

Abstract
Structural studies of polio- and closely related viruses have provided a series of snapshots along their cell entry pathways. Based on the structures and related kinetic, biochemical, and genetic studies, we have proposed a model for the cell entry pathway for polio- and closely related viruses. In this model a maturation cleavage of a capsid protein precursor locks the virus in a metastable state, and the receptor acts like a transition-state catalyst to overcome an energy barrier and release the mature virion from the metastable state. This initiates a series of conformational changes that allow the virus to attach to membranes, form a pore, and finally release its RNA genome into the cytoplasm. This model has striking parallels with emerging models for the maturation and cell entry of more complex enveloped viruses such as influenza virus and HIV.
Keywords: virus structure, conformational changes, spring-loaded trap, virus-receptor interactions, virus-membrane interactions
INTRODUCTION
In many ways simple nonenveloped viruses such as poliovirus and closely related picornaviruses present ideal models for understanding how viruses enter cells and initiate infection. As a result of years of study, arising first from a focus on development of poliovirus vaccines and subsequently from their emergence as model systems, this group of viruses is exceptionally well characterized biochemically and genetically. Receptors for a number of these viruses have been identified, and the viruses are amenable to genetic and structural characterization. However, despite years of characterization the mechanism that allows poliovirus (or any nonenveloped virus) to cross a membrane and release its genome into the cell for replication remains poorly understood. Paradoxically, this process is better understood for enveloped viruses whose structural organization is more complex. Indeed, in many ways the entry process for enveloped viruses is simpler. On a strictly topological basis the nucleocapsid of an enveloped virus is already in the same compartment as the cytoplasm, and this topological equivalence can be realized physically provided that there is a mechanism for facilitating fusion of the viral envelope with a cell membrane. In contrast, the nucleocapsid (virion) of a nonenveloped virus is topologically outside the cell and remains there even if it is internalized in a cellular vesicle. At some point the entire virion, or at least the viral genome, must cross a membrane.
SOME COMMON THEMES IN VIRAL ENTRY
Although there are significant differences in cell entry pathways among viruses, several common themes are emerging from the study of a wide variety of viruses. These common themes are the consequence of a central problem faced by all viruses in the passage from cell-to-cell or from host-to-host. Thus, in order to withstand the rigors of the extracellular environment, the virion must be stable to a variety of insults, which may include extremes of pH, ionic strength, and temperature, and the presence of proteases. The virion must be inert and not adhere to surfaces or to other cells until it reaches its target tissue and encounters its specific receptor. However, once the virion reaches its target cell and encounters the appropriate trigger(s), it must be capable of undergoing structural changes that allow it to cross a membrane and deliver its genome to the appropriate compartment of the cell to initiate replication.
A Link Between Assembly and Entry
For many viruses the solution to this problem involves a link between the final stages of assembly and the initiation of cell entry—an attractive model because it emphasizes the role of the virion as an intermediate (linking assembly and entry) in a cycle. Thus, the final stage of assembly for many viruses involves proteolytic processing of a structural protein (generally a surface glycoprotein for enveloped viruses or a capsid protein of a noneveloped virus). By first folding the structure as a precursor and subsequently introducing a covalent alteration, it becomes possible for the lowest energy structure of the product to differ from the lowest energy structure of the precursor. However, once the precursor has folded, the lowest energy structure of the mature glycoprotein (for an enveloped virus) or the intact mature virus (for the nonenveloped viruses) may not be accessible owing to energy barriers that block the pathway between the two structural forms. The protein or virus particle is then said to be kinetically trapped in a metastable state. When the virus encounters the appropriate trigger (either receptor binding, acidification of an endosome, or both), the protein (or virion) is released from the metastable state and proceeds to a lower-energy state that exposes hydrophobic sequences that allow it to attach to membranes. In enveloped viruses the membrane attachment facilitates fusion of the viral envelope with the cell membrane. In nonenveloped viruses the hydrophobic sequences must either generate a pore or disrupt a cellular membrane to facilitate entry.
