


Abstract
Bulky carcinogen-DNA adducts commonly cause replicative polymerases to stall, leading to a switch to bypass polymerases. We have investigated nucleotide incorporation opposite the major adduct of 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine (PhIP) in the DinB family polymerase, Dpo4, using molecular modeling and molecular dynamics (MD) simulations. PhIP, the most prevalent heterocyclic aromatic amine formed by cooking of proteinaceous food, is mutagenic in mammalian cells and is implicated in mammary and colon tumors. Our results show that the dG-C8-PhIP adduct can be accommodated in the spacious major groove Dpo4 open pocket, with Dpo4 capable of incorporating dCTP, dTTP or dATP opposite the adduct reasonably well. However, the PhIP ring system on the minor groove side would seriously disturb the active site, regardless of the presence and identity of dNTP. Furthermore, the simulations indicate that dATP and dTTP are better incorporated in the damaged system than in their respective mismatched but unmodified controls, suggesting that the PhIP adduct enhances incorporation of these mismatches. Finally, bulky C8-dG adducts, situated in the major groove, are likely to impede translocation in this polymerase (Rechkoblit et al. (2006), PLoS Biol., 4, e11). However, N2-dG adducts, which can reside on the minor groove side, appear to cause less hindrance when in this position.
INTRODUCTION
DNA polymerases of the Y-family have in recent years been shown to play a predominant role in synthesis past DNA bulky lesions, such as those derived from polycyclic aromatic chemicals present in tobacco smoke, automobile exhaust, and broiled meat and fish (1–5). High fidelity replicative DNA polymerases are usually impeded by such damage, leading to a switch to one or more low fidelity bypass polymerases for translesion synthesis (6–11).
The model Y-family polymerase, DNA polymerase IV (Dpo4), is from the archaeon bacterium Sulfolobus solfataricus P2. It is a member of the DinB family (12), of which human pol κ is also a member (13). It has been extensively investigated by X-ray crystallography in binary complexes with primer/template DNA and in ternary complexes in the presence of dNTP, both with and without DNA damage (14–23). These structures have revealed a spacious, water accessible active site that is capable of accommodating two templating bases, in contrast to high fidelity replicative polymerases whose ternary complexes show tight fit of the nascent base pair with exclusion of solvent (24). Dpo4 has an open pocket on the major groove side of the template, as well as a smaller open space on the minor groove side; this is in contrast to replicative polymerases, which only have an open pocket on the major groove side, while the minor groove side is packed with protein–DNA interactions critical for polymerase fidelity (25–29). In addition like other Y-family polymerases, Dpo4 has a unique little finger domain, also called wrist or polymerase associated domain (PAD) at the C-terminus (15,30,31). The flexibility of this little finger domain is believed to play an important role in accommodating specific types of DNA lesions (14,15,21). Crystal structures of Dpo4 binary and ternary complexes also reveal that the little finger domain plays a key role in translocation (23).
Recently, it has been suggested that the DinB family polymerases may be specifically suited for bypass of N2-dG adducts (32). The presence of a conserved ‘steric gate’, usually phenylalanine or tyrosine, is shown to be crucial in bypass of N2-dG minor groove adducts (32). However, the DinB polymerases may be less well suited for bypassing C8-dG bulky adducts. Such adducts would normally be expected, from their position of substitution on guanine, to reside in the major groove of double-stranded DNA. However, rotation of the modified guanine from anti to syn would place the adduct in the minor groove area, in a position roughly similar to an N2-substituted guanine. Despite this formal possibility, experimental primer extension data for two C8-dG adducts derived from 2-acetylaminofluorene (AAF), namely N-(deoxyguanosin-8-yl)-2-acetylaminofluorene (dG-C8-AAF) with Dpo4 (12) and N-(deoxyguanosin-8-yl)-2-aminofluorene (dG-C8-AF) with Escherichia coli DinB and human pol κΔc (33), indicates that these adducts cause polymerase stalling or blockage, with only small amounts of primer extension beyond the lesion site. A recent molecular dynamics (MD) study from our group has provided structural rationale for the case of dG-C8-AAF in Dpo4 (34). However, this adduct contains an acetyl group, which adds to the steric hindrance in C8 adducts.
Here, we investigate a C8-dG adduct (Figure 1a) derived from the most prevalent heterocyclic aromatic amine formed by cooking proteinaceous food, 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine (PhIP) (35–43). This substance causes predominantly G to T transversions in mammalian cells, with some G to A transitions, and a few deletions (44–52). In a previous molecular modeling and MD study of this adduct in the Pol α family replicative polymerase RB69, we found considerable active site distortion caused by this lesion at the templating base or in the first double-stranded extension position, when partnered by C or A (53). These results suggested that the replicative polymerase would be stalled by the lesion, affording opportunity for switch to one or more bypass polymerases. We now use this dG-C8-PhIP adduct, N2-(2′-deoxyguanosin-8-yl)-PhIP, as a model to study the structural feasibility of accommodating a C8-dG lesion lacking the acetyl group in Dpo4, employing Dpo4 type I (16) and type II (15) crystal structures as initial models (Figure 1).
Figure 1.

(a) Structure and numbering scheme of the dG-C8-PhIP adduct. Torsion angle definitions are as follows: χ, O4′-C1′-N9-C4; α′, N9-C8-[PhIP]N2-[PhIP]C2; β′, C8-[PhIP]N2-[PhIP]C2-[PhIP]N1; γ′, [PhIP]C5-[PhIP]C6-[PhIP]C1′-[PhIP]C2′. (b) dG-C8-PhIP-modified DNA sequence used in G·dNTP. Models are derived from a Type I crystal structure of Dpo4 (PDBID: 1S0M) (16). (c) dG-C8-PhIP-modified DNA sequence used in −1 deletion studies. Models are derived from a Type II crystal structure of Dpo4 (PDBID: 1JXL) (15).
Our results show that this dG-C8 adduct, like dG-C8-AAF, if accommodated on the Dpo4 minor groove side pocket, would cause serious distortions to the active site region. However, the PhIP ring system can be accommodated in the spacious major groove pocket of this polymerase, and Dpo4 appears capable of incorporating dCTP, dTTP or dATP reasonably well. In addition, our results show that the PhIP-modified lesion produces less distorted structures for anti-G*·anti-dTTP and anti-G*·syn-dATP than for their respective unmodified controls (anti-G·anti-dTTP and anti-G·syn-dATP). These findings may be relevant to observed mutagenic behavior of PhIP in inducing G to T transversions and G to A transitions in mammalian systems (44–52), if the human DinB polymerase pol κ is involved in the error-prone incorporation opposite the lesion, and its structural properties prove to be similar to those of its prokaryotic homolog Dpo4. Finally, based on Dpo4 binary and ternary complex structures (23), we suggest that translocation may be seriously inhibited by bulky dG-C8 adducts positioned on the major groove side of the DNA duplex region, while N2-dG adducts residing on the minor groove side (54) would be less hindering.
COMPUTATIONAL METHODS
Molecular modeling
G·dNTP initial models. The crystal structure of Dpo4 polymerase with 10R(+)-cis-anti-benzo[a]pyrene(BP)-N6-dA located on the major groove side of the modified DNA (BP-2 complex) (16) was used to obtain initial structures for the G·dNTP models (PDB ID: 1S0M) (55). Of the many type I structures of Dpo4, the BP-2 complex has the most reaction-ready active site, since it contains two metal ions in the active site and the 3′ hydroxyl group at the primer end. This structure was further remodeled, as described in previous work from our group (34), to achieve ideal Mg2+ coordination (Supplementary Table S1) and an O3′-Pα distance of 3.1 Å, in the reaction-ready range.
The DNA sequence was then adjusted to match the sequence in the adenomatous polyposis coli (Apc) gene mutational hotspot for PhIP, codon 635 (Figure 1b), with the guanine selected for PhIP modification situated at the templating position opposite the incoming dNTP. The initial models for dynamics were then constructed by locating structures with minimal crowding between the PhIP moiety and the polymerase, by rotating α′ and β′ at 10° intervals, in combination (Figure 1a). γ′ was initiated at 26° as in the NMR solution structure (56). Both anti and syn conformations of the glycosidic torsion χ were investigated, with χ adjusted to achieve optimal hydrogen bonding and stacking in the nascent base pair for each χ domain. The torsion angles in the initial models are summarized in Supplementary Table S2.
