrates into the membrane when the concave surface makes tight contact with the membrane. This possibility was investigated using tryptophan fluorescence, which is sensitive to hydrophobicity of the microenvironment around the indole moiety. The A66W mutant showed 10-nm blueshift of the fluorescence peak in a liposome-dose-dependent and saturable manner, while F202W, a control mutant in which Phe202 on the convex surface was mutated to Trp, did not show any shift (Figure 4A and Supplementary Figure 5). The amount of the blueshift was greater than that observed in 50% DMSO or 50% methanol, indicating that the indol moiety was in a highly hydrophobic environment.
Figure 4.
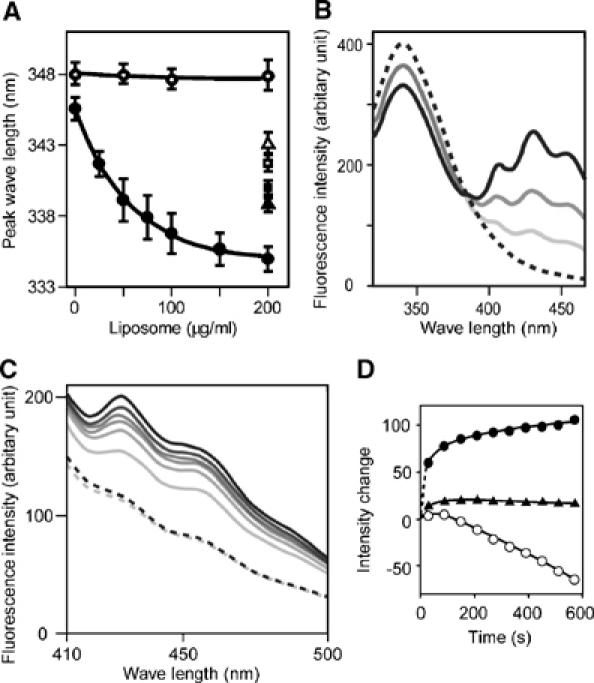
Tryptophan fluorescence blueshift and FRET assays. (A) Tryptophan fluorescence emission peak when excited at 280 nm was observed in different concentration of liposome. A66W (•), F202W control mutant (○), A66W alone in 50% DMSO (▴), in 50% MeOH (▪), F202W alone in 50% DMSO (▵), in 50% MeOH (□), 140 μg/ml protein for all measurements. Mean and s.d. (N=4–11). The dose dependency is significant (P≪0.001) for the A66W mutant but insignificant (P>0.8) for the F202W mutant (one-way ANOVA). DMSO and MeOH were used as blueshift inducer for tryptophan. (B) Dose-dependent FRET efficiency from the A66W tryptophan to DPH incorporated in liposomes was examined by the changes of fluorescence. Fluorescence spectrum of A66W (100 μg/ml) with the control liposome (200 μg/ml) excited at 280 nm (hatched). Pale to dark solid curves represent DPH:lipid weight ratios of 1:2000, 1:1000 and 1:500 in the same condition. (C) Time-dependent increase in the FRET efficiency from either A66W (pale to dark solid lines, from 30 to 570 s) or F202W tryptophan (pale and dark hatched lines, at 30 and 570 s) to DPH incorporated in liposomes. DPH:lipid weight ratio is 1:500. (D) The intensity changes at the 430-nm peak are plotted against time. A66W (•), F202W (▴) excited at 280 nm and A66W (○) excited at 360 nm.
To determine whether this blueshift was caused by the insertion of the indol moiety into the hydrophobic core of the lipid bilayer, we made fluorescence resonance energy transfer (FRET) assays using diphenyl-hexatriene (DPH) as the acceptor probe. DPH has been shown to insert specifically in the nonpolar interior of the membrane and not to alter the membrane structure and dynamics (Repáková et al, 2005). DPH liposomes did not affect liposome binding and tubulation (Figure 3 and Supplementary Figure 4). A66W but not F202W showed effective FRET from the 340-nm tryptophan fluorescence (donar) to the DPH fluorescence (acceptor) peaked at 430 nm (Figure 4B and C). It was not caused by changes in the fluorescence property of DPH itself possibly accompanied by tubulation/vesiculation of liposomes (Figure 4D and Supplementary Figure 6). These data suggest that the indol ring of 66W penetrates into the hydrophobic core of the membrane and that the remaining residues of the ridge, about 8 Å in height, appear to be embedded in the layer of lipid head-groups of the contacting membrane leaflet. These results confirmed that the ridge is contacting membrane and that the convex is not contacting membrane surface.
To provide further support for the membrane insertion of the ridge in the wild-type EndA1-BAR, we made a mutant with amphiphysin/arfaptin shape and examined its tubulation activity. The mutant (ΔApp), in which the entire appendage (Q59–Q88) was replaced with a helical stretch (AHLSSLLQ) derived from arfaptin2 sequence (A152–Q159, Y155S), show the crystal structure of a canonical BAR-domain dimer as designed (Figure 5A and Supplementary Figure 7). The ΔApp could bind to liposomes (Figure 3) and cause tubulation to a lesser extent than the wild type and amphiphysin-BAR (Figure 5D and Supplementary Figure 4). As the diameter of the tubules reflects the membrane curvature if the section of the tube is circle, we measured the diameter of the tube to compare the curvature of the EndA1-BAR and its mutant-induced tubes. Despite the higher curvature of the concave surface, the ΔApp dimer induced larger diameter tubules than the wild type did, indicating a positive contribution of the wild-type hydrophobic ridge to drive membrane curvature. Taken all together, the hydrophobic ridge penetrates into the interfacial leaflet of the lipid bilayer when the concave surface is in contact with the membrane and promotes membrane curvature formation.
