

Abstract
Simultaneous ablation of the two known activators of plasminogen (Plg), urokinase-type (uPA) and the tissue-type (tPA), results in a substantial delay in skin wound healing. However, wound closure and epidermal re-epithelialization are significantly less impaired in uPA;tPA double-deficient mice than in Plg-deficient mice. Skin wounds in uPA;tPA-deficient mice treated with the broad-spectrum matrix metalloproteinase (MMP) inhibitor galardin (N-[(2R)-2-(hydroxamido-carbonylmethyl)-4-methylpentanoyl]-L-tryptophan methylamide) eventually heal, whereas skin wounds in galardin-treated Plg-deficient mice do not heal. Furthermore, plasmin is biochemically detectable in wound extracts from uPA;tPA double-deficient mice. In vivo administration of a plasma kallikrein (pKal)-selective form of the serine protease inhibitor ecotin exacerbates the healing impairment of uPA;tPA double-deficient wounds to a degree indistinguishable from that observed in Plg-deficient mice, and completely blocks the activity of pKal, but not uPA and tPA in wound extracts. These findings demonstrate that an additional plasminogen activator provides sufficient plasmin activity to sustain the healing process albeit at decreased speed in the absence of uPA, tPA and galardin-sensitive MMPs and suggest that pKal plays a role in plasmin generation.
Keywords: fibrinolysis; plasma kallikrein; plasmin; uPA-, tPA- and plasminogen-deficient mice; wound healing
Introduction
Confined proteolytic degradation of the extracellular matrix by the concerted action of the serine protease plasmin and members of the matrix metalloprotease (MMP) family is considered to play a key role in a variety of physiological and pathological processes involving tissue remodeling and cell migration, including cancer invasion (Danø et al, 2005; Egeblad and Werb, 2002). A role for the plasmin precursor plasminogen (Plg) in tissue remodeling processes has been conclusively demonstrated by studies of skin wound healing (Rømer et al, 1996), cancer metastasis (Bugge et al, 1998), post-lactational mammary gland involution (Lund et al, 2000) and placental development (Solberg et al, 2003) in mice with a disrupted Plg gene. Plasmin is formed from Plg by cleavage catalyzed by Plg activators. Two physiologically active mammalian Plg activators are known, the urokinase-type (uPA) and the tissue-type (tPA) (Danø et al, 1985; Collen and Lijnen, 2005). uPA is secreted as an inactive precursor, pro-uPA, which can bind to the uPA receptor (uPAR), a glycolipid-anchored membrane protein with three-fold finger domains (Llinas et al, 2005). Pro-uPA is activated to uPA by plasmin while bound to uPAR, and receptor-bound uPA can activate Plg (Ploug, 2003). Concomitant binding of pro-uPA and Plg to cell surfaces strongly accelerates plasmin generation owing to an increased efficiency of the reciprocal activation of the two proenzymes (Ellis et al, 1991). tPA-directed plasminogen activation is accelerated by concomitant binding of tPA and Plg to fibrin (Carmeliet et al, 1994; Collen and Lijnen, 2005). Preferential sites for the uPA and tPA pathways of Plg activation are therefore surfaces of uPAR-expressing cells and fibrin deposits, respectively. In accordance, the primary established functions of uPA are within tissue remodeling processes (Danø et al, 1999), whereas those of tPA are within vascular thrombolysis (Collen and Lijnen, 2005). Despite this divergence in their basic biological functions, uPA and tPA can to some extent serve as mutual, functional substitutes, as has been observed in gene-deficient mice (Carmeliet et al, 1994; Bugge et al, 1996a). In addition to uPA and tPA, a few other serine proteases such as the blood coagulation factors XI and XII and plasma kallikrein are capable of activating plasminogen in test tubes (Danø et al, 1985). However, the physiological impact of such activators has not been thoroughly demonstrated in vivo during, for example, skin wound healing. In mammary gland involution, it was suggested that plasma kallikrein (pKal) is a Plg activator during adipocyte differentiation (Selvarajan et al, 2001).
During healing of skin wounds, the migrating leading-edge keratinocytes express uPA and uPAR. tPA is more scarcely expressed but has been detected in a few keratinocytes late in the re-epithelialization of human wounds (Rømer, 2003). In Plg-deficient mice there is a pronounced delay in wound healing characterized by a decreased rate of migration of keratinocytes from the wound edges and an accumulation of fibrin in front of the leading-edge keratinocytes, suggesting that the delay is owing to a diminished ability of these cells to proteolytically dissect their way through the extracellular matrix (Rømer et al, 1996). This interpretation is supported by the observation that skin wound healing is restored in mice deficient in both plasminogen and fibrin (Bugge et al, 1996b). Although delayed, complete wound healing is eventually achieved in all Plg-deficient mice. Several MMPs, including MMP3, MMP9 and MMP13, are expressed in murine leading-edge keratinocytes (Madlener et al, 1998; Lund et al, 1999), and wound healing is severely impaired in transgenic mice with human collagenase-1-resistant collagen I (Beare et al, 2003). Treatment of wild-type mice with a broad-spectrum MMP inhibitor, galardin (N-[(2R)-2-(hydroxamido-carbonylmethyl)-4-methylpentanoyl]-L-tryptophan methylamide), causes a delay in wound healing time, but all wounds do eventually heal. However, when Plg-deficient mice are treated with galardin, healing is completely arrested, demonstrating that protease activity is essential for wound healing and that there is a functional overlap between the two classes of matrix-degrading proteases in this process (Lund et al, 1999).
In order to study the relative contribution of the individual plasminogen activators during wound healing, we have examined the impact on skin repair of single deficiencies in uPA, tPA and Plg as well as double deficiency in uPA and tPA. We now provide in vivo biochemical and genetic evidence to demonstrate first a functional overlap between uPA and tPA, and second the existence of at least one additional Plg activator contributing to wound healing. Further experimental evidence points to plasma kallikrein as the possible third plasminogen activator with in vivo activity during skin wound healing.
Results
Functional overlap between uPA and tPA in wound healing
We first examined the effect on wound healing of single deficiencies in uPA and tPA and double deficiency in both activators. Standardized 20 mm long, full thickness, incisional wounds were generated in wild-type mice (n=30), uPA-deficient mice (n=13), tPA-deficient mice (n=17) and mice double-deficient for uPA and tPA (n=13). All mice were F2-generation siblings derived from backcrossing single-deficient mice into the C57Bl/6J strain for 16 generations. The wounds were examined grossly by visual inspection, and the wound lengths measured three times a week. Immediately after surgery, the wounds were spindle-shaped with well-separated incision edges and the underlying muscle fascia was exposed. By the second day post-wounding, the wounds were covered by a dehydrated wound crust, which was gradually lost as healing progressed. Lesions were scored as fully healed when there was a complete loss of the wound crust and closure of the incision interface with restoration of the epidermal covering. Immediately after the wounds were scored as healed by gross inspection, the wounded skin was removed for histological examination. Healing was in all cases verified by the presence of an intact multilayered epidermis (see Figure 3).
Figure 3.
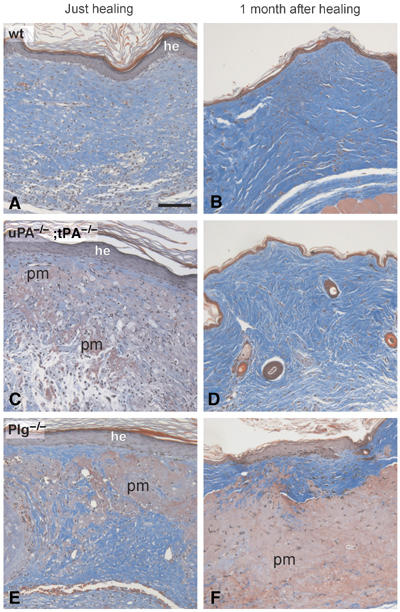
Analysis of skin wounds in wild-type, uPA;tPA double-deficient and Plg-deficient mice at the time of healing and 1 month post-healing Trichrome-stained sections of wounds from wild-type (A, B), uPA;tPA double-deficient (C, D) and Plg-deficient mice (E, F). Sections of tissue isolated just after healing reveal an intact, but hyperplastic epidermis (he), In wild-type mice (A), the provisional matrix has been completely replaced by granulation tissue, whereas in uPA;tPA double-deficient and Plg-deficient mice, larger areas of nondegraded provisional matrix (pm) were interspersed in the granulation tissue (C, E). At 1 month after healing, wild-type (B) and uPA;tPA double-deficient (D) mice all reveal a normal-looking epidermis and dermis, whereas in the Plg-deficient mice the epidermis is hyperplastic and the dermis is abnormal with abundant accumulations of undegraded provisional matrix (F). Bar: 60 μm.