For the G*·dATP mismatch, three starting models were obtained. Two of them featured the PhIP-modified guanine (G*) anti and the PhIP rings on the major groove side of the template, with the incoming nucleotide dATP either anti or syn. A syn-dATP was investigated since it has been observed opposite a lesion in a Dpo4 crystal structure (17). A high resolution NMR solution structure containing a dG-C8-PhIP in an 11mer duplex shows a base-displaced intercalation model (56). In this conformation, the modified guanine is syn and displaced into the major groove, while the PhIP rings intercalate within the DNA duplex. However, the PhIP rings in such a base-displaced intercalation conformation would occupy the position of the incoming nucleotide in Dpo4. A PhIP-modified syn-guanine model could be built with the PhIP rings on the minor groove side of the dNTP, which does allow the accommodation of the dNTP at the active site. With a syn-G* as the template, only anti-dATP is favorable, since the bases in the syn-G*·syn-dATP pair are too far apart for hydrogen bonding. Hydrogen bonding in the nascent base pair is an important consideration in building the initial models, since Dpo4 as well as other DinB family polymerases, such as Dbh and pol κ rely more on hydrogen bonding for catalytic efficiency than the high fidelity polymerases (57,58).
In the G*·dCTP/dTTP/dGTP series, the G*s were all constructed in the same anti conformation as in the G*·dATP mismatch. A syn-G* was eliminated based on results for the syn-G*·anti-dATP simulation; these show that a syn-G* generated much more disturbance to the polymerase active site than an anti one, as described in Results, and these disturbances are essentially independent of the dNTP. In the G*·dCTP model, the incoming dCTP is anti, in order to form Watson–Crick hydrogen bonds with the anti-G*. In the G*·dTTP model, the dTTP is anti to achieve a wobble pair with the anti-G*. A wobble paired T·dGTP has been observed in a Dpo4 ternary complex (18). A syn-dTTP can form no hydrogen bonds with anti-G* and syn pyrimidines have scarcely been observed (59). In the G*·dGTP model, a syn-dGTP was employed, since an anti-dGTP could not form hydrogen bonds with anti-G* in the polymerase, and would collide with either the anti-G* or the minor groove side protein residues.
As an unmodified control for all the simulations, an unmodified anti-G opposite a Watson–Crick paired anti-dCTP was used. Additional controls containing an unmodified anti-G opposite anti-dATP for the G*·dATP mismatch, anti-dTTP for the G*·dTTP mismatch and syn-dGTP for the G*·dGTP mismatch were also obtained. Stereoviews of the active sites of the initial models are shown in Supplementary Figure S1.
−1 deletion initial models. The crystal structure of the Dpo4 type II ternary complex (PDB ID: 1JXL) (15) was used to obtain the starting models for the −1 deletion structures (Figure 1). In this crystal structure, the coordinates of the first base at the 5′ end of the template could not be resolved, probably due to its flexible conformation outside the polymerase. We therefore modeled it into the structure, using the conformation of an analogous terminal base in the Dpo4 type I structure (PDB ID: 1JX4). Hydrogen atoms absent in the crystal structures were added by the AMBER suite. The dideoxy group at the 3′ end of the primer was replaced by a hydroxyl group. The Ca2+ ion, residing in the position of the nucleotide binding metal ion, was replaced by a Mg2+ ion, and repositioned for proper octahedral coordination with water molecules and amino acid residues (Supplementary Table S1). The DNA sequence was also remodeled to match the sequence in the Apc gene mutational hotspot codon 635 (Figure 1c). The PhIP moiety was linked to the unpartnered guanine in the active site, while the incoming dCTP paired with the guanine on the 5′ side of the adduct. Starting models were obtained using the same approach as for G*·dNTP models, by rotating torsion angles α′ and β′ at 10° intervals, in combination. Structures were selected for anti and syn domains of χ based on minimal steric close contacts and optimal stacking. The torsion angles in these starting models are summarized in Supplementary Table S2 and stereoviews of the active sites of the initial models are shown in Supplementary Figure S1.
Force field parameterization
Parameters for the anti-dG-C8-PhIP adduct and the incoming anti-dCTP, anti-dATP, syn-dATP and anti-dTTP were the same as in earlier work (53,54). The parameters for the syn-dG-C8-PhIP adduct and the syn-dGTP were obtained using the same method described previously (53). Supplementary Tables S3 and S4 shows the AMBER atom type, connection type and partial charge assignment for these cases.
MD simulation and data analyses
Simulations were carried out using the SANDER module of the AMBER 6.0 MD software package (University of California, San Francisco), the Cornell et al. force field (60) and the parm99 parameter set (61). Electrostatic interactions were approximated by the particle mesh Ewald method (62), and a 10 Å cutoff was applied to Lennard–Jones interactions. All bonds involving hydrogen atoms were constrained by the SHAKE algorithm (63) with a tolerance of 0.0005 Å, and a 2 fs time-step was used in the dynamics simulation. Periodic boundary conditions were applied, and all MD simulations were carried out under constant temperature, 300 K, with a temperature coupling parameter of 4.0 ps and at constant atmospheric pressure with a 1.0 ps coupling parameter. The translational motion of the center of mass was removed every 1 ps. No obvious overall rotation of the system was observed during the simulation; thus, energy leakage from internal motion to global rotation through the ‘flying ice cube effect’ did not happen here (64).
Fifteen Na+ ions were added to each system for neutralization using the LEaP module of AMBER, followed by 600 steps of steepest descent (SD) and 400 steps of conjugate gradient (CG) to relax the added Na+ ions and crystal waters. Then ∼15 000 TIP3P water molecules were added to solvate each system creating a rectangular periodic box containing a total of ∼50 000 atoms. The systems were further minimized and heated up to 300 K. The equilibration protocol was conducted in the same manner as in earlier work (53) and the details are given in Supplementary Data. Production MD simulation was carried out for 3 ns.
The PTRAJ, ANAL and CARNAL modules of AMBER were employed to analyze the trajectories. The root-mean-square deviations (RMSDs) of each system were calculated relative to the starting structures as shown in Supplementary Figure S2. The overall structure and the active site region of the protein–DNA complexes became reasonably stable after ∼500 ps production MD simulation in each system. Therefore, the last 2.5 ns of MD simulation were used for structural analyses. Stereoviews of unmodified and modified systems after the total 3 ns of production MD simulations are shown in Supplementary Figure S3, and the active sites of these systems are shown in Figures 2 and 3, and Supplementary Figure S4.
Figure 2.

The active sites after 3 ns MD in the G·dNTP models. The Dpo4 DNA polymerase, shown in ribbon representation, is colored as follows: thumb is in dark orange, palm in lime, finger in yellow, and little finger in blue; the DNA ternary complex is shown in stick representation. The primer and template DNA strands are colored in gray, the dCTP is colored in cyan, dATP in magenta, dTTP in marine, dGTP in pink, the template guanine is colored in green and the PhIP moiety is in red. Mg2+ ions A and B, shown in spheres representation, are colored in orange.
Figure 3.
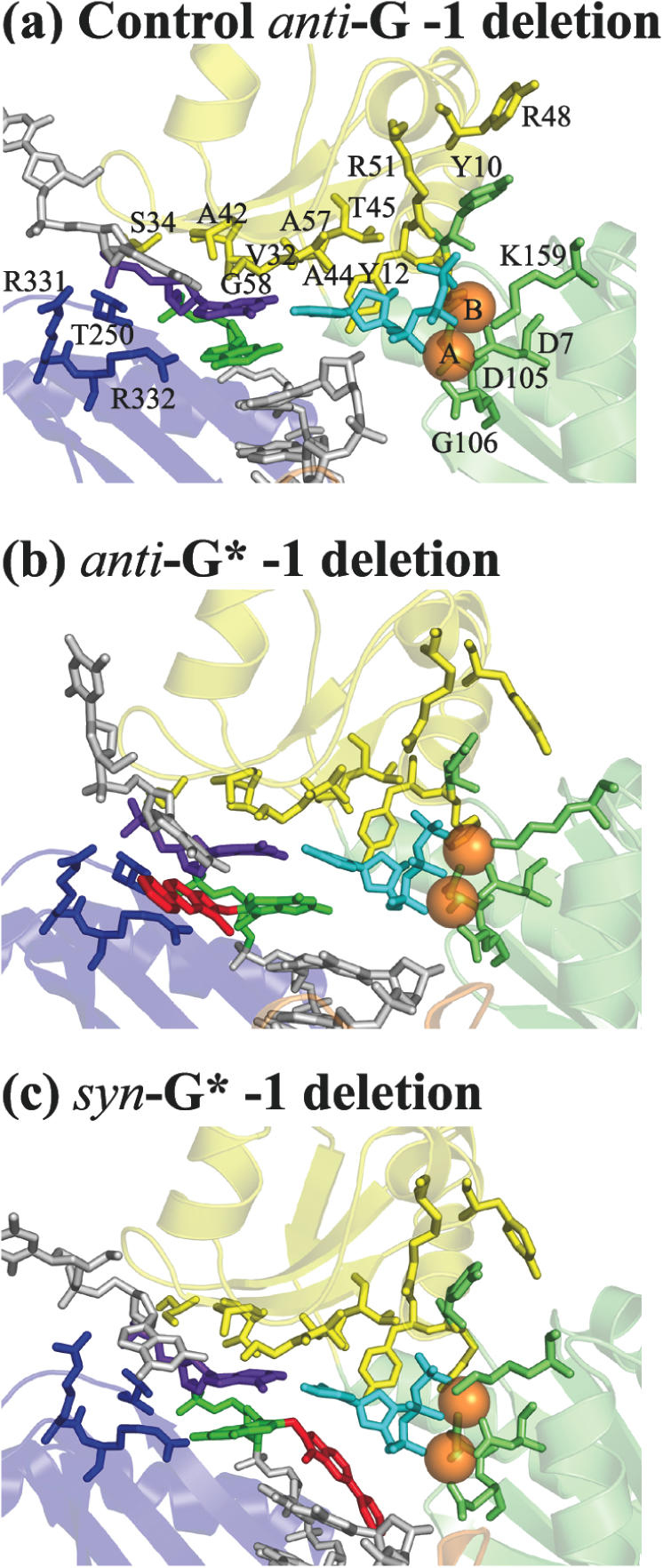
The active sites after 3 ns MD in −1 deletion models. The protein and two Mg2+ ions are colored the same as in Figure 2. The template guanine is colored in purple; the dCTP is in cyan; the template guanine without partner is in green, and the PhIP moiety is in red.