Figure 5.
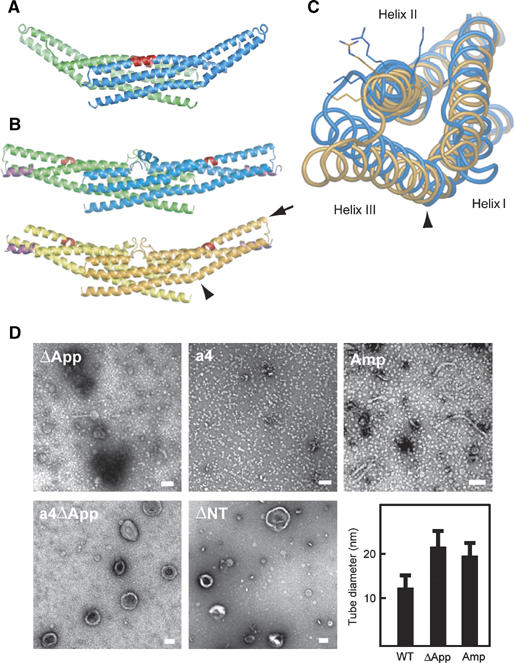
Distinct liposome tubulation induced by endophilin-A1 BAR domain mutants. (A) Ribbon representation of a mutated EndA1-BAR dimer lacking the entire appendages (ΔApp, PDB ID: 1X04). The entire appendage (Q59–Q88) was replaced with a helical stretch (AHLSSLLQ) derived from arfaptin2 sequence (A152–Q159, Y155S). Red, mutated segment. (B) Ribbon representation of the a4 mutant with swinging arms (PDB ID: 2D4C). One helical pitch was inserted into the helix II in the proximal portion of the extending arm (QSAL was inserted between I154 and Q155). Two dimers in the asymmetrical unit are shown separately. Red, inserted segment; magenta, helix 0. The bending patterns of the helix II and III varies among four monomers. An obvious kink in the helix III remains in the orange monomer (arrowhead, also in (C)). The residual curvature in the blue–green dimer is provided by the intersection of the monomers. (C) Superimposition of the a4 mutant monomer (orange one in (B)) and the wild-type monomer (blue) in the core region. A view from the distal end along the helix II (arrow in (B)) shows the maximum structural difference in these arms. Side chains of K171, 173 and R174 are shown. The helix III rotates 12° counterclockwise and shift 6 Å relative to the helix II at the distal end of the arm. The helix 0 and the core region are omitted. (D) Negatively stained liposome tubules induced by the BAR domains of endophilin mutants and amphiphysin. ΔApp, 7 μM, incubated for 10 min; a4, 7 μM, 10 min; a4ΔApp, 28 μM, 10 min; ΔNT, 21 μM, 10 min; Amp, 7 μM, 10 min. Note that a4, a4ΔApp, and ΔNT do not induce liposome tubulation. Scale, 100 nm. The bar graph shows tubule diameter (mean and s.d.).
The BAR domain is rigid enough to impose its intrinsic curvature on membrane
A simple model for the concave surface-driven mechanism is that each BAR domain dimer acts as a molecular mold that impresses its curved surface on the membrane. This model suggests that the membrane curvature approximately mirrors the curvature of the concave surface. Indeed, the diameters of tubules induced by amphiphysin, ΔApp (Figure 5D), SS and SSQ mutants (Figure 2) are compatible with the model-based prediction (see Supplementary Table II for statistical analysis). However, this model has an assumption that the dimer should be rigid enough to overcome the bending resistance of the membrane (Nossal and Zimmerberg, 2002; Farsad and De Camilli, 2003). To examine whether the molecular mold mechanism is feasible, we developed a straight BAR domain by inserting one helical pitch into the helix II in the proximal portion of the extending arm (QSAL is inserted between I154 and Q155). This mutation (a4) would compensate the unequal lengths between helix II and III in the arm, a common feature of the known BAR domain structures, and let the curved arm into a straight one. Although the a4 mutant was designed simply to straighten the curvature of the domain, the structure solved at 2.4 Å resolution shows that it actually has the very interesting property of a flexible arm rather than a rigid one (Figure 5B). Four monomers in the asymmetrical unit show deviation in the bending angles of arms. The blue and the green monomers have straight arms while the orange monomer shows a bending pattern similar to the wild type and the yellow monomer is an intermediate. The structural deviation almost exclusively occurs in the helix kink regions (Supplementary Figure 8), indicating that the arm can swing at least from the bend-free straight position to nearly the wild-type position.
The a4 mutant allowed us to examine how flexibility of the crescent-shaped main body of the BAR dimer affects the membrane curvature formation. The insertion of one helical pitch slightly distorts relative position of the helix II and III (Figure 5C), but does not largely rearrange the spatial positions of the residues on the concave surface of the arm (Supplementary Figure 8). Indeed, the a4 mutant and its appendage-lacking derivative (a4ΔApp) retained normal liposome binding activity (Figure 3). The a4 mutant vesiculated liposomes without any tubulation, while a4ΔApp lost these membrane-deforming activities (Figure 5D and Supplementary Figure 4). The concave surface-induced membrane deforming activity appeared to be lost in the a4 mutant, while the appendage's membrane insertion remained active. These results suggested that the rigidity of the crescent dimer structure is essential for liposome tubulation but not for vesiculation, although appendage insertion induces the vesiclulation.