The mean times to complete healing for mice with single deficiencies in either uPA or tPA was 18.2±2.7 and 17.6±2.8 days, respectively, which were similar to the healing time of 16.9±3.2 days in wild-type mice (Table I; Figure 1A). In mice double-deficient for uPA and tPA, the mean healing time increased to 31.2±11.3 days, a significant delay in skin wound healing compared to the wild-type mice (P=0.0006) and also to the uPA (P=0.0008) or tPA (P=0.0007) single-deficient mice (Table I; Figure 1A). The combined ablation of uPA and tPA thus results in a pronounced impairment of wound healing, whereas there is virtually no effect on wound healing of deficiency in either of the two genes alone. We conclude that the presence of either uPA or tPA is required to maintain full wound healing capacity and that there is a functional overlap between the two.
Table 1.
Effect of uPA, tPA and Plg single deficiencies, uPA;tPA double-deficiency and galardin treatment on wound healing time
| Group | Genotype | Number of backcrossings | Treatment | Number of mice | Mean healing time (days) | s.d. | P-value |
|---|---|---|---|---|---|---|---|
| 1 | Wild-type | 16 | None | 30 | 16.9 | 3.2 | |
| 2 | uPA−/−; tPA+/+ | 16 | None | 13 | 18.2 | 2.7 | 2 versus 1 NSa |
| 3 | uPA+/+; tPA−/− | 16 | None | 17 | 17.6 | 2.8 | 3 versus 1 NSa |
| 4 | uPA−/−; tPA−/− | 16 | None | 13 | 31.2 | 11.3 | 4 versus 1 0.0006 |
| 5 | Wild-type | 21 | None | 24 | 16.8 | 2.2 | |
| 6 | Plg−/− | 21 | None | 20 | 42.9 | 15.6 | 6 versus 5 0.0001; 6 versus 4 0.03 |
| 7 | Wild-type | 16 | Mock | 16 | 17.0 | 3.7 | |
| 8 | Wild-type | 16 | Galardinb | 10 | 22.3 | 5.5 | 8 versus 7 0.008 |
| 9 | uPA−/−; tPA−/− | 16 | Mock | 13 | 30.2 | 10.0 | 9 versus 7 0.0004 |
| 10 | uPA−/−; tPA−/− | 16 | Galardinb | 7 | 45.9 | 6.9 | 10 versus 9 0.002 |
| 11 | Wild-type | 21 | Mock | 15 | 19.0 | 2.9 | |
| 12 | Wild-type | 21 | Galardinb | 15 | 24.9 | 3.8 | 12 versus 11 0.0001 |
| 13 | Plg−/− | 21 | Mock | 14 | 50.9 | 19.3 | 13 versus 11 0.0001; 13 versus 9 0.002 |
| 14 | Plg−/− | 21 | Galardinb | 12 | >70 daysc | 14 versus 13 0.0001; 14 versus 10 0.0001 | |
| Not significant (P>0.05). | |||||||
| 100 mg/kg daily. | |||||||
| No wounds healed at end of galardin treatment. | |||||||
| Abbreviations: Plg, plasminogen; tPA, tissue-type plasminogen activator; uPA, urokinase-type plasminogen activator. |
Figure 1.

Time course for healing of full-thickness skin wounds in wild-type, tPA-deficient, uPA-deficient, uPA;tPA double-deficient and Plg-deficient mice. (A) The percentage fraction of mice with complete gross healing is plotted versus time after the incision. A modest and nonsignificant delay was observed in tPA single-deficient and uPA single-deficient mice compared to wild-type mice, whereas a significant delay was observed in uPA;tPA double-deficient mice. In Plg-deficient mice, there was an even more pronounced delay and the healing occurred significantly later than in the uPA;tPA double-deficient mice. (B) The average wound length is plotted versus time after incision. The average wound length of wild-type, uPA-deficient and tPA-deficient mice was indistinguishable until re-epithelialization. In contrast, the wound length of uPA;tPA double-deficient mice was significantly increased compared to either wild-type or uPA- or tPA single-deficient mice. The average wound length of Plg-deficient mice was significantly increased compared to uPA;tPA double-deficient mice. (C) Examples of the progress of wound repair in wild-type, uPA;tPA double-deficient and Plg-deficient mice. (D) Galardin or mock treatment of wild-type, uPA;tPA double-deficient and Plg-deficient mice. The percentage fraction of wounds healed is plotted versus time after the incision. The wild-type and uPA;tPA double-deficient mice treated with galardin exhibited a delay in healing compared to mock-treated mice but all wounds eventually healed. In contrast, galardin treatment of Plg-deficient mice completely blocked healing. However, this arrest was reversible as cessation of galardin treatment in these mice at day 70 resulted in completion of healing, so that the wounds were completely healed within 28 days.
Skin wound healing is less impaired in mice double deficient for uPA and tPA compared to Plg-deficient mice
We next compared the efficiency of the skin wound healing in the uPA;tPA double-deficient mice with that in Plg-deficient mice. For this purpose, we used Plg gene-targeted mice backcrossed into the C57Bl/6J strain for 21 generations. These heterozygous Plg gene-deficient mice were interbred, resulting in an F1 generation of mice, of which the homozygous Plg-deficient mice (n=20) and their wild-type siblings (n=24) were used in the experiment.
The mean wound healing time in the Plg-deficient mice was 42.9±15.6 days, whereas the mean healing time in the control group of wild-type mice was 16.8±2.2 days (Table I; Figure 1A) in accordance with previous studies (Rømer et al, 1996). The mean healing time in the Plg-deficient mice was also significantly longer (P=0.03) than the 31.2±11.3 days observed in the uPA;tPA double-deficient mice as shown in Figure 1A. Importantly, the healing time in the two control groups of wild-type siblings corresponding to the uPA;tPA double-deficient and the Plg-deficient mice, respectively, were indistinguishable (16.9±3.2 and 16.8±2.2 days). In a separate experimental setup, wild-type mice obtained by breeding of MMP-9;Plg and MMP3;Plg double-heterozygous mice exhibited similar healing times (17.0±3.2 and 17.8±3.6 days). Notably, these were indistinguishable from the healing times in the background strain C57Bl/6J (16.3±2.8 days). This emphasize that the observed difference between the uPA;tPA double-deficient and Plg-deficient mice is not caused by random genetic differences between the various lines of gene-targeted mice, but is clearly ascribed to a role of Plg in wound healing that is not entirely dependent on the presence of the cognate activators uPA and tPA.
Following wounding, the average wound lengths of wild-type, uPA single-deficient and tPA single-deficient mice were indistinguishable until completion of re-epithelialization (Figure 1B). As opposed to this, the average wound length in uPA;tPA double-deficient mice was significantly increased compared to either wild-type or uPA- or tPA single-deficient mice (P<0.001). Furthermore, the average wound length of Plg-deficient mice was significantly increased compared to uPA;tPA double-deficient mice (P<0.02) (Figure 1B).