Molecular modeling was carried out with Insight II 97.0 (Accelrys, Inc., a subsidiary of Pharmacopeia, Inc.). Figures of structures were prepared with PyMOL (DeLano Scientific, LLC.).
RESULTS
Initial models
Our initial models (see Computational Methods) investigated both anti and syn conformations of the adduct in the active site of Dpo4, partnered with dATP or unpaired in the −1 deletion simulations. In addition, anti-G* opposite anti-dCTP, anti-dTTP or syn-dGTP were investigated using results from dATP to guide selection of the starting structures. Production MD simulations were carried out for 3 ns and the ensembles of structures derived from the last 2.5 ns trajectory were analyzed.
Structural analyses
A number of structural criteria were evaluated in each modified system and compared to the undamaged control simulation. These include: (i) the number and occupancy of the hydrogen bonds in the nascent base pair (Supplementary Figure S5 and Supplementary Tables S5 and S10); (ii) the distance between C1′ of the template and C1′ of the incoming nucleotide (dNTP), normally near 10.8 Å in a Watson–Crick pair (65) (Supplementary Table S7); (iii) stacking interactions between the nascent base pair and the primer-terminus base pair (Figures 2 and 3, and Supplementary Tables S8 and S9); (iv) the frequency of sampling a near reaction-ready distance (3.1 to 3.5 Å) between O3′ of the primer-terminus and Pα of the dNTP (Supplementary Table S7); (v) angle O3′ (primer 3′ end)-Pα(dNTP)-O3α(dNTP), ideally 180° for in-line attack of O3′ on Pα (66) (Supplementary Table S7); (vi) the chelation of Mg2+ ions (Figures 2 and 3); (vii) the distance between the two Mg2+ ions (Supplementary Table S7) and (viii) hydrogen bonds between the nascent base pair and the polymerase. These are summarized in Tables 1 and 2, using a distortion scoring function to evaluate the lesion impact on the polymerase active site (34). While we analyze the structural features individually, we realize there is interdependence among them; how they affect each other remains unclear but their composite quality sheds light on a given model's structural feasibility. While scoring has subjective criteria elements, it reflects our current stage for evaluating polymerase active site distortions. We hypothesize that the more distorted the active site is, the less efficient the nucleotidyl transfer reaction will be.
Table 1.
Composite evaluation of structural distortion in G·dNTP models
| Control | anti-G*·dCTP | anti-G·syn-dATP | anti-G*·anti-dATP | anti-G*·syn-dATP | syn-G*·anti-dATP | anti-G·anti-dTTP | anti-G*·anti-dTTP | anti-G·syn-dGTP | anti-G*·syn-dGTP | |
|---|---|---|---|---|---|---|---|---|---|---|
| Pα-O3′a | √ | √ | × | √ | √ | × | × | √ | √ | × |
| HB geometryb | √ | √ | ×× | ×× | × | ××× | ×× | × | ×× | ×× |
| C1′-C1′c | √ | √ | √ | × | √ | × | √ | √ | × | × |
| Key amino acids-DNA interactiond | √ | ×× | ××× | ××× | × | ××× | × | × | ×× | ×× |
| Base pair stackinge | √ | √ | √ | √ | √ | × | √ | √ | √ | √ |
| In-line attackf | √ | √ | √ | √ | √ | √ | √ | √ | √ | √ |
| Mg2+ ions distanceg | √ | √ | √ | √ | √ | √ | √ | √ | √ | √ |
| Mg2+ ions octahedral coordinationh | √ | √ | √ | √ | √ | √ | √ | √ | √ | √ |
| Composite scorei | 0 | −2 | −6 | −6 | −2 | −9 | −4 | −2 | −5 | −6 |
aOne cross denotes a decrease in occupancy compared to the control by more than 15%, two crosses denote a decrease by >30%, and three denote a decrease by >45%. See Supplementary Table S7 for details.
bEach cross denotes a decrease in occupancy compared to the control by >10%, or a loss of one hydrogen bond. See Supplementary Table S6 for details.
cOne cross denotes an increase in C1′–C1′ distance compared to the control (10.9 Å) by more than 1 Å. See Supplementary Table S7 for details.
dEach cross denotes a decrease in occupancy compared to the control by >10%, or a loss of one stacking interaction. See Supplementary Table S6 for details.
eOne cross denotes a disruption of stacking interaction between the nascent and the primer-terminus base pairs, indicated by diminished coplanarity of the base pairs determined visually, and a weakened van der Waals interaction energy between them compared to the control (−15.1 kcal/mol) by ≥5.0 kcal/mol. See Figure 2 and Supplementary Table S8 for details.
fOne cross denotes occupancy ≤50%. See Supplementary Table S7 for details.
gOne cross denotes occupancy ≤90%. See Supplementary Table S7 for details.
hNo disruptions of the chelation interactions involving the two Mg2 ions are observed (the distance between chelating atom and Mg2 ≥2.5 Å is considered to be a disruption). See Figure 2 for details.
iScore is based on the unmodified control, which was assigned a base score of 0. Each cross penalizes the base score by 1.
Table 2.
Composite evaluation of structural distortion in the −1 deletion models
| Control | anti-G* −1 deletion | syn-G* −1 deletion | |
|---|---|---|---|
| Pα-O3′a | √ | ×× | ××× |
| HB geometryb | √ | √ | √ |
| C1′-C1′c | √ | √ | √ |
| Key amino acids-DNA interactiond | √ | ××× | ×××× |
| Base stackinge | √ | √ | × |
| In-line attackf | √ | √ | × |
| Mg2 ions distanceg | √ | √ | √ |
| Mg2 ions octahedral coordinationh | √ | √ | √ |
| Composite scorei | 0 | −5 | −9 |
aOne cross denotes a decrease in occupancy compared to the control by >15%, two crosses denote a decrease by >30%, and three denote a decrease by >45%. See Supplementary Table S7 for details.
bEach cross denotes a decrease in occupancy compared to the control by >10%, or a loss of one hydrogen bond. See Supplementary Table S11 for details.
cOne cross denotes an increase in C1′–C1′ distance compared to the control (10.8 Å) by >1 Å. See Supplementary Table S7 for details.
dEach cross denotes a decrease in occupancy compared to the control by >10%, or a loss of one stacking interaction. See Supplementary Table S11 for details.
eOne cross denotes a disruption of the stacking interactions between G4 and A5, indicated by diminished coplanarity of the bases determined visually and a weakened van der Waals interaction energy between them compared to the control (−5.3 kcal/mol) by ≥2.5 kcal/mol. See Figure 3 and Supplementary Table S9 for details.
fOne cross denotes occupancy ≤50%. See Supplementary Table S7 for details.
gOne cross denotes occupancy ≤90%. See Supplementary Table S7 for details.
hNo disruptions of the chelation interactions involving the two Mg2 ions are observed (the distance between chelating atom and Mg2 ≥2.5 Å is considered to be a disruption). See Figure 3 for details.
iScore is based on the unmodified control, which was assigned a base score of 0. Each cross penalizes the base score by 1.