Roles for the amphipathic helix 0 of the N-BAR domain
The structure of a short amphipathic helix (helix 0) characterizing the N-BAR (Peter et al, 2004) can be resolved in the a4 mutant structure due to its tight crystal packing (Figures 5B and 6). The helix 0 is disordered in the wild type (Figure 6) and the ΔApp structures. The helix 0 has been suggested to be helical only when the amphiphysin BAR domain binds to liposomes (Peter et al, 2004). The helix 0 displays the hydrophobic branch of T14, V17 and V21 on one side, while K12, K16 and E19 on the other side (Figure 6). The helix 0 is connecting with the Helix I by a flexible linker G23–G24–A25. Consistent with the previous report (Farsad et al, 2001), truncation of the helix 0 (ΔNT) resulted in loss of liposome binding activity (Figure 3) and consequently abolished the tubulation (Figure 5D). In contrast, all the helix 0-containing mutants, including the A66D and the a4ΔApp showed intact liposome binding activity irrespective of their tubulation or vesiculation activities. These results indicate that the helix 0 in the endA1-BAR is critical for liposome binding and that the membrane binding of endA1-BAR via helix 0 is not sufficient to induce tubulation or vesiculation.
Figure 6.
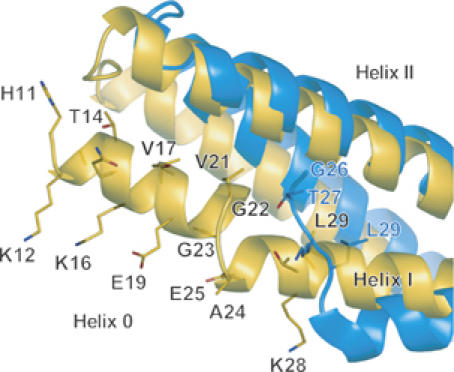
Close-up of helix 0 in an a4 mutant monomer (orange). The same superimposition as in Figure 5C but viewed from the side and displays the helix 0. The helix 0 is disordered in the wild-type structure (blue). The side chains of N-terminal residues are shown (H11KATQKVSEKVGGAEGTKL29 in the a4 and G26TKL in the wild type). The amphipathic helix 0 is stabilized by hydrophobic interactions with the helix II and III and also by hydrogen bonds with a symmetrical molecule.
BAR domain induces tubular membrane deformation in vivo
To explore the significance of the helix 0, the rigid crescent mold, and the appendage of endophilin-A1 BAR domain in vivo, we further examined the membrane deformation activity of endophilin-A1 BAR domain in cells (Figure 7). Human umbirical vascular endothelial cells (HUVECs) expressing endophilin-A1 lacking SH3 domain (residues 1–296, hereafter, EndA1-BAR296), which was C-terminally tagged with enhanced green fluorescence protein (EGFP), exhibited intracellular fibrous structure similar to those induced by other BAR domain-containing molecules (Kamioka et al, 2004; Itoh et al, 2005). Notably, these structures developed from the periphery toward the center of the cells dynamically and disappeared reversibly in living cells (Figure 7E and Supplementary Movie 1). Furthermore, these GFP-marked structures were co-localized with in vivo biotin-labeled membrane (Figure 7D), indicating that EndA1-BAR296-induced fibrous structure seems to be a membrane invagination originated from the plasma membrane. These structures were found in other cells we tested (Figure 7C). In clear contrast, ΔApp, ΔNT and a4 were incapable of inducing membrane deformation in cells, indicating the importance of helix 0, the rigid crescent shape, and the appendage of BAR domain for membrane deformation in vivo.
Figure 7.
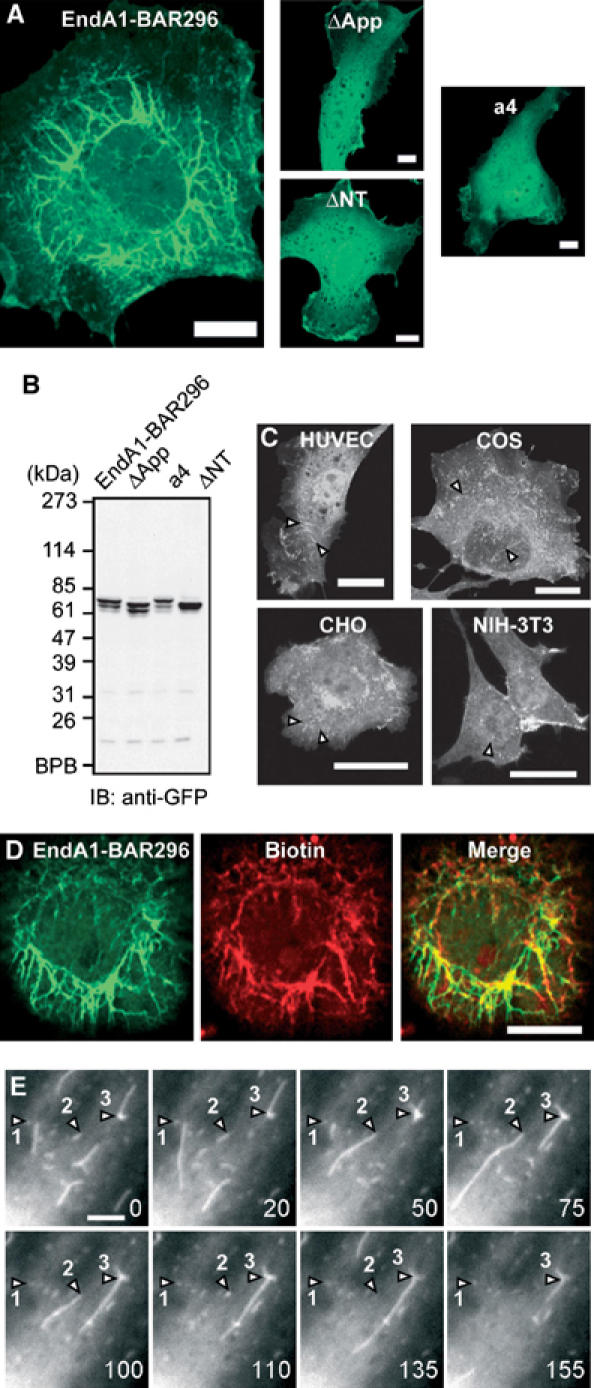
Endophilin A1 BAR domain induces membrane tubulation in vivo. (A) HUVECs were transfected with plasmids expressing C-teminally EGFP-tagged EndA1-BAR296 (amino acid 1–296 of endophilin-A1), ΔApp, a4, and ΔNT. Cells were GFP-imaged on an epifluorescnce microscope (Olympus IX-71). Fibrous structures were observed exclusively in EndA1-BAR296-expressing cells. Scale, 10 μm. (B) Protein expression of the EndA1-BAR296 and the mutants tagged with EGFP in transfected 293T cells were examined by immunoblotted with anti-GFP antibody. (C) Cells indicated were similarly transfected to (A). Arrowheads indicate the fibrous structures. Scale, 20 μm. (D) Live HUVECs expressing EGFP-tagged EndA1-BAR296 were biotinylated with sulfo-NHS-biotin for 10 min and chased for further 10 min. Covalently bound biotin was visualized using Alexa633-streptavidine. Fluorescnece images for EGFP (left), Alexa633 (center), and merge (right) are shown. Scale, 10 μm. (E) A time lapse images of HUVECs expressing EGFP-tagged EndA1-BAR296 were obtained at the time point (seconds) after the observation (Supplementary Movie 1). EGFP-marked structure grows from the cell periphery towards the center of the cell. Notably, both extension and retraction of GFP-marked structure is observed (numbered arrow heads indicate each extending/retracting structure). Scale, 5 μm.