In the presence of galardin, wound healing is delayed in uPA and tPA double-deficient mice but arrested in Plg-deficient mice
We have previously demonstrated that the MMP inhibitor galardin delays wound healing in wild-type mice and that healing in Plg-deficient mice is completely blocked by galardin treatment (Lund et al, 1999). We now compared the effect of galardin on skin wound healing in uPA;tPA double-deficient mice and Plg-deficient mice. For each of the genotypes, we used mock-treated wild-type mice that were siblings to the gene-deficient mice as controls. By daily administration of galardin at a dose of 100 mg/kg, healing was further delayed in the uPA;tPA double-deficient mice (from 30.2±10.0 days in mock-treated mice to 45.9±6.9 days in galardin-treated mice). However, all the galardin-treated uPA;tPA double-deficient mice did eventually heal. In contrast, there was a complete arrest of the healing in the galardin-treated Plg-deficient mice for the treatment period of 70 days (Table I and Figure 1D). Notably, there was no difference in the wound healing times between the two mock-treated wild-type control groups or between the two galardin-treated wild-type controls (Table I). These findings indicate that the higher wound healing capability in uPA;tPA double-deficient mice in comparison with Plg-deficient mice is not dependent on a galardin-sensitive protease.
Interestingly, the Plg-deficient mice that had been treated with galardin for 70 days were still capable of healing following discontinuation of galardin treatment, and the tissue repair processes were completed within 28 days (see Figure 1D), indicating that the effect of galardin is due to a transient inhibition, and not to permanent defects in the wound-healing program independent of direct protease inhibition.
Pronounced histological differences between skin wounds in uPA;tPA double-deficient and Plg-deficient mice
We next analyzed histological sections of wound sites from mice of the various genotypes at day 10 after incision (Figure 2), at the time point when the wounds were grossly scored as healed, and 1 month after healing (Figure 3). Wounds in wild-type mice obtained from the breeding of both heterozygous Plg+/− mice and heterozygous uPA+/−;tPA+/− mice were indistinguishable with respect to gross healing as well as histology at all time points post-wounding (data not shown).
Figure 2.

Analysis of skin wounds in wild-type, tPA-deficient, uPA-deficient, uPA;tPA double-deficient and Plg-deficient mice at day 10 after incision. (A) H&E-stained sections of wounds from wild-type (a, b), tPA-deficient (c, d), uPA-deficient (e, f), uPA;tPA double-deficient (g, h) and Plg-deficient mice (i, j). The boxes in a, c, e, g and i are shown in higher magnification in b, d, f, h, and j. Sections of wild-type (a, b) and tPA-deficient mouse wounds (c, d) show a complete multilayered epidermal barrier underneath the wound crust and a newly formed granulation tissue with a small isolated Islets of provisional matrix remaining. A section from a uPA-deficient mouse (e, f) reveals the leading-edge keratinocytes underneath the wound crust (f). A section of a uPA;tPA double-deficient mouse (g, h) reveals migrating keratinocytes surrounded by provisional matrix. Accumulation of amorphous material is observed both in front of, and underneath, the keratinocytes. Similar, but more pronounced accumulations are seen in the sections from Plg-deficient mice (i and j). The straight black arrows in (e, g, i, f, h and j) point to the tip of the leading-edge keratinocytes. (B) Tissue sections of day 7 wounds from wild-type (a) uPA;tPA double-deficient (b) and Plg-deficient (c) mice were stained by double immunofluorescence for cytokeratin 8 (red) and fibrin(ogen) (green). Accumulation of immunoreactivity for fibrinogen was observed underneath the keratinocytes in wild-type (a) and uPA;tPA double-deficient mice (b). In Plg-deficient mice, similar but more pronounced accumulations are seen underneath as well as in front of the keratinocytes (c). The straight white arrows point to the tip of the leading-edge keratinocytes. Bar: Aa, c, e, g and i ∼140 μm; Ab, d, f, h and j ∼35 μm.
At 10 days after wounding, the newly formed epidermal layer underneath the wound crust had completely covered the wound gap in the majority of wild-type and tPA-deficient mice (Figure 2A; panels a–d). The wound crust was still retained, which explains why these wounds were not yet scored as healed by gross inspection. The epidermal layer had not covered the wound field in most of the uPA-deficient mice, but complete epidermal closure was occasionally observed at this time point (Figure 2A; panels e and f). In contrast, none of the wounds in uPA;tPA double-deficient or Plg-deficient mice were covered by a new epidermal layer at day 10 post-wounding. (Figure 2A; panels g–j). The provisional matrix was almost completely degraded and replaced by newly formed granulation tissue in wild-type and tPA-deficient mice at day 10 (Figure 2A; panels a–d). The formation of granulation tissue was less pronounced in the uPA-deficient mice and almost completely absent in the area beneath the epidermal outgrowths in the uPA;tPA double-deficient and Plg-deficient mice (Figure 2A; panels h and j). All Plg-deficient mice and most of the mice single deficient in uPA or double deficient in uPA and tPA showed keratinocyte outgrowths that were markedly blunt-ended (Figure 2A; panels f, h and j). In the Plg-deficient mice, the keratinocyte outgrowth was covered by a dense layer of provisional matrix (Figure 2A; panels i and j), with an accumulation of fibrin(ogen) in front of the keratinocytes that was not seen to the same extent in the other genotypes, although considerable amounts of fibrin(ogen) accumulates beneath the epidermal outgrowth in uPA;tPA double-deficient mice (Figure 2B; panels a–c).
At the time of gross healing, which occurred from 12 to 80 days after incision dependent on the genotype of the mice, the newly formed epidermal layer covering the wound was thickened and multilayered both in wild-type mice and in the various protease-deficient mice (Figure 3A, C and E and data not shown). However, at the time of healing, the overall morphology of the provisional matrix and granulation tissue in the dermis differed between the genotypes. In wild-type, uPA single-deficient and tPA single-deficient mice, the entire provisional matrix was replaced by a well-organized granulation tissue (Figure 3A and B and data not shown). In contrast, in both uPA;tPA double-deficient and Plg-deficient mice, large areas of nondegraded provisional matrix were interspersed with foci of apparently normal vessel- and collagen-rich granulation tissue (Figure 3C and E).
At 1 month after completion of re-epithelialization, the area with regenerated tissue could not be distinguished from the adjacent noninjured epidermis in wild-type, uPA-deficient, tPA-deficient and uPA;tPA double-deficient mice (Figure 3B and D, and data not shown), whereas Plg-deficient mice still revealed clusters of nondegraded provisional matrix interspersed in areas of granulation tissue (Figure 3F).
Attenuated defect in keratinocyte re-epithelialization in uPA;tPA double-deficient versus Plg-deficient mice
To assess the impact of uPA-deficiency, tPA-deficiency, uPA;tPA double deficiency and Plg-deficiency on re-epithelialization by morphometric analysis, we calculated the percentage fraction of the wound width covered by keratinocytes from both sides of the wound. At 10 days after wounding, the re-epithelialization was lower in the mice with single deficiency for uPA (55±7%), double-deficiency for uPA and tPA (53±4%) and deficiency for Plg (45±7%), than in both wild-type mice (88±4%) and tPA-deficient mice (92±5%) (Figure 4A). At day 21 after incision, the fraction of the wound width covered by keratinocytes was significantly lower in Plg-deficient mice (76±8%) than in uPA;tPA-deficient mice (93±4%; t-test P=0.001) (Figure 4B). In all other genotypes, the wounds were completely re-epithelialized at this time point.
Figure 4.