G·dNTP models
Unmodified control of anti-G·anti-dCTP. In the unmodified control simulation, anti-G Watson–Crick pairs with an incoming anti-dCTP. Views of this complex and its active site after 3 ns production MD simulation are shown in Supplementary Figures S3a and S4a, and Figure 2a. The active site remains essentially undisturbed throughout the simulation. The Watson–Crick hydrogen bonds in the nascent base pair have occupancies >90% (Supplementary Table S5); the C1′–C1′ distance in the nascent base pair remains normal (10.9 ± 0.1 Å); the chelation of the two Mg2+ ions is preserved (Figure 2a); the distance between the two Mg2+ has an average value of 3.7 ± 0.1 Å; the Pα–O3′ distance samples the near reaction-ready range frequently, during 78.7% of the time; and the in-line attack angle is 167.1 ± 5.1°, close to the ideal value of 180°. These active site features are similar in polymerase ternary complex crystal structures (67,68). In the active site of Dpo4 polymerase, the flat face of the nascent base pair is topped by protein residues with unusually small and hydrophobic side chains: Val32, Ala42, Ala44, Ala57 and Gly58 (Figure 2a). These residues form the ‘ceiling’ of the dNTP binding pocket (25). The ‘floor’ of this pocket is considered to be the flat face of the base pair at the primer terminus (25). The geometry of the dNTP binding pocket remains normal here (Figure 2a). A highly conserved aromatic residue in the DinB family (Tyr12 in Dpo4, Phe12 in Dbh and Phe13 in DinB) is tightly packed on the sugar ring of the dNTP. This has the effect of placing the dNTP properly for catalysis and acting as a ‘steric gate’ to prevent binding of ribo-NTPs through steric exclusion of a 2′-OH (32,69). In our unmodified control simulation, the proper position of Tyr12 is preserved. Polymerase residues Tyr10, Phe11, Tyr12, Ser34, Thr45, Arg51, Lys159, Thr250 and Arg331 form hydrogen bonds with the nascent base pair in the unmodified control simulation (Supplementary Table S6).
Syn-G*·anti-dATP minor groove. The syn-G*·anti-dATP minor groove model is highly disturbed. With G* syn, the PhIP rings are on the minor groove side and inserted between the nascent base pair and the base pair at the primer terminus (Figure 2f). The location of the PhIP rings in the syn-G*·anti-dATP simulation disrupts the flat face of the primer-terminus base pair, and also causes poor stacking interactions between the nascent and the primer-terminus base pairs, as shown by energetic consideration: a partial energetic assessment of differential stacking interactions can be obtained from van der Waals interactions between bases for the same sequences. In the syn-G*·anti-dATP simulation, this interaction between the nascent and the primer-end base pairs is only −2.3 ± 0.5 kcal/mol, significantly weaker than −16.8 ± 0.9 kcal/mol in the anti-G*·syn-dATP, or −15.8 ± 0.9 kcal/mol in the anti-G*·anti-dATP case. The syn-G*·anti-dATP also manifests an enlarged C1′–C1′ distance (12.2 ± 0.4 Å), and a less frequently sampled near reaction-ready Pα-O3′ distance (59.8%). The hydrogen bond in the nascent base pair in the syn-G*·anti-dATP initial model, between O6 of the syn-G* and N6 of the anti-dATP, is ruptured during equilibration and remains broken throughout the simulation. To achieve the hydrogen bond in the initial model, the PhIP moiety was positioned in a crowded region near the ceiling of the dNTP binding pocket, which proved unfavorable (Supplementary Figure S1f). These disturbances, triggered by the syn-G* adduct, are essentially independent of the specific dNTP.
Anti-G*·anti-dATP and anti-G*·syn-dATP major groove. The anti-G*·anti-dATP and anti-G*·syn-dATP major groove models are more disturbed than the unmodified control, but less than the syn-G*·anti-dATP minor groove model. In the anti-G*·syn-dATP complex, the phenyl ring of the PhIP is directed towards the 3′ end of the template, along the template backbone (Figure 2e), while in the anti-G*·anti-dATP complex, it is oriented toward the 5′ end of the template (Figure 2d). A ∼180° difference in α′ is responsible for this difference in orientation (Supplementary Figure S6). With an incoming syn-dATP, one full and one bifurcated hydrogen bond are formed in the nascent base pair (Supplementary Figure S5 and Supplementary Table S5), with a close to normal C1′–C1′ distance (11.1 ± 0.2 Å). On the other hand, with the incoming nucleotide anti, the C1′–C1′ distance is significantly enlarged (12.7 ± 0.2 Å), and the full hydrogen bond between O6 of G* and N6 of anti-dATP has an occupancy 20% lower than for syn-dATP (Supplementary Table S5). A bifurcated hydrogen bond is formed between the little finger residue Arg332 and the PhIP rings only in the anti-G*·syn-dATP simulation (Supplementary Table S6). The frequencies of sampling the near reaction-ready Pα-O3′ distance and the geometries of the binding pocket are not dramatically different in these two major groove complexes (Supplementary Table S7 and Figure 2).
Anti-G*·anti-dTTP and anti-G*·syn-dGTP major groove. The anti-G*·anti-dTTP major groove model is little distorted, while the anti-G*·syn-dGTP major groove structure is very distorted. At this stage, the syn-G* was eliminated due to poor accommodation of the minor groove positioned PhIP regardless of dNTP, as revealed in the syn-G*·anti-dATP simulation (Figure 2f and Table 1). Therefore, only anti-G* was employed to build the models for G*·dTTP and G*·dGTP. With the incoming dTTP anti, the anti-G* forms a wobble pair with the anti-dTTP (Supplementary Figure S5). During the anti-G*·anti-dTTP simulation, the two hydrogen bonds in the wobble nascent base pair are highly occupied (>94% of the time); the C1′–C1′ distance has a normal average value of 10.8 ± 0.2 Å; the Pα-O3′ distance remains in the near reaction-ready range almost all the time (93.0%), and the nascent base pair is well stacked with the base pair at the primer-terminus. In the G*·G mismatch model, the incoming dGTP is syn and forms two hydrogen bonds with the anti-G* (Supplementary Figure S5 and Supplementary Table S5). However, the C1′–C1′ distance is enlarged to 11.9 ± 0.3 Å, and a somewhat less frequently sampled near reaction-ready Pα-O3′ distance is noted (68.1%).
Anti-G*·anti-dCTP major groove. With anti-G* opposite a Watson–Crick paired dCTP in the active site, the structural features are comparable to those of the unmodified control. In this anti-G*·anti-dCTP simulation, the Watson–Crick hydrogen bonds in the nascent base pair are highly occupied (>95% of the time); the C1′–C1′ distance has a normal average value of 10.9 ± 0.9 Å; the nascent base pair stacks well with the base pair at the primer-terminus, and the near reaction-ready Pα-O3′ distance is frequently sampled (82.4% of the time). However, two protein–DNA interactions have lower hydrogen bond occupancies in comparison to the unmodified control (Supplementary Table S6).
Controls of anti-G·syn-dATP, anti-G·anti-dTTP and anti-G·syn-dGTP. These control models containing G·dATP, G·dTTP and G·dGTP mismatches without PhIP modification show distortions at the active sites in comparison to the Watson–Crick G·dCTP paired unmodified control (Table 1). In the control of anti-G·syn-dATP, although the C1′–C1′ distance has a close to normal value of 11.2 ± 0.2 Å (Supplementary Table S7), the full and the bifurcated hydrogen bonds in the nascent base pair have lower occupancies (<90% of the time, Supplementary Table S5), and the near reaction-ready Pα-O3′ distance is less frequently sampled (61.4% of the time, Supplementary Table S7). In the control of anti-G·anti-dTTP, the C1′–C1′ distance has a normal value of 10.8 ± 0.2 Å (Supplementary Table S7). However, one of the two hydrogen bonds in the nascent pair is less frequently occupied (80.2% of the time, Supplementary Table S5), and the Pα-O3′ distance sampled the near reaction-ready range during only 59.7% of the time (Supplementary Table S5). In the control of anti-G·syn-dGTP, the C1′–C1′ distance is enlarged to 11.9 ± 0.3 Å and one of the two hydrogen bonds in the nascent base pair is poorly occupied (only 46.6% of the time, Supplementary Table S5). However, the near reaction-ready Pα-O3′ distance is frequently sampled (76.7% of the time, Supplementary Table S5).