Discussion
The endophilin-A1 BAR domain dimer consists of three sub-modules: the crescent-shaped main body, the helix 0 and the unique appendage. We tried to understand the functional roles for these sub-modules in the membrane curvature formation. In this study by determining the structure of endopholin-A1 BAR domain and developing mutants that were critical for the sub-module structure, we have explored the roles of sub-modules.
Here, we show that the structural rigidity of the crescent-shaped main body is critical for membrane tubulation. The BAR dimer is sufficiently rigid to overcome the bending resistance of the membrane and to be scaffolds for the tubulation (McMahon and Gallop, 2005; Zimmerberg and Kozlov, 2006). The insertion of one helical-pitch into the helix II at distal to the kink brings flexibility to the dimer (a4 mutant). The relative position of the three helices in the mutant arm was not changed in a4 mutant irrespective of the bend levels (Supplementary Figure 8). The mutant arm behaves as a rigid body and its structure changes only in the vicinity of the helix kinks when it swings. Therefore, it is unlikely that the flexibility of the mutant dimer can be a result of weakened inter-helix interactions in the arm. Moreover, we could not find any specific structural features in the kink region that might explain the flexible hinge in the swinging-arm mutant as well as the rigid bend in the wild-type BAR dimers of endophilin, amphiphysin, and arfaptin.
In this study, for the first time we could determine the structure of the N-terminal amphipathic helix (helix 0) using a swinging-arm mutant. Our mutant and previous mutation analyses indicated that the N-terminal helical sequence of endophilin-A1 is indispensable for liposome binding (Farsad et al, 2001), whereas that of amphiphysin is important but not essential for liposome binding and tubulation (Peter et al, 2004). The BAR domain of endophilin-A1 is an acidic polypeptide and the cluster of positive charge at the distal end of the arm is not prominent (Figure 1A). This property can explain the critical role for the helix 0 of the EndA1-BAR in liposome binding by providing additional basic residues. The helix 0 structure suggests that K12, K16 and possibly K8 are in a suitable position for cooperation with the positive charge cluster at the distal end. The amphipathic nature of the helix 0 implies that it can also insert into the membrane and facilitate the membrane curvature formation (Peter et al, 2004; Gallop and McMahon, 2005; McMahon and Gallop, 2005). Loss of the membrane-deforming activities of the A66D mutant (Figure 2) and the a4ΔApp mutant (Figure 5D) accounts for the additional mechanism for membrane deformation in addition to the membrane insertion of the helix 0.
The N-BAR of endophilins has one additional step to tubulate membrane. Here, we show that the hydrophobic ridge of the endophilin-specific appendage is inserted into the contacting membrane surface. Our data suggested that the entire ridge of the wild-type BAR domain, about 8 Å in height, is embedded in the layer of lipid head-groups of the contacting membrane leaflet. The embedding of the ridge into the membrane is consistent with the local spontaneous curvature mechanism that is reported very recently (Zimmerberg and Kozlov, 2006). As a protruding structure found in epsin1 induces liposome tubulation by being inserted to one leaflet of the lipid bilayer (Ford et al, 2002), the penetration of the hydrophobic ridge can drive the positive curvature by causing asymmetrical expansion of the surface area between two leaflets as shown in Figure 8 (Farsad and De Camilli, 2003).