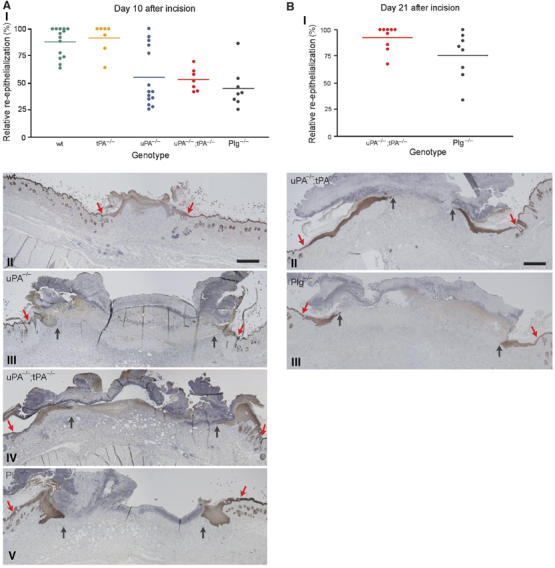
Morphometric analysis of wound re-epithelialization in wild-type, tPA-deficient, uPA-deficient, uPA;tPA double-deficient and Plg-deficient mice. (A) Scatter plot of the relative re-epithelialization of wounds day 10 after incision in wild-type mice, tPA-deficient mice, uPA-deficient mice, uPA;tPA double-deficient mice and Plg-deficient mice (AI). The relative re-epithelialization was determined as the distance from the wound edge to the front of the leading-edge keratinocytes, divided by the distance between the two wound edges. Examples of keratin-stained sections of day 10 wounds from wild-type mice (AII), uPA-deficient mice (AIII), uPA;tPA double-deficient mice (AIV) and Plg-deficient mice (AV). The red arrows mark the wound edge and black arrows point to the tip of the leading-edge keratinocytes. Bar: II–V ∼60 μm. (B) Scatter plot of the relative re-epithelialization of wounds day 21 after incision in uPA;tPA double-deficient mice and Plg-deficient mice (BI). Examples of keratin-stained sections of day 21 wounds from uPA;tPA double-deficient mice (BII) and Plg-deficient mice (BIII). Red arrows mark the wound edge and black arrows identify the tip of the leading-edge keratinocytes. Bar: II–III ∼60 μm.
Wound extracts contain a fibrinolytic activity corresponding to pKal
Because previous studies have suggested a role for plasma kallikrein (pKal) in plasminogen activation during adipocyte differentiation (Selvarajan et al, 2001), we next analyzed purified pKal and wound extracts for fibrinolytic activities by fibrin/Plg overlay zymography. As shown in Figure 5A, there was a concentration-dependent degradation of fibrin by purified human pKal. The fibrinolytic activity of 0.5 μg pKal is comparable to that of 0.5 ng of purified murine uPA. pKal showed lower fibrinolytic activity in the absence of Plg, which indicates that pKal has some Plg-activator activity (Figure 5A).
Figure 5.
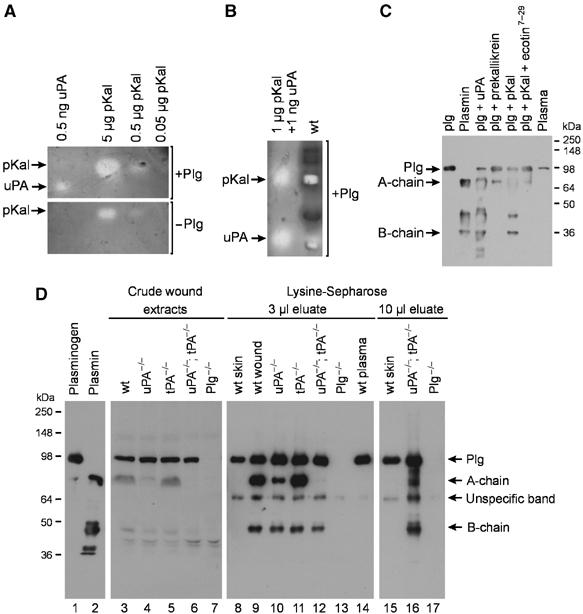
Detection of fibrinolytic activities and Plg/plasmin in wound extracts. (A) Fibrin/Plg overlay zymography of purified human pKal and murine uPA and without Plg. Note that the size of the lysis zones generated by pKal decreases in the absence of Plg. (B) Prolonged development of a zymogram with 1 μg of human pKal and 0.1 ng murine uPA in one lane and wild-type 7-day-old incisional wound extract in the second lane reveals activity comigrating with uPA and pKal in the wild-type wound extract. (C) Western blot analysis of 1 μg mouse Plg, 1 μg mouse plasmin and 1 μl of mouse plasma as controls. Generation of plasmin was detected in 1 μg Plg activated for 2 h at 37°C either by 10 ng uPA, 0.1 μg prekallikrein, 0.1 μg pKal or 0.1 μg pKal in the presence of Ecotin7–29. (D) Western blot analysis for Plg and plasmin. 1 μg mouse Plg (lane 1), 1 μg mouse plasmin (lane 2). Lanes 3–7: 16 μl of crude wound extract prepared from 7-day-old incisions from either wild-type mice (lane 3), uPA-deficient mice (lane 4), tPA-deficient mice (lane 5), uPA;tPA double-deficient mice (lane 6) and Plg-deficient mice (lane 7). Lanes 8–14: 3 μl of eluate pool from Lysine-Sepharose columns, wild-type mice skin (lane 8), wild-type mice wounds (lane 9), uPA-deficient mice (lane 10), tPA-deficient mice (lane 11), uPA;tPA double-deficient mice (lane 12), Plg-deficient mice (lane 13) and plasma from wild-type mice (lane 14). Lanes 15–17: 10 μl of eluate pool from Lysine-Sepharose columns, wild-type mice skin (lane 15), uPA;tPA double-deficient mice (lane 16) and Plg-deficient mice (lane 17).
In wound extracts from wild-type mice, fibrinolytic activities with electrophoretic mobilities corresponding to purified uPA and pKal were detected after prolonged development of the fibrin/Plg zymograms (Figure 5B). After standard development of the zymograms, uPA activity was found in extracts from tPA-deficient and Plg-deficient mice, whereas virtually no uPA was visible in the wild-type wounds, suggesting an upregulation of uPA in tPA- and Plg-deficient wounds (Figure 6A). The extracts of all genotypes contained a substantial amount of fibrinolytic activity with an electrophoretic mobility corresponding to purified pKal (Figures 5B and 6A). No other fibrinolytic activity was observed. In zymograms without Plg, all fibrin-degrading activities in the extracts were absent (data not shown).
Figure 6.
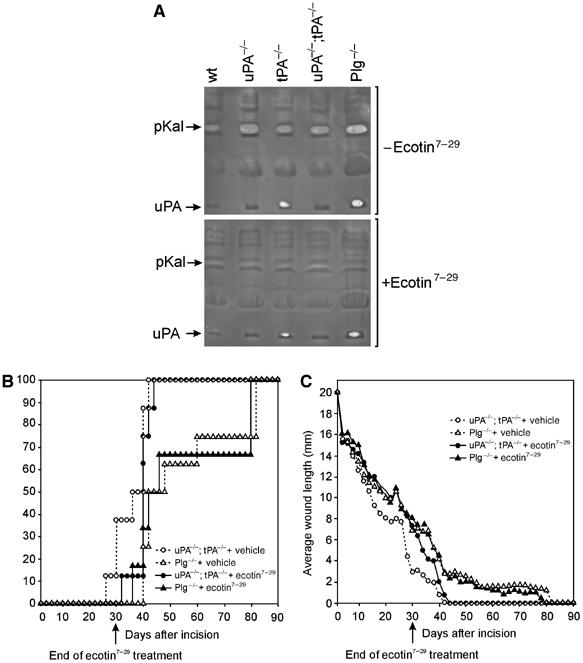
The pKal inhibitor ecotin7–29 inhibits plasmin-mediated fibrinolysis and the healing of uPA;tPA double-deficient wounds. (A) Extracts of 7-day wounds from wild-type, uPA-deficient, tPA-deficient, uPA;tPA double-deficient and Plg-deficient mice (pooled from two mice per genotype) were subjected to fibrin/Plg overlay zymography with or without the serine protease inhibitor ecotin7–29. Note the complete inhibition of activity corresponding to pKal by ecotin7–29, whereas the uPA activity is not inhibited. (B) uPA;tPA double-deficient and Plg-deficient mice were treated with the serine protease inhibitor ecotin7–29 or vehicle for 30 days (n=6–8). The percentage fraction of mice with complete gross healing is plotted versus the time after the incision. (C) The wound length plotted versus the time after the incision. Until the end of treatment at day 30 after the incision, the wound length kinetics in ecotin7–29-treated uPA;tPA double-deficient mice were indistinguishable from vehicle- or ecotin7–29-treated Plg-deficient mice.