−1 Deletion models
Anti-G*, syn-G* and unmodified control. In these models, the damaged template guanine has been skipped and the incoming dCTP pairs with the guanine on the 5′ side of the adduct (Figure 1c). In comparison to the unmodified −1 control system, the two adduct systems, the anti-G* −1 deletion and syn-G* −1 deletion are significantly distorted. In the anti-G* −1 deletion model, the PhIP moiety is positioned on the major groove side, near-perpendicular to the template strand (Figure 3b); in the syn-G* −1 deletion model, the PhIP moiety is positioned on the minor groove side (Figure 3c). Views of these ternary complexes and their active sites after 3 ns production MD simulations are shown in Supplementary Figures S3 and S4, and Figure 3. For the syn-G* −1 deletion system, the PhIP rings are reoriented through rotation of α′ and β′ after about 320 ps of MD (Supplementary Figure S7) to avoid crowding between the distal phenyl ring and Lys78. This rearrangement results in a favorable environment around the PhIP ring system. The phenyl ring is in van der Waals contact with hydrophobic residue Ile104; the imidazo ring N3 and N4 atoms have favorable electrostatic interactions with the amino group of Lys78 (actual hydrogen bonds are formed ∼9% of the time). As shown in Supplementary Figure S8, the PhIP moiety is pocketed by the protein residues, the nascent base pair and the primer-terminus base pair.
Watson–Crick hydrogen bonds in the nascent base pair are preserved in all three simulations (Supplementary Table S10). The C1′–C1′ distances are also normal in all cases (Supplementary Table S7), as is the chelation of the two Mg2+ ions (Figure 3). Compared to the unmodified control, the primer end is repositioned towards the 5′ direction and away from the active site in the anti-G* and syn-G*adduct simulations (Supplementary Figure S9); this is more prominent in the syn-G* −1 deletion simulation, where the terminal primer base is shifted by about 3 Å. The backbone of the primer strand is concomitantly shifted, accompanied by small rotation of the thumb domain, which maintains contacts with the relocated backbone phosphate groups of the primer strand. In order to retain the B-DNA geometry, the backbone of the template strand is also adjusted, followed by small rotation of the little finger domain in order to maintain contacts with the same phosphate groups of the template strand. A similar relocation of the primer terminal base was observed in a Dpo4 crystal structure containing the 10R(+)-cis-anti-[BP]-N6-dA adduct, with BP aromatic rings intercalated between the nascent and the primer terminal base pair (16). Here, the primer-terminus was positioned >10 Å away from the incoming dNTP. In both the anti-G* and syn-G* −1 deletion simulations, the dCTP loses its proper stacking contact with the ‘steric gate’ Tyr12 from the finger domain, and its hydrogen bonding with the finger residue Arg51. The separation of the dCTP from the finger domain could be a result of hydrogen bonds formed between the dCTP and the adduct (Supplementary Table S11). The Pα-O3′ distance is also affected by this primer side relocation. In the unmodified control system for the −1 deletion, the near reaction-ready Pα-O3′ distance is sampled during the whole simulation (51.4%, Supplementary Figure S10k). In the anti-G* −1 deletion model, this distance is only achieved in the 380–480 ps time frame, and comes close to 4 Å occasionally after this time range (Supplementary Figure S10l). However, in the syn-G* −1 deletion case, the distance has an average value of 7.5 ± 0.7 Å throughout the simulation, and does not approach closer than 5.5 Å (Supplementary Figure S10m). In the syn-G* −1 deletion system, the stacking between the PhIP-modified syn-G and its 3′-neighbour is also disrupted (Figure 3c). Thus, the −1 deletion simulations suggest various distortions likely to hinder nucleotide incorporation.
DISCUSSION
Our simulations suggest that regardless of the incoming dNTP or whether the damaged base has a partner, the dG-C8-PhIP adduct is not likely to be accommodated in the minor groove side pocket of the Dpo4 DinB family polymerase (Tables 1 and 2). When situated in this position, with the G* in the syn conformation, the bulky aromatic rings disrupt the geometry of the active site, particularly on the primer side (Figure 2f and Figure 3c). With the G* anti, however, the aromatic rings fit well on the major groove side. With dCTP opposite the anti-G*, Watson–Crick pairing is preserved and simulations show only modest distortions compared to the unmodified control (Supplementary Table S5 and Table 1). An incoming dTTP affords a well-formed wobble pair with only small distortions. In the case of the dATP, greater distortions are observed, with syn-dATP providing the more favorable structure, which contains hydrogen bonds employing the Hoogsteen edge of the dATP. Incoming dGTP was most distorted because even the syn conformation for dGTP caused enlargement of the nascent base pair. Furthermore, in the case of structures with no partner opposite the lesion, the −1 deletion models, the PhIP aromatic rings can again reside in the large major groove side open space; however, the active site region is notably distorted, and the approach of the primer-terminus to the α-phosphate of dNTP is inhibited by the PhIP moiety, particularly by its protruding methyl group (Supplementary Figure S9 and Supplementary Table S7).
In contrast, an earlier study of the 10S(+)-trans-anti-[BP]-N2-dG showed that this adduct can be accommodated reasonably well on the minor groove side of the Dpo4, irrespective of incoming dNTP, with only modest enzyme perturbation including opening of the little finger and some small rearrangement of active site region residues (54). In this case, the modified dG adopts the anti conformation. The major groove position with the N2-dG adduct in syn conformation also provides a good accommodation for the BP ring system. Experimental kinetic studies revealed promiscuous nucleotide incorporation for this case. Running-start primer extension experiments indicated that the damage can be bypassed to a significant extent (54).
Figure 4 illustrates the differential accommodation of the C8 and N2-dG adducts in the Dpo4 minor groove, showing why the C8 adduct is not well positioned there, while the N2 adduct is, regardless of dNTP (see Supplementary Figure S11 for stereoview). Specifically, this N2 adduct ring system is directed 5′ along the template strand in our models; however, the C8 adduct rings are oriented 3′ along the template strand, protrude to the primer side and thereby disrupt the active site directly where the nucleotidyl transfer reaction is to take place. Perhaps C8-dG bulky adducts, such as dG-C8-PhIP and dG-C8-AAF might less readily allow nucleotide incorporation by Dpo4, in part because of their poor accommodation in the enzyme minor groove side pocket; thus, only one site is available to harbor the lesion, while N2 adducts, such as 10S(+)-trans-anti-[BP]-N2-dG can reside in either major or minor groove side pockets.
Figure 4.

Minor groove positioned adducts in Dpo4. The view is into the minor groove side with the major groove in back. (a) syn-dG-C8-PhIP·anti-dATP after 3 ns MD simulation and (b) anti-dG-N2-BP·syn-dATP as in Figure 3b of (54). The carcinogen-modified guanine is green; the PhIP moiety is red; the BP moiety is brown; the incoming dATP is magenta, and the primer-terminus base pair is yellow. The protein is colored the same as in Figure 2.
Additionally, our structures suggest that translocation may be difficult for such major groove positioned multi-ringed adducts. Specifically, a recent crystal structure of Dpo4 has suggested that translocation starts with movement of the little finger during nucleotide binding, followed by thumb movement during the chemical reaction (23). The replication cycle involves a screw-like counterclockwise rotation/translocation of the polymerase along the DNA helix axis (viewed in the 5′ to 3′ direction of the template strand). This threading of the DNA through the polymerase places the next templating base in the active site, awaiting entry of its complementary dNTP. Prior study of the dG-C8-AAF adduct suggested that the preferred 3′-directed orientation of the fluorenyl ring system along the template strand, in the major groove, would impede the rotation of the little finger (34); this impediment is not due to the acetyl group. The current study of the dG-C8-PhIP adduct suggests a similar impact on translocation. As shown in Figure 5a and b, in the insertion position the adduct in the anti conformation is enveloped by the little finger without collision (see Supplementary Figure S12 for stereoview). However, once in the post-insertion site, the adduct would be in collision with the little finger, indicating that translocation to the post-insertion site would be difficult. Very large rearrangement of the PhIP adduct would be required to move it to another orientation that allows translocation. Additional bypass polymerases may be required to further extend beyond the lesion (4). Furthermore, it appears that N2 adducts, such as 10S(+)-trans-anti-[BP]-N2-dG, would be less likely to impede translocation by colliding with the little finger, when in the anti conformation and placed in the minor groove side polymerase pocket (Figure 5c and d). The number of rings in the adduct together with their orientation will determine the extent of these effects on translocation and this remains to be elucidated.
Figure 5.

Major groove positioned anti-dG-C8-PhIP adducts and minor groove positioned anti-dG-N2-BP adducts in Dpo4. For (a) and (b), the view is into the major groove with the minor groove in back, while for (c) and (d), the view is into the minor groove with the major groove in back. (a) anti-dG-C8-PhIP at the insertion position. (b) Modeled anti-dG-C8-PhIP at the post-insertion position with linkage site torsion angles α′, β′ and γ′ same as in (a); collisions between the PhIP rings and the little finger domain are indicated. (c) anti-dG-N2-BP adduct at insertion position opposite anti-dCTP as in Figure 3a of (54). (d) Modeled anti-dG-N2-BP at the post-insertion position in same linkage conformation as (c). The color code is the same as in Figure 4.