The Influenza Hemagglutinin as an Example
Perhaps the best-characterized model for this link between assembly and cell entry is the influenza A virus hemagglutinin [reviewed in (100, 117)]. In influenza A, the hemagglutinin glycoprotein spike provides the site for binding to its cellular receptor (sialic acid residues on glycoproteins and glycolipids on the cell surface) and serves as the fusion protein. The hemagglutinin is first synthesized as a precursor HA0 and is cleaved late in assembly to form two chains, HA1 and HA2. The newly generated N terminus of HA2 begins with a string of nonpolar amino acids, “the fusion peptide,” that ultimately facilitates fusion of the viral envelope with the cell membrane. When the virus binds to its cellular receptor, it is taken up into endosomes. Upon acidification of the endosomes, the hemagglutinin undergoes a massive conformational change that results in the exposure of the fusion peptide, its insertion into the membrane of the endosome, the fusion of the viral envelope with the endosome, and the release of the nucleocapsid into the cytoplasm. Thus, for influenza the receptor plays a single role, namely to concentrate virus at the surface of susceptible cells, and the trigger that releases the hemagglutinin from its metastable state is acidification of the endosome. In other enveloped viruses the receptor also serves as the trigger that induces the conformational changes required for entry, in which case the virus envelope may fuse directly with the plasma membrane.
High-resolution structures of ectodomains of HA0 (21), the mature HA (118), and of the fusogenic form of HA2, which is produced by proteolytic removal of HA1 from HA subsequent to acidification (18), have been solved in Don Wiley's laboratory at Harvard University. The hemagglutinin is a trimer that is held together largely by coil-coil interactions between long helices in the HA2 subunit (Figure 1). The globular head of the trimer comprises much of the HA1 chain and contains the receptor-binding sites. In HA0 and HA at neutral pH, the fusion peptide is located at the base of the molecule near the site of attachment to the viral envelope. The fusion peptide is contained in an exposed loop in HA0 (Figure 1a), and upon cleavage to produce HA the newly freed fusion peptide is inserted between the helices and is not exposed (Figure 1b). In mature HA at neutral pH, the fusion peptide is located nearly 100Å away from the receptor-binding site (Figure 1b). Acidification induces a massive conformational rearrangement that moves both the fusion peptide and the C terminus of the ectodomain of HA2 (which in the intact protein is anchored in the viral envelope) to a point near the top (cell-proximal end) of the molecule (Figure 1c). At this point the fusion peptide and the cell membrane to which it is attached, and the C terminus of HA2 and the viral envelope to which it is attached, are in close proximity, allowing fusion of the two membranes to proceed. The detailed mechanism of induction of fusion is still poorly understood.
Figure 1.
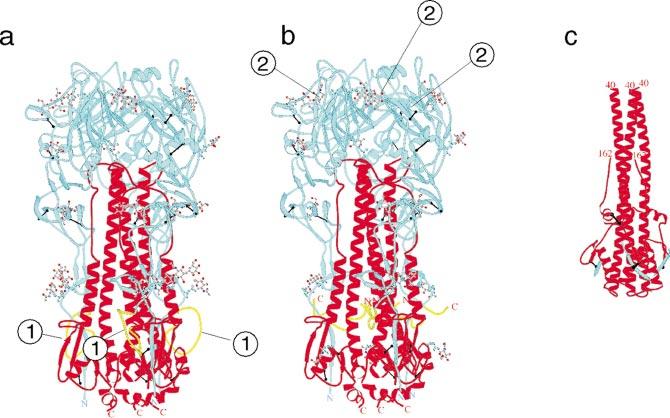
Structural changes on maturation and acidification of the influenza A virus hemagglutinin. The hemagglutinin is a homotrimer. (a) The monomer is initially synthesized as a single chain HA0.(b)HA0 is subsequently processed late in assembly to form two chains, HA1 (blue) and HA2 (red). (c) Upon acidification in early endosomes during entry, HA undergoes a significant conformational alteration to a fusogenic form. The newly generated amino terminus of HA2 (yellow) facilitates fusion and is called the fusion peptide. In HA0 the fusion peptide is in an exposed loop near the base of the molecule (indicated by the 1 in Figure 1a). (b) In the mature HA the fusion peptide is buried. The fusion peptide is located near the bottom of the molecule, which would be close to the viral membrane, and some 100Å away from the receptor-binding site (indicated by 2 in Figure 1b). In the fusogenic form (c) the fusion peptide and the C terminus of HA2 (which anchors the molecule in the viral membrane) and the fusion peptide (which is believed to insert into the cell membrane) are both near the top of the molecule. This would bring the viral membrane and cell membrane into close proximity. Figure reproduced from Skehel & Wiley (100), with permission.
The Model May be General
The HA of influenza A is the only protein where both the metastable and fusogenic forms of the glycoprotein have been characterized structurally. However, similar conformational changes have been proposed for the envelope glycoproteins of a number of other viruses based on structures of analogues of the fusogenic form (5, 20, 32, 61, 71, 72, 113, 114, 123). Using poliovirus as an example, we demonstrate that similar mechanisms in which receptor binding releases virus from a metastable state and exposes hydrophobic sequences may also occur in nonenveloped viruses.