We used a modified form of the bacterial serine protease inhibitor ecotin7–29 that is highly selective for pKal and has no inhibitory effect towards, for example, plasmin, uPA, tPA or six other closely related serine proteases, for all of which the Ki is 4–7 orders of magnitude higher (Stoop and Craik, 2003). The activity corresponding to pKal in wound lysates was completely inhibited by ecotin7–29, whereas the inhibitor did not block the activity ascribed to either uPA or the corresponding plasmin activity generated by uPA (Figure 6A). To demonstrate directly that pKal can activate Plg, we next analyzed Plg conversion in vitro by Western blot analysis. Both uPA and pKal convert Plg to two-chain plasmin, whereas only trace amounts of Plg are cleaved by prekallikrein. The Plg activation by pKal was not caused by trace impurities in the enzyme preparation as it was inhibited by the pKal-specific ecotin7–29 (Figure 5C).
Detection of plasmin in wound extracts from uPA;tPA double-deficient mice
Western blot analysis reveals comparable levels of Plg in the crude extracts from wild-type, uPA single-deficient, tPA single-deficient and uPA;tPA double-deficient mice. As expected, no Plg was detected in Plg-deficient mice (Figure 5D, lanes 3–7). Similar amounts of plasmin A- and B-chains were found in wound extracts prepared from wild-type mice and tPA-deficient mice, whereas only low levels of plasmin chains were observed in the crude extract of uPA-deficient mice. However, in uPA;tPA double-deficient mice, no plasminogen conversion was directly detectable in the crude extracts (Figure 5D, lanes 3–7). To improve sensitivity, we subsequently performed a one-step enrichment of Plg/plasmin in wound extracts by lysine-Sepharose affinity chromatography. This provided the required sensitivity that enabled the detection of trace amounts of the individual plasmin chains present in some of the genotypes. As evident in Figure 5D, lanes 12 and 16, both A- and B-chains of plasmin were indeed detectable in extracts from uPA;tPA double-deficient mice demonstrating that Plg can be converted in vivo independently of uPA and tPA. Importantly, neither A- nor B-chains were detectable when extract from noninjured skin or plasma from wild-type mice were tested (Figure 5D, lanes 8, 14 and 15), emphasizing that Plg is activated in vivo during active tissue remodeling, and that no conversion occurs during the extraction or purification procedures.
The serine protease inhibitor ecotin7–29 exacerbates the healing impairment in uPA;tPA double-deficient mice
To examine whether pKal has a physiological role during wound healing, we next treated wounded uPA;tPA-deficient or Plg-deficient mice systemically for 30 days with either vehicle (PBS) or ecotin7–29. Ecotin7–29 treatment of the Plg-deficient mice had no effect on the average wound length (Figure 6C). In contrast, ecotin7–29 treatment of uPA;tPA double-deficient mice significantly delayed wound healing compared to vehicle control, as measured by the average wound length at the end of treatment 30 days post-wounding (P<0.03) (Figure 6C). In fact, the wound lengths of ecotin7–29-treated uPA;tPA double-deficient mice, mock-treated Plg-deficient mice and ecotin7–29-treated Plg-deficient mice were indistinguishable, which indicates that ecotin7–29 blocks virtually all remaining plasminogen activation in the uPA;tPA double-deficient mice. At the end of ecotin7–29 treatment at day 30 none of the uPA;tPA double-deficient wounds (0%) were healed as opposed to 3 out of 8 healed wounds (38%) in the PBS-treated uPA;tPA double-deficient mice (Figure 6B). Immediately after discontinuation of the treatment, a rapid completion of healing was observed in the ecotin7–29-treated uPA;tPA double-deficient mice.
Discussion
Skin wound healing is impaired in Plg-deficient mice (Rømer et al, 1996). Through a direct comparison of Plg deficiency versus uPA;tPA double deficiency, we now show that the healing in mice that lack both uPA and tPA is significantly less impaired as compared to Plg-deficient mice. This difference is based on observations of attenuated impairments in gross healing times as well as re-epithelialization in the uPA;tPA double-deficient mice. We have found that gross skin wound healing in both uPA single-deficient and tPA single-deficient mice is indistinguishable from the healing of wild-type mice. In contrast, the concomitant ablation of both uPA and tPA caused a significant delay in the healing, in agreement with our previous findings (Bugge et al, 1996a). These results clearly demonstrate a functional redundancy between uPA and tPA in skin wound healing.
The cellular architecture both during and after complete re-epithelialization of the wounds revealed a significant histological difference between uPA;tPA double-deficient and Plg-deficient mice. The higher residual healing capability of uPA;tPA double-deficient mice shows that Plg is playing a role in the healing process even in the absence of the two cognate Plg activators, uPA and tPA. Plg-deficient mice accumulate abundant fibrin, which disturbs the re-epithelialization process in particular (Rømer et al, 1996; Lund et al, 1999) and skin wound healing is rescued in double-deficient mice (Bugge et al, 1996b). This implies that the impaired healing in Plg-deficient mice is caused primarily by impaired fibrinolysis as a consequence of insufficient plasmin generation. Collectively, these data reveal that sufficient levels of plasmin must be generated despite the uPA;tPA deficiency in these mice, thus implying the presence of an additional Plg activator in these wounds. Accordingly, we have indeed demonstrated the presence of plasmin in wounds from uPA;tPA double-deficient mice thus providing the first biochemical evidence for an additional plasminogen activator in vivo.
A functional overlap exists between Plg and MMPs during skin wound healing (Lund et al, 1999) and trophoblast implantation (Solberg et al, 2003). We therefore compared the effect of MMP inhibitor treatment on the healing of skin wounds in Plg-deficient mice and uPA;tPA double-deficient mice. In agreement with our previous study we find that treatment with the MMP inhibitor galardin arrests healing in Plg-deficient mice. In uPA;tPA double-deficient mice, galardin treatment delays the healing process, whereas all the wounds do eventually heal with a mean healing time of 46 days. Two separate conclusions can be drawn from these findings.
Firstly, the further delay of healing in the uPA;tPA double-deficient mice by blocking MMP activity shows that MMPs contribute to fibrinolysis either directly or indirectly via Plg activation. In further consolidation of this interpretation, we found that galardin treatment of Plg;fibrinogen double-deficient wounds cause only a moderate delay in healing time (unpublished results) as opposed to the complete block of healing observed in the galardin-treated Plg-deficient mice (Lund et al, 1999). It remains however to be elucidated which MMP is responsible for fibrin degradation in vivo. MMP3, MMP8, MMP9, MMP12, MMP13 and MT1-MMP all have fibrinolytic activities in vitro (Bini et al, 1996; Hiller et al, 2000; Lelongt et al, 2001; Hotary et al, 2002), but no severe phenotypes after incisional wound healing are reported in the single-deficient mice tested so far; MMP3, MMP9 or MMP13 (Bullard et al, 1999; Mohan et al, 2002; Hartenstein et al, 2006). This suggests that the fibrinolytic potential of these MMPs should be tested in vivo in mice double-deficient in Plg and each of these MMPs. Secondly, the pharmacological blocking of MMP-mediated fibrinolysis in uPA;tPA double-deficient mice is not sufficient to arrest the healing process, demonstrating that these mice, in contrast to the galardin-treated Plg-deficient mice, still possess some residual fibrinolytic potential. This provides evidence of the existence of at least one Plg activator in addition to uPA and tPA, which is not galardin-sensitive.