Figure 8.
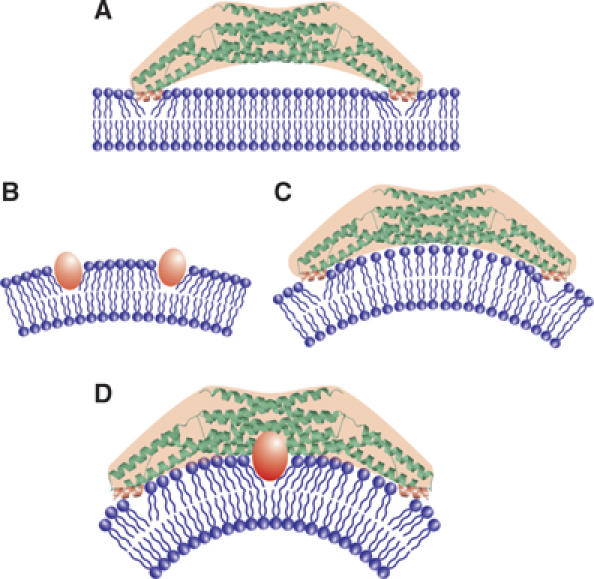
Two potential mechanisms for driving membrane curvature by endophilin-A1. (A) Kissing adhesion of an N-BAR domain on planar lipid bilayer. The helix 0 is essential for the membrane binding. Membrane insertion of the helix 0 is supposed. (B) Insertion of hydrophobic portions of macromolocules into one leaflet can create bilayer surface discrepancy that causes membrane curvature. (C) The simple N-BAR domain, such as amphiphysin and ΔApp, induces membrane curvature by impressing the concave surface onto the membrane. The rigidity of the molecule is required for this mechanism. (D) To drive membrane curvature, the endophilin N-BAR domain uses both the rigid crescent shape-mediated deformation and the insertion of hydrophobic ridge on the concave surface in addition to kissing adhesion of N-BAR to membrane surface.
We further explored the importance of the ridge, rigid crescent shape, and helix 0 in cells. We for the first time showed that N-BAR domain induced membrane invaginations originated from plasma membrane, although other BAR-containing molecules have been reported to induce similar invaginations (Itoh et al, 2005). Neither mutant that lacked either the ridge or the helix 0 nor flexible mutant formed the tubular invaginations in cells, indicating the significance of these sub-module structure in cells as suggest by in vitro studies. We constructed a series of endorphin-A1-EGFP expression plasmids to delineate the domain for the membrane invagination. Full-length endophilin-expressing cells did not show any tubular formation. Because endophilin consists of BAR domain and an SH domain, SH3-binding molecule such as dynamin may inhibit the extension of membrane invagination. This possibility has been suggested in the membrane invagination found in FBP17 and amphiphysin (Kamioka et al, 2004; Itoh et al, 2005).
Collectively, EndA1-BAR uses two newly identified mechanisms to drive positive membrane curvature in addition to the essential binding capacity of helix 0 to the membrane: one by the scaffold mechanism common to the BAR domains and the other by the local spontaneous curvature mechanism caused by the membrane insertion of the ridge (Figure 8D). The ridge, which occupies the bottom of the concave lipid-binding surface, may not work until the main body of the BAR dimer localizes itself to a curved membrane. The ridge then inserts into the bilayer roughly perpendicular to the main body, and thus both deformations will occur in the same direction.
Materials and methods
Protein expression and purification by CRECLE
cDNAs encoding BAR domains (amphiphysin1, 1–239; endophilin-A1, 1–247; endophilin-B1, 1–246 in amino-acid residues) were amplified by PCR from a human brain cDNA library. Recombinant proteins were expressed in Escherichia coli as GST-fusions using the pGEX6p3 vector, purified by glutathione-Sepharose, cleaved from the GST-tag using Prescission protease (Amersham Biosciences), and further purified by ion-exchange chromatography (Yamagishi et al, 2004). The final polypeptide contained an artificial linker sequence of GPLGS at the N-terminus. EndA1-BAR proteins except for F202W and a4 mutants were purified by crystallization during Prescission protease cleavage. The method, crystallization by regulated cleavage of large hydrophilic tag (CRECLE), was as follows. Purified GST fusions were concentrated to 20–30 mg/ml in an elution buffer (20 mM glutathione, 100 mM Tris–HCl, pH 8.0, 10 mM DTT, 1 mM EDTA, 1 mM EGTA) and then cleaved by a low concentration of prescission protease (1 U/mg protein or less) at 4°C. Slow increase in the tag-free protein concentration might be suitable for crystallization and more than a half of EndA1-BAR protein could be recovered as 20–100 μm microcrystals. They were washed with a low-salt buffer (20 mM HEPES, pH 7.4, 2 mM DTT, 0.2 mM EDTA, 0.2 mM EDTA) and resolved into a high-salt buffer (350 mM NaCl in the low-salt buffer) and used for further analyses.