THE LIFE CYCLE OF POLIO- AND RELATED VIRUSES
Poliovirus is a member of the picornavirus family, which includes a number of significant pathogens of humans (e.g., rhinoviruses, Coxsackieviruses, echoviruses, enteroviruses, and hepatitis A virus) and livestock (e.g., foot-and-mouth disease viruses). The viruses are small (˜300Å in diameter) and consist entirely of an icosahedral protein coat that encapsidates a single-stranded plus-sense RNA genome. Despite their simple organization, the viruses have a complex life cycle [reviewed in (92)].
Assembly
PROTEOLYTIC PROCESSING IS ASSOCIATED WITH VIRAL ASSEMBLY A brief summary of the poliovirus life cycle is shown in Figure 2. Because the process is cyclic we may start at any point. We choose viral assembly as our starting point to develop the parallel with the model described above. Upon release into the cytoplasm, the viral RNA is translated in a single open reading frame to produce a polyprotein that is processed cotranslationally by viral proteases to yield the viral proteins (57, 87). The polyprotein is myristoylated at its N terminus (22). An early cotranslational cleavage of the polyprotein by the viral 2A protease releases a precursor protein myristoyl-P1 from the N terminus of the polyprotein. This cleavage may occur in cis. The P1 protein contains all the capsid protein sequences. Subsequent cleavage of P1 by the viral 3CD protease produces the capsid proteins VP1 and VP3 and the immature capsid protein myristoyl-VP0. This cleavage is associated with the assembly of the proteins into a pentameric assembly intermediate, which spontaneously assembles into empty capsid containing 60 copies each of VP0, VP3, and VP1. The empty capsids and pentamers are apparently in equilibrium within the cell, and therefore it has not been possible to determine whether RNA is encapsidated by pentamers or by insertion into the preformed empty capsids. Regardless, the encapsidation appears to be tightly linked to RNA replication, as there is an absolute dependence of encapsidation on de novo synthesis of progeny RNA (77, 78). Encapsidation leads to the formation of a precursor, called the provirion, that contains the RNA and 60 copies each of VP0, VP3, and VP1. The processing of the immature protein myristoyl-VP0 to yield myristoyl-VP4 and VP2 is associated with encapsidation of the RNA. There is no known protease requirement for this cleavage, and it is thought to be autocatalytic, depending only on the capsid proteins themselves and perhaps the viral RNA. The cleavage of VP0 to form the virion is associated with a significant increase in the stability of the particle. The mechanism of release of the virus from the cell is unclear.
Figure 2.
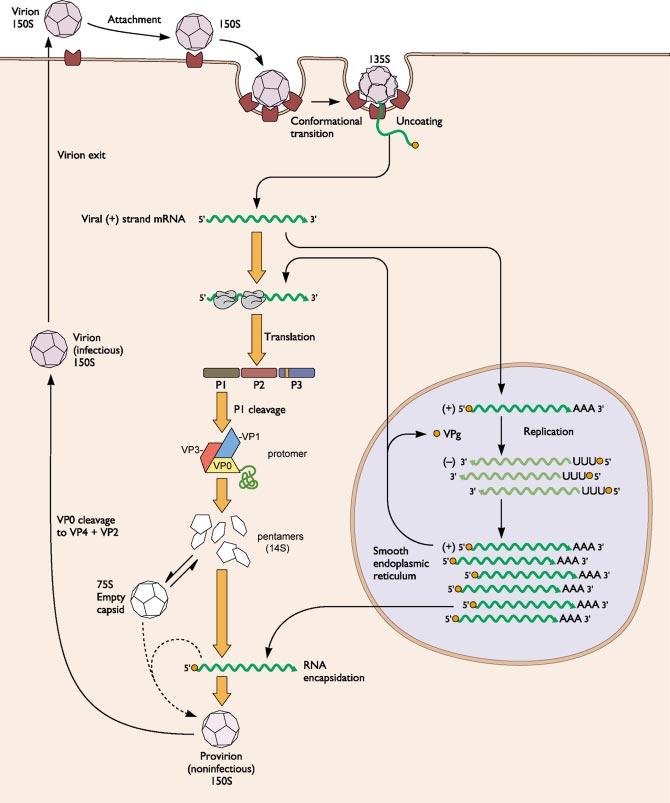
The life cycle of poliovirus and related picornaviruses. Infection is initiated by attachment to receptor, which induces conformational changes in the virus that facilitate translocation of the viral RNA into the cytoplasm where it is replicated to yield progeny RNAs and translated to yield viral proteins. Translation produces a long polyprotein that is processed by viral proteases. Assembly of the virus is linked to processing of the polyprotein and proceeds through a series of intermediates including a protomer, a pentamer, an empty capsid, a provirion, and ultimately the virus. Adapted from Principles of Virology, (S.J. Flint, V.R. Racaniello, L.W. Enquist, A.M. Skalka, & R.M. Krug) with permission.