Primer extension studies of dG-N2-AAF and dG-C8-AAF with hpol κ have shown that the frequency of bypass of dG-N2-AAF is at least 4 orders of magnitude higher than for dG-C8-AAF, and it was proposed that the observed difference may be due to the different linkage site (70). This is consistent with our suggested poorer accommodation on the minor groove side of C8 than N2 adducts. However, we do not know exactly how structurally similar pol κ and Dpo4 ternary complexes are, since to date, only a crystal structure of the apo-pol κ catalytic core is available (71).
Furthermore, our results show that the PhIP-modified lesion produces less distorted structures for G*·dTTP and G*·dATP (anti-G*·anti-dTTP and anti-G*·syn-dATP) than for their respective unmodified controls (anti-G·anti-dTTP and anti-G·syn-dATP) (Table 1). The structural origin of this interesting phenomenon is at least partly in the formation of hydrogen bonds between the PhIP rings and the little finger residue Arg332, which stabilize the anti conformation in the adduct (Figure 2 and Supplementary Table S6). These findings would be relevant to observed mutagenic behavior of PhIP in inducing G to T transversions and G to A transitions in mammalian systems (44–52), if error-prone incorporation opposite the lesion involved the human DinB polymerase pol κ, and its structural properties prove similar to those of its prokaryotic homolog Dpo4.
In conclusion, our modeling and MD simulations for dG-C8-PhIP suggest that the adduct would increase the infidelity of Dpo4 and hinder translocation by the enzyme. We hope that the hypotheses resulting from our modeling studies will provide useful suggestions for future experimental investigations.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR online.
Acknowledgments
This work was supported by NIH grant CA75449 (S.B.), CA46533 (D.J.P.), and Ruth L. Kirschstein National Research Service Award F32 GM069152 (O.R.). Funding to pay the Open Access publication charges for this article was provided by NIH grant CA75449 (S.B.).
Conflict of interest statement. None declared.
REFERENCES
- 1.Zhang Y., Yuan F., Wu X., Wang M., Rechkoblit O., Taylor J.S., Geacintov N.E., Wang Z. Error-free and error-prone lesion bypass by human DNA polymerase κ in vitro. Nucleic Acids Res. 2000;28:4138–4146. doi: 10.1093/nar/28.21.4138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.McCulloch S.D., Kokoska R.J., Chilkova O., Welch C.M., Johansson E., Burgers P.M., Kunkel T.A. Enzymatic switching for efficient and accurate translesion DNA replication. Nucleic Acids Res. 2004;32:4665–4675. doi: 10.1093/nar/gkh777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lehmann A.R. Replication of damaged DNA by translesion synthesis in human cells. FEBS Lett. 2005;579:873–876. doi: 10.1016/j.febslet.2004.11.029. [DOI] [PubMed] [Google Scholar]
- 4.Prakash S., Johnson R.E., Prakash L. Eukaryotic translesion synthesis DNA polymerases: specificity of structure and function. Annu. Rev. Biochem. 2005;74:317–353. doi: 10.1146/annurev.biochem.74.082803.133250. [DOI] [PubMed] [Google Scholar]
- 5.Wagner J., Gruz P., Kim S.R., Yamada M., Matsui K., Fuchs R.P., Nohmi T. The dinB gene encodes a novel E.coli DNA polymerase, DNA pol IV, involved in mutagenesis. Mol. Cell. 1999;4:281–286. doi: 10.1016/s1097-2765(00)80376-7. [DOI] [PubMed] [Google Scholar]
- 6.Cordonnier A.M., Fuchs R.P. Replication of damaged DNA: molecular defect in Xeroderma pigmentosum variant cells. Mutat. Res. 1999;435:111–119. doi: 10.1016/s0921-8777(99)00047-6. [DOI] [PubMed] [Google Scholar]
- 7.Pagès V., Fuchs R.P. How DNA lesions are turned into mutations within cells? Oncogene. 2002;21:8957–8966. doi: 10.1038/sj.onc.1206006. [DOI] [PubMed] [Google Scholar]
- 8.Becherel O.J., Fuchs R.P. Mechanism of DNA polymerase II-mediated frameshift mutagenesis. Proc. Natl Acad. Sci. USA. 2001;98:8566–8571. doi: 10.1073/pnas.141113398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Napolitano R., Janel-Bintz R., Wagner J., Fuchs R.P. All three SOS-inducible DNA polymerases (Pol II, Pol IV and Pol V) are involved in induced mutagenesis. EMBO J. 2000;19:6259–6265. doi: 10.1093/emboj/19.22.6259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fuchs R.P., Koffel-Schwartz N., Pelet S., Janel-Bintz R., Napolitano R., Becherel O.J., Broschard T.H., Burnouf D.Y., Wagner J. DNA polymerases II and V mediate respectively mutagenic (−2 frameshift) and error-free bypass of a single N-2-acetylaminofluorene adduct. Biochem. Soc. Trans. 2001;29:191–195. doi: 10.1042/0300-5127:0290191. [DOI] [PubMed] [Google Scholar]
- 11.Friedberg E.C., Lehmann A.R., Fuchs R.P. Trading places: how do DNA polymerases switch during translesion DNA synthesis? Mol. Cell. 2005;18:499–505. doi: 10.1016/j.molcel.2005.03.032. [DOI] [PubMed] [Google Scholar]
- 12.Boudsocq F., Iwai S., Hanaoka F., Woodgate R. Sulfolobus solfataricus P2 DNA polymerase IV (Dpo4): an archaeal DinB-like DNA polymerase with lesion-bypass properties akin to eukaryotic polη. Nucleic Acids Res. 2001;29:4607–4616. doi: 10.1093/nar/29.22.4607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ohashi E., Ogi T., Kusumoto R., Iwai S., Masutani C., Hanaoka F., Ohmori H. Error-prone bypass of certain DNA lesions by the human DNA polymerase κ. Genes Dev. 2000;14:1589–1594. [PMC free article] [PubMed] [Google Scholar]
- 14.Ling H., Boudsocq F., Woodgate R., Yang W. Snapshots of replication through an abasic lesion; structural basis for base substitutions and frameshifts. Mol. Cell. 2004;13:751–762. doi: 10.1016/s1097-2765(04)00101-7. [DOI] [PubMed] [Google Scholar]
- 15.Ling H., Boudsocq F., Woodgate R., Yang W. Crystal structure of a Y-family DNA polymerase in action: a mechanism for error-prone and lesion-bypass replication. Cell. 2001;107:91–102. doi: 10.1016/s0092-8674(01)00515-3. [DOI] [PubMed] [Google Scholar]
- 16.Ling H., Sayer J.M., Plosky B.S., Yagi H., Boudsocq F., Woodgate R., Jerina D.M., Yang W. Crystal structure of a benzo[a]pyrene diol epoxide adduct in a ternary complex with a DNA polymerase. Proc. Natl Acad. Sci. USA. 2004;101:2265–2269. doi: 10.1073/pnas.0308332100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ling H., Boudsocq F., Plosky B.S., Woodgate R., Yang W. Replication of a cis-syn thymine dimer at atomic resolution. Nature. 2003;424:1083–1087. doi: 10.1038/nature01919. [DOI] [PubMed] [Google Scholar]
- 18.Vaisman A., Ling H., Woodgate R., Yang W. Fidelity of Dpo4: effect of metal ions, nucleotide selection and pyrophosphorolysis. EMBO. J. 2005;24:2957–2967. doi: 10.1038/sj.emboj.7600786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zang H., Goodenough A.K., Choi J.Y., Irimia A., Loukachevitch L.V., Kozekov I.D., Angel K.C., Rizzo C.J., Egli M., Guengerich F.P. DNA adduct bypass polymerization by Sulfolobus solfataricus DNA polymerase Dpo4: analysis and crystal structures of multiple base pair substitution and frameshift products with the adduct 1,N2-ethenoguanine. J. Biol. Chem. 2005;280:29750–29764. doi: 10.1074/jbc.M504756200. [DOI] [PubMed] [Google Scholar]
- 20.Trincao J., Johnson R.E., Wolfle W.T., Escalante C.R., Prakash S., Prakash L., Aggarwal A.K. Dpo4 is hindered in extending a G·T mismatch by a reverse wobble. Nature Struct. Mol. Biol. 2004;11:457–462. doi: 10.1038/nsmb755. [DOI] [PubMed] [Google Scholar]