A previous report suggested that pKal is a Plg activator during adipocyte differentiation (Selvarajan et al, 2001). We have now confirmed in vitro that purified human pKal has Plg-activating activities and, in addition, has some Plg-independent fibrinolytic activity. We observed an activity corresponding to pKal in murine wound extracts, and collectively these data suggest that pKal may be involved in fibrin degradation during skin wound healing both directly and through activation of Plg. To test this hypothesis, we treated uPA;tPA double-deficient mice with a mutated pKal-selective form of the serine protease inhibitor ecotin (Stoop and Craik, 2003), which decreased the healing rate, as measured by the wound length, to a level that was indistinguishable from vehicle-treated Plg-deficient wounds. In contrast, treatment of Plg-deficient mice with ecotin7–29 did not lower their healing capacity further. This result suggests that ecotin7–29 does not inhibit any rate-limiting proteases unrelated to plasminogen activators and the data do not support a biological role of plasminogen independent of its conversion to plasmin in wound healing. Ecotin7–29 completely blocks the activity of pKal, but not the activity of uPA or tPA in wound extracts as determined by fibrin/Plg zymography (Figure 6A and unpublished results). Thus, pKal most likely contributes to skin wound healing through Plg-dependent fibrinolysis.
Prekallikrein, the precursor of pKal is found at high concentrations (40–50 μg/ml) compared to 2–5 ng/ml of pro-uPA in plasma, which may enable its role as a Plg activator in vivo despite its relatively poor catalytic efficiency as Plg activator in vitro (Colman, 1969). The finding that pKal may play a significant role in Plg activation during skin wound healing suggests that pKal also has important and as yet unidentified physiological functions as a Plg activator in other tissue remodeling processes. Although our results obtained by ecotin7–29 treatment of uPA;tPA double-deficient mice strongly suggest that pKal is an important Plg activator during skin wound healing, we cannot rule out a role for additional Plg activators. Ecotin7–29 may inhibit other unidentified serine proteases with Plg activator activity, although our overlay zymography strongly support the conclusion that pKal is the additional Plg activator in wound healing. A definitive test of the role of pKal in skin wound healing and other tissue remodelling processes will be possible when pKal-deficient mice are available for generation of mice with triple-deficiency for uPA, tPA and pKal. Skin repair in these mice and Plg-deficient mice can then be compared directly.
We find it unlikely that the differences between the healing observed in uPA;tPA double-deficient and Plg-deficient mice can be explained by bacterial infection of the wounds and thereby contamination with prokaryotic Plg activators such as streptokinase or staphylokinase (Sun et al, 2004), as no signs of infection have been observed either macroscopically or microscopically in any mice in the experiments. Furthermore, we have observed a similar difference between the phenotypes of uPA;tPA double-deficient and Plg-deficient mice in a nonlesional tissue remodelling process, that is, post-lactational mammary gland involution, which is less impaired in uPA;tPA double-deficient than in Plg-deficient mice (unpublished results).
The demonstration of a third Plg activator, which at least partially accounts for the difference in wound healing time between uPA;tPA-deficient and Plg-deficient mice, has important implications for the understanding of Plg activation during normal and pathological tissue remodelling. It is well established that Plg deficiency results in serious physiological consequences in both humans and mice although with different degrees of penetrance (Bugge et al, 1995; Schuster et al, 1997; Drew et al, 1998). In several fundamental physiological processes involving normal as well as pathological tissue remodelling, Plg activation has been shown to play a pivotal role, and it will be interesting to define the individual and combined significance of uPA, tPA and pKal.
Numerous studies have revealed that uPA and uPAR are expressed either by invading cancer cells or by stromal cells in their vicinity (Johnsen et al, 1998). The ability of skin squamous carcinoma cells to mimic the ‘invasive' phenotype of re-epithelializing keratinocytes with regard to their expression of components of the Plg activation system and MMPs is likely to reflect a general mechanism employed by invading cancer cells. Our demonstration of the involvement of at least three Plg activators and one or more MMPs in wound healing may thus have important implications for a therapeutic approach aiming at blocking invasion and metastasis in cancer. In order to obtain a complete inhibition of cancer invasion, it will thus be necessary to use combinations of protease inhibitors.
Materials and methods
Animals and animal treatment
uPA and tPA gene-targeted mice of a mixed 129/Black Swiss background (Carmeliet et al, 1994) backcrossed to C57Bl/6J (Panum Institute, Copenhagen) mice for 16 generations were crossed, followed by interbreeding of the double heterozygous offspring. Plg gene-targeted 129/Black Swiss mice (Bugge et al, 1995) were backcrossed into C57BL/6J (Panum Institute, Copenhagen) for 21 generations. The plasminogen genotype was determined as described (Lund et al, 1999). The uPA genotype was determined by multiplex PCR using the following three primers. muPA-7p (CTC CCG TGG CTG GGT AGT GG) hybridizing to position 7719–7738 in the mouse uPA gene (GenBank: M17922), and muPA-4m (AGA GGA CGG TCA GCA TGG GAA C) hybridizing to position 8029–8008 generate a 311 bp product specific for the endogenous allele. muPA-4m and mPGK-2m (GCC TTG GGA AAA GCG CCT C) hybridizing to position 1092–1074 in the mouse phosphoglycerate kinase-1 promoter (GenBank: X15339) generate a ∼186 bp product specific for the targeted allele. The tPA genotype was determined by multiplex PCR using the following three primers; mtPA-3m (GTC TGT TCT TCC TCT CCG GGG AC) hybridizing to position 1673–1695 in the mouse tPA gene (GenBank: AC121835), and mtPA-4p (CTC ACA CCC TTG GCA GGC TG) hybridizing to position 1984–1965 generate a 312 bp product specific for the endogenous allele. mtPA-3m and mPGK-2m generate a ∼243 bp product specific for the targeted allele.
All mice used for experiments were males between 6 and 8 weeks old at the start of the experiment. Wound healing experiments of uPA;tPA-deficient mice and Plg-deficient mice was carried out at the same time and in the same room. Investigators unaware of the mouse genotype performed all experimental evaluations, tissue isolations and microscopic analyses. The MMP inhibitor galardin was synthesized as described (Grobelny et al, 1992). Galardin inhibits the enzymatic activity of a number of MMPs, including MMP2, -3, -9 and -14 (as described in Lund et al, 1999). Galardin was formulated as a 20 mg/ml slurry in 4% carboxymethylcellulose (CMC) in PBS and was administered daily i.p. at 100 mg/kg body weight. Mock treatment included 4% CMC in PBS. The same batch of galardin was used for the treatment of uPA-, tPA- and Plg-deficient mice. A mutated form of ecotin rendered highly specific for plasma kallikrein (ecotin7–29) was isolated from a phage display library as described (Stoop and Craik, 2003). Samples used for animal injections were diluted in PBS, and administered i.p. at a dose of 10 mg/kg/day (two daily injections for 30 days).
Tissue preparation
Incisional skin wounds were generated in 6- to 8-week-old mice and tissues were removed for histological analysis as described previously (Lund et al, 1999). A total of 133 mice were wounded and tissue isolated at day 10 post-wounding from 18 wild-type mice, 10 tPA-deficient, 17 uPA-deficient, 13 uPA;tPA double-deficient mice and nine Plg-deficient mice. At the time of healing, we isolated tissue from 10 wild-type, eight tPA-deficient, 11 uPA-deficient, six uPA;tPA double-deficient mice and five Plg-deficient mice. At 1 month post-healing, we isolated tissue from seven wild-type, five tPA-deficient, four uPA-deficient, five uPA;tPA double-deficient mice and five Plg-deficient mice. Animal care at The Department of Experimental Medicine, University of Copenhagen and Rigshospitalet, Copenhagen, Denmark was in accordance with the institutional and national guidelines.