Protein crystallization
Seleno-methionine (S-Met) derivatives of the EndA1-BAR domain and its appendage-less mutant (ΔApp) were produced in B834(DE3)pLysS cells using Overnight Express Autoinduction System 2 (Novagen). To make X-ray grade crystals in a cryo-ready condition, modified high salt buffer (50 mM HEPES, pH 7.4, 300 mM NaCl, 100 mM KI, 28% ethylene glycol, 5% glycerol, 25 mM DTT) was used. Crystals of 1 mm size were formed by dialysis against 50 mM CHES, pH 9.5, 260 mM NaCl, 28% ethylene glycol, 5% glycerol, 25 mM DTT, 0.4% benzamidine·HCl at 4°C and were flush frozen at 100 K. Crystals could also be grown by vapor diffusion from a similar protein solution using distilled water as the bath solution. The crystals were equilibrated in 50 mM HEPES, pH 7.4, 150 mM NaCl, 25 mM DTT, 0.4% benzamidine·HCl, 5% PEG 8000 and the saturated amount of xylitol as a cryoprotectant. Some of the crystals were soaked with 0.5 mM oleoyl-L-α-lysophosphatidic acid (Sigma) or malonyl-CoA (Sigma) for 4 days with daily change for the substrates. The a4 mutant crystals were grown by sitting-drop vapour diffusion using a bath solution containing 100 mM HEPES, pH 7.2, 200 mM calcium acetate, 10 mM DTT and 20% (w/v) PEG3350 at 20°C and then flush frozen after brief immersion in the same solution containing 16% DMSO. The wild type and the ΔApp mutant crystals belong to the same space group I41 and contain one monomer molecule in the asymmetric unit (Supplemental Figure 1). The a4 crystal belongs to P21 and contains two dimers in the asymmetric unit.
Structural determination
The EndA1-BAR structure was determined using the multiple anomalous dispersion (MAD) method. Multiple-wavelength X-ray diffraction data sets were collected from a single Se-Met crystal (crystal I) at SPring-8 beamline BL44B2 (Supplementary Table I). Single wavelength data sets of another crystal (crystal II) and of a ΔApp crystal used for the refinement were collected at BL45PX. The data set for the a4 mutant was collected at BL38B1. All diffraction data sets were collected at 90 K and were processed using HKL2000 suite (Otwinowski and Minor, 1997). The seven positions out of 10 expected selenium atoms were identified by SOLVE (Terwilliger and Berendzen, 1999). The initial phases calculated by SOLVE with a figure of merit of 0.59 at 3.2 Å resolution were further improved by RESOLVE (Terwilliger, 1999). The density modified MAD map (Supplementary Figure 1) had sufficient quality to trace the polypeptide chain except for the N-terminus and the loop region of the appendage. The model was built with TURBO-FRODO (Roussel and Cambillau, 1996) and refined to the resolutions of 3.1 Å by CNS (Brunger et al, 1998). The final model includes 210 residues (residues 26–71 and 84–247), and has an R factor of 23.6% (Rfree of 26.4%). The ΔApp structure was solved by molecular replacement by MOLREP in the CCP4 suite (CCP4, 1994) and refined to the resolution of 2.9 Å by CNS. The simulated annealing omit electron density map calculated by CNS confirmed the continuous α-helical structure of the replaced region as designed (Supplementary Figure 7). The final model includes 200 amino- acid residues and has an R factor of 23.8% (Rfree of 26.9%). The a4 mutant structure was solved by molecular replacement using the central core of the EndA1-BAR as a starting model and the arms were manually built (Supplementary Figure 7). The structure was refined to the resolution of 2.4 Å by CNS with an R factor of 21.5% (Rfree of 26.9%). Main-chain dihedral angles of all non-glycine residues of these three models lie in allowed regions of the Ramachandran plot, with 94.3% for the EndA1-BAR, 94.1% for the ΔApp mutant, and 96.4% for the a4 mutant in most-favored regions, respectively. Graphical representations were prepared using the programs TURBO-FRODO, MOLSCRIPT (Kraulis, 1991), RASTER3D (Merritt and Bacon, 1997), GRASP (Nicholls et al, 1991) and Pymol (DeLano, 2002).
Liposome binding and tubulation assays
Liposome sedimentation assay and tubulation assay were as earlier described (Peter et al, 2004 see also McMahon lab protocols: http://www2.mrc-lmb.cam.ac.uk/NB/McMahon_H/group/techniqs/techniqs.htm) with slight modifications. Briefly, Folch fraction 1 (Sigma) was used as the lipid source and liposome suspension, 1 mg/ml in liposome buffer (20 mM HEPES, pH 7.4, 150 mM NaCl, 1 mM DTT) was made by sonication. Freshly purified BAR domain proteins were diluted at about 1 mg/ml in the liposome buffer and ultracentrifuged at 400 000 g for 10 min just before use. No crystallization occurred at this or lower concentrations. For sedimentation assays, 20 μg proteins were mixed with 25 or 75 μg liposomes in 100 μl of the liposome buffer, incubated for 10 min on ice and ultracentrifuged at 200 000 g for 10 min. For tubulation assays, 400 μg/ml proteins were mixed with an equal volume of 400 μg/ml liposomes, left for 10 s to 30 min at room temperature, and then processed for negative staining. Judging from the liposome sedimentation and the tryptophan fluorescence assays, this protein to lipid ratio ensured nearly saturated protein–liposome binding. Magnification was calibrated using a grating replica of 2160/mm.