- 21.Boudsocq F., Kokoska R.J., Plosky B.S., Vaisman A., Ling H., Kunkel T.A., Yang W., Woodgate R. Investigating the role of the little finger domain of Y-family DNA polymerases in low fidelity synthesis and translesion replication. J. Biol. Chem. 2004;279:32932–32940. doi: 10.1074/jbc.M405249200. [DOI] [PubMed] [Google Scholar]
- 22.Zang H., Irimia A., Choi J.Y., Angel K.C., Loukachevitch L.V., Egli M., Guengerich F.P. Efficient and high fidelity incorporation of dCTP opposite 7,8-dihydro-8-oxodeoxyguanosine by Sulfolobus solfataricus DNA polymerase Dpo4. J. Biol. Chem. 2006;281:2358–2372. doi: 10.1074/jbc.M510889200. [DOI] [PubMed] [Google Scholar]
- 23.Rechkoblit O., Malinina L., Cheng Y., Kuryavyi V., Broyde S., Geacintov N.E., Patel D.J. Stepwise translocation of Dpo4 polymerase during error-free bypass of an oxoG lesion. PLoS Biol. 2006;4:e11. doi: 10.1371/journal.pbio.0040011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rothwell P.J., Waksman G. Structure and mechanism of DNA polymerases. Adv. Protein Chem. 2005;71:401–440. doi: 10.1016/S0065-3233(04)71011-6. [DOI] [PubMed] [Google Scholar]
- 25.Kool E.T. Active site tightness and substrate fit in DNA replication. Annu. Rev. Biochem. 2002;71:191–219. doi: 10.1146/annurev.biochem.71.110601.135453. [DOI] [PubMed] [Google Scholar]
- 26.Steitz T.A. DNA polymerases: structural diversity and common mechanisms. J. Biol. Chem. 1999;274:17395–17398. doi: 10.1074/jbc.274.25.17395. [DOI] [PubMed] [Google Scholar]
- 27.Franklin M.C., Wang J., Steitz T.A. Structure of the replicating complex of a pol α family DNA polymerase. Cell. 2001;105:657–667. doi: 10.1016/s0092-8674(01)00367-1. [DOI] [PubMed] [Google Scholar]
- 28.Morales J.C., Kool E.T. Functional hydrogen-bonding map of the minor groove binding tracks of six DNA polymerases. Biochemistry. 2000;39:12979–12988. doi: 10.1021/bi001578o. [DOI] [PubMed] [Google Scholar]
- 29.Kunkel T.A. DNA replication fidelity. J. Biol. Chem. 2004;279:16895–16898. doi: 10.1074/jbc.R400006200. [DOI] [PubMed] [Google Scholar]
- 30.Silvian L.F., Toth E.A., Pham P., Goodman M.F., Ellenberger T. Crystal structure of a DinB family error-prone DNA polymerase from Sulfolobus solfataricus. Nature Struct. Biol. 2001;8:984–989. doi: 10.1038/nsb1101-984. [DOI] [PubMed] [Google Scholar]
- 31.Trincao J., Johnson R.E., Escalante C.R., Prakash S., Prakash L., Aggarwal A.K. Structure of the catalytic core of S.cerevisiae DNA polymerase η: implications for translesion DNA synthesis. Mol. Cell. 2001;8:417–426. doi: 10.1016/s1097-2765(01)00306-9. [DOI] [PubMed] [Google Scholar]
- 32.Jarosz D.F., Godoy V.G., Delaney J.C., Essigmann J.M., Walker G.C. A single amino acid governs enhanced activity of DinB DNA polymerases on damaged templates. Nature. 2006;439:225–228. doi: 10.1038/nature04318. [DOI] [PubMed] [Google Scholar]
- 33.Suzuki N., Ohashi E., Hayashi K., Ohmori H., Grollman A.P., Shibutani S. Translesional synthesis past acetylaminofluorene-derived DNA adducts catalyzed by human DNA polymerase κ and Escherichia coli DNA polymerase IV. Biochemistry. 2001;40:15176–15183. doi: 10.1021/bi010702g. [DOI] [PubMed] [Google Scholar]
- 34.Wang L., Broyde S. A new anti conformation for N-(deoxyguanosin-8-yl)-2-acetylaminofluorene (AAF-dG) allows Watson–Crick pairing in the Sulfolobus solfataricus P2 DNA polymerase IV (Dpo4) Nucleic Acids Res. 2006;34:785–795. doi: 10.1093/nar/gkj479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gooderham N.J., Murray S., Lynch A.M., Edwards R.J., Yadollahi-Farsani M., Bratt C., Rich K.J., Zhao K., Murray B.P., Bhadresa S., et al. Heterocyclic amines: evaluation of their role in diet associated human cancer. Br. J. Clin. Pharmacol. 1996;42:91–98. doi: 10.1046/j.1365-2125.1996.37513.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sinha R., Gustafson D.R., Kulldorff M., Wen W.Q., Cerhan J.R., Zheng W. 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine, a carcinogen in high-temperature-cooked meat, and breast cancer risk. J. Natl. Cancer Inst. 2000;92:1352–1354. doi: 10.1093/jnci/92.16.1352. [DOI] [PubMed] [Google Scholar]
- 37.Garner R.C. The role of DNA adducts in chemical carcinogenesis. Mutat. Res. 1998;402:67–75. doi: 10.1016/s0027-5107(97)00283-2. [DOI] [PubMed] [Google Scholar]
- 38.Wakabayashi K., Nagao M., Esumi H., Sugimura T. Food-derived mutagens and carcinogens. Cancer Res. 1992;52:2092s–2098s. [PubMed] [Google Scholar]
- 39.Boobis A.R., Lynch A.M., Murray S., de la Torre R., Solans A., Farre M., Segura J., Gooderham N.J., Davies D.S. CYP1A2-catalyzed conversion of dietary heterocyclic amines to their proximate carcinogens is their major route of metabolism in humans. Cancer Res. 1994;54:89–94. [PubMed] [Google Scholar]
- 40.Kadlubar F., Kaderlik R.K., Mulder G.J., Lin D., Butler M.A., Teitel C.H., Minchin R.F., Ilett K.F., Friesen M.D., Bartsch H., et al. Metabolic activation and DNA adduct detection of PhIP in dogs, rats, and humans in relation to urinary bladder and colon carcinogenesis. Princess Takamatsu. Symp. 1995;23:207–213. [PubMed] [Google Scholar]
- 41.Buonarati M.H., Turteltaub K.W., Shen N.H., Felton J.S. Role of sulfation and acetylation in the activation of 2-hydroxyamino-1-methyl-6-phenylimidazo[4,5-b]pyridine to intermediates which bind DNA. Mutat. Res. 1990;245:185–190. doi: 10.1016/0165-7992(90)90048-o. [DOI] [PubMed] [Google Scholar]
- 42.Frandsen H., Grivas S., Andersson R., Dragsted L., Larsen J.C. Reaction of the N2-acetoxy derivative of 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine (PhIP) with 2′-deoxyguanosine and DNA. Synthesis and identification of N2-(2′-deoxyguanosin-8-yl)-PhIP. Carcinogenesis. 1992;13:629–635. doi: 10.1093/carcin/13.4.629. [DOI] [PubMed] [Google Scholar]
- 43.Lin D., Kaderlik K.R., Turesky R.J., Miller D.W., Lay J.O., Jr, Kadlubar F.F. Identification of N-(Deoxyguanosin-8-yl)-2-amino-1-methyl-6-phenylimidazo [4,5-b]pyridine as the major adduct formed by the food-borne carcinogen, 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine, with DNA. Chem. Res. Toxicol. 1992;5:691–697. doi: 10.1021/tx00029a016. [DOI] [PubMed] [Google Scholar]
- 44.Carothers A.M., Yuan W., Hingerty B.E., Broyde S., Grunberger D., Snyderwine E.G. Mutation and repair induced by the carcinogen 2-(hydroxyamino)-1-methyl-6-phenylimidazo[4,5-b]pyridine (N-OH-PhIP) in the dihydrofolate reductase gene of Chinese hamster ovary cells and conformational modeling of the dG-C8-PhIP adduct in DNA. Chem. Res. Toxicol. 1994;7:209–218. doi: 10.1021/tx00038a015. [DOI] [PubMed] [Google Scholar]
- 45.Gooderham N.J., Zhu H., Lauber S., Boyce A., Creton S. Molecular and genetic toxicology of 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine (PhIP) Mutat. Res. 2002;506–507:91–99. doi: 10.1016/s0027-5107(02)00155-0. [DOI] [PubMed] [Google Scholar]
- 46.Burnouf D., Miturski R., Nagao M., Nakagama H., Nothisen M., Wagner J., Fuchs R.P. Early detection of 2-amino-1-methyl-6-phenylimidazo (4,5-b)pyridine(PhIP)-induced mutations within the Apc gene of rat colon. Carcinogenesis. 2001;22:329–335. doi: 10.1093/carcin/22.2.329. [DOI] [PubMed] [Google Scholar]