Computer-assisted morphometry
Keratinocyte migration was measured microscopically on tissue sections stained immunohistochemically with anti-keratin IgG (Lund et al, 1999). The length of the epidermal tongue was measured as the distance between the tip of the leading-edge keratinocytes and the edge of the wound, defined as the point in the zone of proliferation where a shift from two to three layers of keratinocytes could be identified. The relative migration was determined as the sum of the lengths of both epidermal tongues divided by the width of the wound, defined as the distance between the wound edges. Indication of this point in the proliferation zone and the tip of the epidermal wedge by image analysis (Olympus AX70 system) were performed by an observer unaware of the genotype of the mice. The mean fraction of migration, the standard error (s.e.) was determined for each group of animals.
Statistical analysis
The SAS® software package (version 8.2; SAS Institute, Cary, NC) was used to manage data and for statistical analysis. The distribution of time to wound closure was found to be normal. Prespecified tests of hypothesis comparing experimental groups to the control group of mice were carried out using two sample t-tests. The assumption of variance homogeneity was tested by the folded F-test and the two sample t-test assuming unequal variances was used if the hypothesis of variance homogeneity was rejected. Incomplete data were discarded. Power calculations demonstrated that at least eight mice should be included in each group in order to detect a difference in time to wound closure of 5 days with 80% power. Plots of time to complete re-epithelialization were carried out using Kaplan–Meier estimates. The level of significance was set at 5%.
Zymography
Wound tissue containing both the wound rim and granulation tissue was lysed in 5 μl of 0.1 M Tris/HCl, 1% Triton X-100, pH 7.4 per mg wet weight of tissue. The extracts were used for fibrin/Plg overlay zymograms with and without plasminogen as described (Lund et al, 1996) and with and without ecotin7–29 (10 μg/ml) followed by incubation at 37°C for 16–28 h or in prolonged development for 40 h. Recombinant mouse pro-uPA (produced by Schneider cells, as described previously (Ploug et al, 2001)) and human plasma kallikrein (Kordia Life Sciences, The Netherlands) were used as controls.
Western blot analysis
Frozen tissue powder containing both the wound rim and granulation tissue (pool of three mice per genotype) was resuspended in 5 μl of 0.1 M Tris/HCl, pH 7.4, 10 μg/ml of aprotinin per mg wet weight of tissue, treated for 2 × 8 min on an Ultrasound bath and the resulting supernatants were collected after centrifugation at 12 000 g for 30 min at 4°C. Samples were reduced and alkylated before SDS–PAGE and transferred to PVDF membranes by electroblotting. Additional binding was blocked by incubation for 1 h at room temperature with 5% nonfat-dried milk in phosphate-buffered saline containing 0.05% Tween-20 (PBS-T). Incubation with the primary antibody was overnight at 4°C with 0.5 μg/ml polyclonal rabbit anti-human plasminogen antibody (DAKO, A0081). After washing in PBS-T (5 × 5 min), membranes were incubated for 1½ h at room temperature with HRP-linked secondary antibody (DAKO, P0217) diluted 1:5000 in blocking buffer. After washing in PBS-T and PBS, HRP activity was detected using enhanced chemiluminescence's reagents (Amersham Biosciences, Hillerød, Denmark). Purified mouse Plg and plasmin (Innovative Research, Inc., Hilltop, MI) were used as controls. A negative control without primary antibody included was carried out in parallel.
One-step affinity purification of Plg/plasmin was accomplished by application of 1500 μl wound extract onto 100 μl lysine Sepharose 4B gel (Amersham Bioscience) settled in a disposable column. After sample application, the columns were washed extensively with more than 10 column volumes of 0.1 M phosphate buffer, pH 7.4. The bound Plg/plasmin were eluted by 250 μl 0.1 M 6-aminohexanoic acid in 0.1 M phosphate buffer, pH 7.4 and fractions of approximately 25 μl were collected. All manipulations were performed at 4°C with buffers containing 10 μg/ml aprotinin.
Double immunofluorescence
Tissue sections were deparaffinized in xylene and hydrated through graded ethanol/water dilutions. Antigen retrieval was carried out by incubation with proteinase K for 15 min at 37°C. Sections were washed in running tap water for 5 min and Tris-buffered saline (TBS: 50 mM Tris, 150 mM NaCl, pH 7.6) for 5 min. Sections were incubated overnight simultaneously with both rabbit anti-mouse fibrin(ogen) antibody (1:2000; Bugge et al, 1995) and rat anti-mouse Troma1 antibody (detects cytokeratin 8; 1:100 Kemler et al, 1981), in TBS containing 0.25% BSA at 4°C. Rabbit anti-fibrin(ogen) antibody was detected with Alexa Fluor 488-linked donkey anti-rabbit antibody (1:200, Molecular Probes, CA, A21206) and rat anti-Troma-1 antibody was detected with Alexa Fluor 594-linked goat anti-rat antibody (1:200, Molecular Probes, A11007) diluted together in TBS containing 0.25% BSA and incubated for 45 min at room temperature. Antibody incubations were followed by washes in TBS. Controls without anti-fibrin(ogen) primary antibody were negative for unspecific staining.
Acknowledgments
We thank Dr Thomas Bugge for the Plg-deficient mice and Dr Peter Carmeliet for the uPA- and tPA-deficient mice. We are grateful to Mette M Andersen, Lotte Frederiksen, Kirsten L Jakobsen, Charlotte Lønborg and John Post for excellent technical assistance. This work was supported financially by the European Commission: Grant number QLG1-CT-2000-01131 and LSHC-CT-2003-503297, European Union research training grant for mammary gland biology RTN-2002-00246, Aage Bangs Foundation, the Danish Biotechnology Program, Centre for Medical Biotechnology, the Danish Cancer Society, the Danish Medical Research Council, the Danish Cancer Research Foundation, Agnes and Poul Friis Foundation and a grant from the National Cancer Institute (USA) P01 CA072006.