Tryptophan fluorescence and FRET assay
Fluorescence emission spectra were recorded with a Hitachi F-4500 fluorescence spectrophotometer (Ohki et al, 2004). For tryptophan fluorescence assays, 140 μg/ml tryptophan-containing mutants were mixed with 0–200 μg/ml liposomes in the liposome buffer, incubated for 3 min, and excited at 280 nm. For FRET assays, DPH-liposomes were made by adding DPH (Molecular Probe) into lipid solution (1:500 to lipid, w:w). The fluorescence of DPH-liposomes (200 μg/ml) excited at 280 nm was scanned from 400 to 500 nm at 1-min intervals. The first measurement of the 430-nm DPH peak was obtained at about 30 s after mixing with mutant proteins (100 μg/ml).
Cell culture, transfection and surface biotinylation
HUVECs were purchased from Kurabo and cultured in HuMedia-EG2 as described previously (Sakurai et al., 2006). 293T cells, CHO cells, Cos7 cells, and NIH-3T3 cells were cultured in DMEM supplemented with 10% fetal bovine serum as described previously (Kamioka et al., 2004). Cells were transfected using LipofectAMINE 2000 (Invitrogen). Live HUVECs were biotinylated with 5 mM sulfo-NHS-biotin (Pierce) in Opti-MEM (Invitrogen) for 10 min. They were washed once with Opti-MEM and chased for 10 min with the normal culture medium, and fixed with 2% formaldehyde after a brief wash with Opti-MEM containing 1/20 volume of Avidin D blocking solution (Vector Laboratory) to reduce the cell surface background staining. HUVECs were permeabilized with cold MeOH and biotin was visualized using Alexa633-streptavidine (Molecular Probe).
Supplementary Material
Supplementary Figure 1
Supplementary Figure 2
Supplementary Figure 3
Supplementary Figure 4
Supplementary Figure 5
Supplementary Figure 6
Supplementary Figure 7
Supplementary Figure 8
Supplementary Table I
Supplementary Table II
Supplementary Movie 1
Supplementary Movie Legend
Acknowledgments
We thank H Nakajima, T Matsu, Y Kawano and H Naitow for technical assistance with SPring-8 beamlines, and H Ago and M Miyano, Structural Biophysics Laboratory, RIKEN Harima Institute at SPring-8, for their helpful advice. This work was supported in part by Grant for Research on Advanced Medical Technology from the Ministry of Health, Labour, and Welfare of Japan, by the Program for Promotion of Fundamental Studies in Health Sciences of the National Institute of Biomedical Innovation (NIBIO), and by Special Coordination Funds for Promoting Science and Technology, Ministry of Education, Culture, Sports, Science and Technology (MEXT) of Japan.
Competing interests statement The authors declare that they have no competing commercial interests in relation to this work.
References
- Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL (1998) Crystallography & NMR system: a new software suite for macromolecular structure determination. Acta Crystallogr D 54: 905–921 [DOI] [PubMed] [Google Scholar]
- Collaborative Computational Project Number 4 (1994) The CCP4 suite: programs for protein crystallography. Acta Crystallogr D 50: 760–763 [DOI] [PubMed] [Google Scholar]
- DeLano WL (2002) The PyMOL User's Manual. DeLano Scientific: San Carlos, CA, USA [Google Scholar]
- Galli T, Haucke V (2004) Cycling of synaptic vesicles: How far? How fast!. Sci STKE 2004: re19. [DOI] [PubMed] [Google Scholar]
- Farsad K, Ringstad N, Takei K, Floyd SR, Rose K, De Camilli P (2001) Generation of high curvature membranes mediated by direct endophilin bilayer interactions. J Cell Biol 155: 193–200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farsad K, De Camilli P (2003) Mechanisms of membrane deformation. Curr Opin Cell Biol 15: 372–381 [DOI] [PubMed] [Google Scholar]
- Ford MG, Mills IG, Peter BJ, Vallis Y, Praefcke GJ, Evans PR, McMahon HT (2002) Curvature of clathrin-coated pits driven by epsin. Nature 419: 361–366 [DOI] [PubMed] [Google Scholar]
- Gallop JL, McMahon HT (2005) BAR domains and membrane curvature: bringing your curves to the BAR. Biochem Soc Symp 72: 223–231 [DOI] [PubMed] [Google Scholar]
- Habermann B. (2004) The BAR-domain family of proteins: a case of bending and binding. EMBO Rep 5: 250–255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Heuvel E, Bell AW, Ramjaun AR, Wong K, Sossin WS, McPherson PS (1997) Identification of the major synaptojanin-binding proteins in brain. J Biol Chem 272: 8710–8716 [DOI] [PubMed] [Google Scholar]
- Itoh T, Erdmann KS, Roux A, Habermann B, Werner H, De Camilli P (2005) Dynamin and the actin cytoskeleton cooperatively regulate plasma membrane invagination by BAR and F-BAR proteins. Dev Cell 9: 791–804 [DOI] [PubMed] [Google Scholar]
- Kamioka Y, Fukuhara S, Sawa H, Nagashima K, Masuda M, Matsuda M, Mochizuki N. (2004) A novel dynamin-associating molecule, formin-binding protein 17, induces tubular membrane invaginations and participates in endocytosis. J Biol Chem 279: 40091–40099 [DOI] [PubMed] [Google Scholar]