- 47.Morgenthaler P.M., Holzhauser D. Analysis of mutations induced by 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine (PhIP) in human lymphoblastoid cells. Carcinogenesis. 1995;16:713–718. doi: 10.1093/carcin/16.4.713. [DOI] [PubMed] [Google Scholar]
- 48.Yadollahi-Farsani M., Gooderham N., Davies D., Boobis A. Mutational spectra of the dietary carcinogen 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine(PhIP) at the Chinese hamsters hprt locus. Carcinogenesis. 1996;17:617–624. doi: 10.1093/carcin/17.4.617. [DOI] [PubMed] [Google Scholar]
- 49.Okonogi H., Ushijima T., Zhang X., Heddle J., Suzuki T., Sofuni T., Felton J., Tucker J., Sugimura T., Nagao M. Agreement of mutational characteristics of heterocyclic amines in lacI of the Big Blue mouse with those in tumor related genes in rodents. Carcinogenesis. 1997;18:745–748. doi: 10.1093/carcin/18.4.745. [DOI] [PubMed] [Google Scholar]
- 50.Okonogi H., Stuart G.R., Okochi E., Ushijima T., Sugimura T., Glickman B.W., Nagao M. Effects of gender and species on spectra of mutation induced by 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine in the lacI transgene. Mutat. Res. 1997;395:93–99. doi: 10.1016/s1383-5718(97)00146-0. [DOI] [PubMed] [Google Scholar]
- 51.Shibutani S., Fernandes A., Suzuki N., Zhou L., Johnson F., Grollman A.P. Mutagenesis of the N-(deoxyguanosin-8-yl)-2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine DNA adduct in mammalian cells. Sequence context effects. J. Biol. Chem. 1999;274:27433–27438. doi: 10.1074/jbc.274.39.27433. [DOI] [PubMed] [Google Scholar]
- 52.Yu M., Jones M.L., Gong M., Sinha R., Schut H.A., Snyderwine E.G. Mutagenicity of 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine (PhIP) in the mammary gland of Big Blue rats on high-and low-fat diets. Carcinogenesis. 2002;23:877–884. doi: 10.1093/carcin/23.5.877. [DOI] [PubMed] [Google Scholar]
- 53.Zhang L., Shapiro R., Broyde S. Molecular dynamics of a food carcinogen-DNA adduct in a replicative DNA polymerase suggest hindered nucleotide incorporation and extension. Chem. Res. Toxicol. 2005;18:1347–1363. doi: 10.1021/tx050132b. [DOI] [PubMed] [Google Scholar]
- 54.Perlow-Poehnelt R.A., Likhterov I., Scicchitano D.A., Geacintov N.E., Broyde S. The spacious active site of a Y-family DNA polymerase facilitates promiscuous nucleotide incorporation opposite a bulky carcinogen-DNA adduct: elucidating the structure-function relationship through experimental and computational approaches. J. Biol. Chem. 2004;279:36951–36961. doi: 10.1074/jbc.M404332200. [DOI] [PubMed] [Google Scholar]
- 55.Berman H.M., Westbrook J., Feng Z., Gilliland G., Bhat T.N., Weissig H., Shindyalov I.N., Bourne P.E. The Protein Data Bank. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Brown K., Hingerty B.E., Guenther E.A., Krishnan V.V., Broyde S., Turteltaub K.W., Cosman M. Solution structure of the 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine C8-deoxyguanosine adduct in duplex DNA. Proc. Natl Acad. Sci. USA. 2001;98:8507–8512. doi: 10.1073/pnas.151251898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mizukami S., Kim T.W., Helquist S.A., Kool E.T. Varying DNA base-pair size in subangstrom increments: evidence for a loose, not large, active site in low-fidelity dpo4 polymerase. Biochemistry. 2006;45:2772–2778. doi: 10.1021/bi051961z. [DOI] [PubMed] [Google Scholar]
- 58.Potapova O., Chan C., Delucia A.M., Helquist S.A., Kool E.T., Grindley N.D., Joyce C.M. DNA polymerase catalysis in the absence of Watson–Crick hydrogen bonds: analysis by single-turnover kinetics. Biochemistry. 2006;45:890–898. doi: 10.1021/bi051792i. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Berman H.M., Olson W.K., Beveridge D.L., Westbrook J., Gelbin A., Demeny T., Hsieh S.H., Srinivasan A.R., Schneider B. The nucleic acid database. A comprehensive relational database of three-dimensional structures of nucleic acids. Biophys. J. 1992;63:751–759. doi: 10.1016/S0006-3495(92)81649-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Cornell W.D., Cieplak P., Bayly C.I., Gould I.R., Merz K.M., Ferguson D.M., Spellmeyer D.C., Fox T., Caldwell J.W., Kollman P.A. A second generation force-field for the simulation of proteins, nucleic-acids, and organic-molecules. J. Am. Chem. Soc. 1995;117:5179–5197. [Google Scholar]
- 61.Cheatham T.E., III, Cieplak P., Kollman P.A. A modified version of the Cornell et al. force field with improved sugar pucker phases and helical repeat. J. Biomol. Struct. Dyn. 1999;16:845–862. doi: 10.1080/07391102.1999.10508297. [DOI] [PubMed] [Google Scholar]
- 62.Darden T., York D., Pedersen L. Particle mesh Ewald: an Nxlog(N) method for Ewald sums in large systems. J. Phys. Chem. 1993;98:10089–10092. [Google Scholar]
- 63.Ryckaert J.P., Ciccotti G., Berendsen H.J.C. Numerical integration of cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes. J. Comput. Phys. 1977;23:327–341. [Google Scholar]
- 64.Harvey S.C., Tan R.K.Z., Cheatham T.E. The flying ice cube: velocity rescaling in molecular dynamics leads to violation of energy equipartition. J. Comput. Chem. 1998;19:726–740. [Google Scholar]
- 65.Barsky D., Kool E.T., Colvin M.E. Interaction and solvation energies of nonpolar DNA base analogues and their role in polymerase insertion fidelity. J. Biomol. Struct. Dyn. 1999;16:1119–1134. doi: 10.1080/07391102.1999.10508321. [DOI] [PubMed] [Google Scholar]
- 66.Brautigam C.A., Steitz T.A. Structural and functional insights provided by crystal structures of DNA polymerases and their substrate complexes. Curr. Opin. Struct. Biol. 1998;8:54–63. doi: 10.1016/s0959-440x(98)80010-9. [DOI] [PubMed] [Google Scholar]
- 67.Doublie S., Ellenberger T. The mechanism of action of T7 DNA polymerase. Curr. Opin. Struct. Biol. 1998;8:704–712. doi: 10.1016/s0959-440x(98)80089-4. [DOI] [PubMed] [Google Scholar]
- 68.Batra V.K., Beard W.A., Shock D.D., Krahn J.M., Pedersen L.C., Wilson S.H. Magnesium-induced assembly of a complete DNA polymerase catalytic complex. Structure. 2006;14:757–766. doi: 10.1016/j.str.2006.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.DeLucia A.M., Grindley N.D., Joyce C.M. An error-prone family Y DNA polymerase (DinB homolog from Sulfolobus solfataricus) uses a ‘steric gate’ residue for discrimination against ribonucleotides. Nucleic Acids Res. 2003;31:4129–4137. doi: 10.1093/nar/gkg417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Yasui M., Dong H., Bonala R.R., Suzuki N., Ohmori H., Hanaoka F., Johnson F., Grollman A.P., Shibutani S. Mutagenic properties of 3-(deoxyguanosin-N2-yl)-2-acetylaminofluorene, a persistent acetylaminofluorene-derived DNA adduct in mammalian cells. Biochemistry. 2004;43:15005–15013. doi: 10.1021/bi048279+. [DOI] [PubMed] [Google Scholar]
- 71.Uljon S.N., Johnson R.E., Edwards T.A., Prakash S., Prakash L., Aggarwal A.K. Crystal structure of the catalytic core of human DNA polymerase κ. Structure (Camb) 2004;12:1395–1404. doi: 10.1016/j.str.2004.05.011. [DOI] [PubMed] [Google Scholar]
- 72.Jorgensen W.L., Chandreskhar J., Madura J.D., Imprey R.W., Klein M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983;79:926–935. [Google Scholar]
- 73.Berendsen H.J.C., Postma J.P.M., Vangunsteren W.F., Dinola A., Haak J.R. Molecular-dynamics with coupling to an external bath. J. Chem. Phys. 1984;81:3684–3690. [Google Scholar]