References
- Beare AHM, O'Kane S, Krane SM, Ferguson MWJ (2003) Severely impaired wound healing in the collagenase-resistant mouse. J Invest Dermatol 120: 153–163 [DOI] [PubMed] [Google Scholar]
- Bini A, Itoh Y, Kudryk BJ, Nagase H (1996) Degradation of cross-linked fibrin by matrix metalloproteinase 3 (stromelysin 1): hydrolysis of the gamma Gly 404-Ala 405 peptide bond. Biochemistry 35: 13056–13063 [DOI] [PubMed] [Google Scholar]
- Bugge TH, Flick MJ, Danton MJ, Daugherty CC, Rømer J, Danø K, Carmeliet P, Collen D, Degen JL (1996a) Urokinase-type plasminogen activator is effective in fibrin clearance in the absence of its receptor or tissue-type plasminogen activator. Proc Natl Acad Sci USA 93: 5899–5904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bugge TH, Flick MJ, Daugherty CC, Degen JL (1995) Plasminogen deficiency causes severe thrombosis but is compatible with development and reproduction. Genes Dev 9: 794–807 [DOI] [PubMed] [Google Scholar]
- Bugge TH, Kombrinck KW, Flick MJ, Daugherty CC, Danton MJ, Degen JL (1996b) Loss of fibrinogen rescues mice from the pleiotropic effects of plasminogen deficiency. Cell 87: 709–719 [DOI] [PubMed] [Google Scholar]
- Bugge TH, Lund LR, Kombrinck KK, Nielsen BS, Holmback K, Drew AF, Flick MJ, Witte DP, Danø K, Degen JL (1998) Reduced metastasis of Polyoma virus middle T antigen-induced mammary cancer in plasminogen-deficient mice. Oncogene 16: 3097–3104 [DOI] [PubMed] [Google Scholar]
- Bullard KM, Lund L, Mudgett JS, Mellin TN, Murphy B, Ronan J, Banda MJ (1999) Impaired wound contraction in stromelysin-1 deficient mice. Ann Surg 230: 260–265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmeliet P, Schoonjans L, Kieckens L, Ream B, Degen J, Bronson R, De Vos R, van den Oord JJ, Collen D, Mulligan RC (1994) Physiological consequences of loss of plasminogen activator gene function in mice. Nature 368: 419–424 [DOI] [PubMed] [Google Scholar]
- Collen D, Lijnen HR (2005) Thrombolytic agents. Thromb Haemost 93: 627–630 [DOI] [PubMed] [Google Scholar]
- Colman RW (1969) Activation of plasminogen by human plasma kallikrein. Biochem Biophys Res Commun 35: 273–279 [DOI] [PubMed] [Google Scholar]
- Danø K, Andreasen PA, Grondahl-Hansen J, Kristensen P, Nielsen LS, Skriver L (1985) Plasminogen activators, tissue degradation, and cancer. Adv Cancer Res 44: 139–266 [DOI] [PubMed] [Google Scholar]
- Danø K, Behrendt N, Hoyer-Hansen G, Johnsen M, Lund LR, Ploug M, Romer J (2005) Plasminogen activation and cancer. Thromb Haemost 93: 676–681 [DOI] [PubMed] [Google Scholar]
- Danø K, Romer J, Nielsen BS, Bjorn S, Pyke C, Rygaard J, Lund LR (1999) Cancer invasion and tissue remodeling—cooperation of protease systems and cell types. Apmis 107: 120–127 [DOI] [PubMed] [Google Scholar]
- Drew AF, Kaufman AH, Kombrinck KW, Danton MJ, Daugherty CC, Degen JL, Bugge TH (1998) Ligneous conjunctivitis in plasminogen-deficient mice. Blood 91: 1616–1624 [PubMed] [Google Scholar]
- Egeblad M, Werb Z (2002) New functions for the matrix metalloproteinases in cancer progression. Nat Rev Cancer 2: 161–174 [DOI] [PubMed] [Google Scholar]
- Ellis V, Behrendt N, Dano K (1991) Plasminogen activation by receptor-bound urokinase. A kinetic study with both cell-associated and isolated receptor. J Biol Chem 266: 12752–12758 [PubMed] [Google Scholar]
- Grobelny D, Ponez L, Galardy RE (1992) Inhibition of human skin fibroblast collagenase, thermolysin and Pseudomonas aeruginosa elastase by peptide hydroxamic acids. Biochemistry 31: 7152–7154 [DOI] [PubMed] [Google Scholar]
- Hartenstein B, Dittrich BT, Stickens D, Heyer B, Vu TH, Teurich S, Schorpp-Kistner M, Werb Z, Angel P (2006) Epidermal development and wound healing in matrix metalloproteinase 13-deficient mice. J Invest Dermatol 126: 486–496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiller O, Lichte A, Oberpichler A, Kocourek A, Tschesche H (2000) Matrix metalloproteinases collagenase-2, macrophage elastase, collagenase-3, and membrane type 1-matrix metalloproteinase impair clotting by degradation of fibrinogen and factor XII. J Biol Chem 275: 33008–33013 [DOI] [PubMed] [Google Scholar]
- Hotary KB, Yana I, Sabeh F, Li XY, Holmbeck K, Birkedal-Hansen H, Allen ED, Hiraoka N, Weiss SJ (2002) Matrix metalloproteinases (MMPs) regulate fibrin-invasive activity via MT1–MMP-dependent and -independent processes. J Exp Med 195: 295–308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnsen M, Lund LR, Rømer J, Almholt K, Danø K (1998) Cancer invasion and tissue remodeling: common themes in proteolytic matrix degradation. Curr Opin Cell Biol 10: 667–671 [DOI] [PubMed] [Google Scholar]
- Kemler R, Brulet P, Schnebelen M T, Gaillard J, Jacob F (1981) Reactivity of monoclonal antibodies against intermediate filament proteins during embryonic development. J Embryol Exp Morphol 64: 45–60 [PubMed] [Google Scholar]
- Lelongt B, Bengatta S, Delauche M, Lund LR, Werb Z, Ronco PM (2001) Matrix metalloproteinase 9 protects mice from anti-glomerular basement membrane nephritis through its fibrinolytic activity. J Exp Med 193: 793–802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llinas P, Le Du MH, Gardsvoll H, Dano K, Ploug M, Gilquin B, Stura EA, Menez A (2005) Crystal structure of the human urokinase plasminogen activator receptor bound to an antagonist peptide. EMBO J 24: 1655–1663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lund LR, Bjørn SF, Sternlicht MD, Nielsen BS, Solberg H, Usher PA, Osterby R, Christensen IJ, Stephens RW, Bugge TH, Dano K, Werb Z (2000) Lactational competence and involution of the mouse mammary gland require plasminogen. Development 127: 4481–4492 [DOI] [PubMed] [Google Scholar]
- Lund LR, Rømer J, Bugge TH, Nielsen BS, Frandsen TL, Degen JL, Stephens RW, Danø K (1999) Functional overlap between two classes of matrix-degrading proteases in wound healing. EMBO J 18: 4645–4656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lund LR, Rømer J, Thomasset N, Solberg H, Pyke C, Bissell MJ, Danø K, Werb Z (1996) Two distinct phases of apoptosis in mammary gland involution: proteinase-independent and -dependent pathways. Development 122: 181–193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madlener M, Parks WC, Werner S (1998) Matrix metalloproteinases (MMPs) and their physiological inhibitors (TIMPs) are differentially expressed during excisional skin wound repair. Exp Cell Res 242: 201–210 [DOI] [PubMed] [Google Scholar]
- Mohan R, Chintala SK, Jung JC, Villar WV, McCAbe F, Russo LA, Lee Y, McCarthy BE, Wollenberg KR, Jester JV, Wang M, Welgus HG, Shipley JM, Senior RM, Fini ME (2002) Matrix metalloproteinase gelatinase B (MMP-9) coordinates and effects epithelial regeneration. J Biol Chem 277: 2065–2072 [DOI] [PubMed] [Google Scholar]
- Ploug M (2003) Structure–function relationships in the interaction between the urokinase-type plasminogen activator and its receptor. Curr Pharm Des 9: 1499–1528 [DOI] [PubMed] [Google Scholar]
- Ploug M, Ostergaard S, Gardsvoll H, Kovalski K, Holst-Hansen C, Holm A, Ossowski L, Danø K (2001) Peptide-derived antagonists of the urokinase receptor. Affinity maturation by combinatorial chemistry, identification of functional epitopes, and inhibitory effect on cancer cell intravasation. Biochemistry 40: 12157–12168 [DOI] [PubMed] [Google Scholar]
- Rømer J (2003) Skin cancer and wound healing: tissue-specific similarities in extracellular proteolysis. APMIS 111 (Suppl 107): 1–36 [Google Scholar]
- Rømer J, Bugge TH, Pyke C, Lund LR, Flick MJ, Degen JL, Dano K (1996) Impaired wound healing in mice with a disrupted plasminogen gene. Nat Med 2: 287–292 [DOI] [PubMed] [Google Scholar]
- Schuster V, Mingers AM, Seidenspinner S, Nussgens Z, Pukrop T, Kreth HW (1997) Homozygous mutations in the plasminogen gene of two unrelated girls with ligneous conjunctivitis. Blood 90: 958–966 [PubMed] [Google Scholar]
- Selvarajan S, Lund LR, Takeuchi T, Craik CS, Werb Z (2001) A plasma kallikrein-dependent plasminogen cascade required for adipocyte differentiation. Nat Cell Biol 3: 267–275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solberg H, Rinkenberger J, Danø K, Werb Z, Lund LR (2003) A functional overlap of plasminogen and MMPs regulates vascularization during placental development. Development 130: 4439–4450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoop AA, Craik CS (2003) Engineering of a macromolecular scaffold to develop specific protease inhibitors. Nat Biotechnol 21: 1063–1068 [DOI] [PubMed] [Google Scholar]
- Sun H, Ringdahl U, Homeister JW, Fay WP, Engleberg NC, Yang AY, Rozek LS, Wang X, Sjobring U, Ginsburg D (2004) Plasminogen is a critical host pathogenicity factor for group A streptococcal infection. Science 305: 1283–1286 [DOI] [PubMed] [Google Scholar]