- Karbowski M, Jeong SY, Youle RJ (2004) Endophilin B1 is required for the maintenance of mitochondrial morphology. J Cell Biol 166: 1027–1039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraulis PJ (1991) MOLSCRIPT: a program to produce both detailed and schematic plots of protein structure. J Appl Crystallogr 24: 946–950 [Google Scholar]
- McMahon HT, Mills IG (2004) COP and clathrin-coated vesicle budding: different pathways, common approaches. Curr Opin Cell Biol 16: 379–391 [DOI] [PubMed] [Google Scholar]
- McMahon HT, Gallop JL (2005) Membrane curvature and mechanisms of dynamic cell membrane remodeling. Nature 438: 590–596 [DOI] [PubMed] [Google Scholar]
- Merritt EA, Bacon DJ (1997) Raster3D: photorealistic molecular graphics. Methods Enzymol 277: 505–524 [DOI] [PubMed] [Google Scholar]
- Modregger J, Schmidt AA, Ritter B, Huttner WB, Plomann M (2003) Characterization of Endophilin B1b, a brain-specific membrane-associated lysophosphatidic acid acyl transferase with properties distinct from endophilin A1. J Biol Chem 278: 4160–4167 [DOI] [PubMed] [Google Scholar]
- Nicholls A, Sharp K, Honig B (1991) Protein folding and association: insights from the interfacial and thermodynamic properties of hydrocarbons. Proteins 11: 281–296 [DOI] [PubMed] [Google Scholar]
- Nossal R, Zimmerberg J (2002) Endocytosis: curvature to the ENTH degree. Curr Biol 12: R770–R772 [DOI] [PubMed] [Google Scholar]
- Ohki T, Mikhailenko SV, Morales MF, Onishi H, Mochizuki N (2004) Transmission of force and displacement within the myosin molecule. Biochemistry 43: 13707–13714 [DOI] [PubMed] [Google Scholar]
- Otwinowski Z, Minor W (1997) Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol 276: 307–326 [DOI] [PubMed] [Google Scholar]
- Peter BJ, Kent HM, Mills IG, Vallis Y, Butler PJ, Evans PR, McMahon HT (2004) BAR domains as sensors of membrane curvature: the amphiphysin BAR structure. Science 303: 495–499 [DOI] [PubMed] [Google Scholar]
- Repáková J, Holopainen JM, Morrow MR, McDonald MC, Capkova P, Vattulainen I (2005) Influence of DPH on the structure and dynamics of a DPPC bilayer. Biophys J 88: 3398–3410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ringstad N, Nemoto Y, De Camilli P (1997) The SH3p4/Sh3p8/SH3p13 protein family: binding partners for synaptojanin and dynamin via a Grb2-like Src homology 3 domain. Proc Natl Acad Sci USA 94: 8569–8574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ringstad N, Nemoto Y, De Camilli P (2001) Differential expression of endophilin 1 and 2 dimers at central nervous system synapses. J Biol Chem 276: 40424–40430 [DOI] [PubMed] [Google Scholar]
- Roussel A, Cambillau C (1996) TURBO-FRODO Manual. Marseille France AFMB-CNRS, Paris, France [Google Scholar]
- Sakurai A, Fukuhara S, Yamagishi A, Sako K, Kamioka Y, Masuda M, Nakaoka Y, Mochizuki N (2006) MAGI-1 is required for Rap1 activation upon cell–cell contact and for enhancement of vascular endothelial cadherin-mediated cell adhesion. Mol Biol Cell 17: 966–976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuske KR, Richmond JE, Matthies DS, Davis WS, Runz S, Rube DA, van der Bliek AM, Jorgensen EM (2003) Endophilin is required for synaptic vesicle endocytosis by localizing synaptojanin. Neuron 40: 749–762 [DOI] [PubMed] [Google Scholar]
- Tarricone C, Xiao B, Justin N, Walker PA, Rittinger K, Gamblin SJ, Smerdon SJ (2001) The structural basis of Arfaptin-mediated cross-talk between Rac and Arf signalling pathways. Nature 411: 215–219 [DOI] [PubMed] [Google Scholar]
- Terwilliger TC (1999) Reciprocal-space solvent flattening. Acta Crystallogr D 55: 1863–1871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terwilliger TC, Berendzen J (1999) Automated MAD and MIR structure solution. Acta Crystallogr D 55: 849–861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verstreken P, Koh TW, Schulze KL, Zhai RG, Hiesinger PR, Zhou Y, Mehta SQ, Cao Y, Roos J, Bellen HJ (2003) Synaptojanin is recruited by endophilin to promote synaptic vesicle uncoating. Neuron 40: 733–748 [DOI] [PubMed] [Google Scholar]
- Weissenhorn W (2005) Crystal structure of the endophilin-A1 BAR domain. J Mol Biol 351: 653–661 [DOI] [PubMed] [Google Scholar]
- Wenk MR, De Camilli P (2004) Protein–lipid interactions and phosphoinositide metabolism in membrane traffic: insights from vesicle recycling in nerve terminals. Proc Natl Acad Sci USA 101: 8262–8269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamagishi A, Masuda M, Ohki T, Onishi H, Mochizuki N (2004) A novel actin-bundling/filopodium-forming domain conserved in insulin receptor tyrosine kinase substrate p53 and missing in metastasis protein. J Biol Chem 279: 14929–14936 [DOI] [PubMed] [Google Scholar]
- Zimmerberg J, Kozlov MM (2006) How proteins produce cellular membrane curvature. Nat Rev Mol Cell Biol 7: 9–19 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1
Supplementary Figure 2
Supplementary Figure 3
Supplementary Figure 4
Supplementary Figure 5
Supplementary Figure 6
Supplementary Figure 7
Supplementary Figure 8
Supplementary Table I
Supplementary Table II
Supplementary Movie 1
Supplementary Movie Legend