Receptor Binding and Cell Entry
RECEPTORS The next round of replication is initiated when the virus encounters its receptor. The receptors for a number of picornaviruses have been identified (11-14, 43, 51, 76, 98, 107). The poliovirus receptor (Pvr) is a CAM-like molecule with three extracellular Ig-like domains (76). There is no known requirement for a coreceptor for poliovirus entry. The cellular function for Pvr is not known, but two paralogs in humans, called Nectin-1 and Nectin-2, are homotrophic adhesion proteins that interact with the actin skeleton through a cytoplasmic protein called afadin (108). Mice lacking the mouse homolog of Nectin-2 show specific defects in spermatogenesis (17). It is not clear whether Pvr plays similar roles, as it lacks sequence motifs in its cytoplasmic domain that mediate interactions with afadin. Interestingly, Pvr, Nectin-1, and Nectin-2 are coreceptors for alpha herpesviruses (38, 99). Several lines of evidence suggest that the first (N-terminal) domain of Pvr is responsible for poliovirus binding and infection. (a) Cells expressing the first domain by itself or as a chimera with other Ig-like proteins are susceptible to infection with poliovirus (58, 80, 96, 97). This functionality is independent of the cytoplasmic domain or the nature of the transmembrane anchor, suggesting that intracellular signaling is not critical for Pvr's function as a receptor. Indeed, the ectodomain fused to a GPI anchor is a functional receptor (M. Chow, personal communication). (b) Mutations in the first Ig-like domain alter virus binding (2, 15, 79).
THE SITE OF CELL ENTRY IS A MYSTERY Although many textbooks state that picornaviruses enter cells by receptor-mediated endocytosis (70), the actual site and mechanism of cell entry remains unknown. Indeed, the characterization of the entry mechanism is complicated by the relatively high particle-to-pfu (plaque-forming-unit) ratio for polio and its relatives (ranging from 102 to 103). Thus, when virus or viral-derived particles are detected in a given compartment during entry it is not clear whether the particles are involved in productive or nonproductive events. As a result, classical biochemical and electron microscopic methods (which “count” particles rather than infectious units) have led to contradictory findings. Early experiments with monensin and lysomotrophic amines (which do measure infectious units) suggested that poliovirus entry requires acidification (70). However, subsequent studies with bafilomycin A demonstrated that poliovirus entry is independent of acidification of endosomes (88) and suggested that the contradictory results of the earlier studies were an artifact of inhibition of downstream events in infection, most likely RNA replication. Similar studies indicate that most enteroviruses and some (8), but not all (91), rhinoviruses enter via mechanisms that do not depend on acidification of endosomes.
Others and we have taken advantage of the recent development of dominant-negative mutants of dynamin (28) to further probe the route of entry of poliovirus and several closely related entero- and rhinoviruses. Dynamin is a large GTPase that plays a key role in “pinching off” coated vesicle to form coated pits during classical clathrin-mediated endocytosis. Recent data suggest that dynamin also is required for internalization via caveoli. DeTulleo & Kirchhausen (30) have shown that expression of a dominant-negative dynamin mutant abrogates the ability of rhinovirus 14 but not poliovirus to initiate infection (30). We have characterized the dependence of several additional enteroviruses including echovirus 1, echovirus 7, Coxsackie B3 (Nancy), Coxsackie B3 (RD), and Coxsackievirus B4 (E.S. Mittler, J.M. Bergelson & J.M. Hogle, manuscript in preparation). These studies showed that closely related viruses differ in their dependence on dynamin for cell entry. Thus, echovirus 1 and the RD strain Coxsackievirus B3 were shown to be dynamin dependent, whereas echovirus 7, the Nancy strain of Coxsackievirus B3 (which is the parent of the RD strain), and Coxsackievirus B4 were not. Dynamin dependence seemed to be correlated with the receptor used, but it was not correlated with the nature of the transmembrane anchor of the receptor or the presence of known clathrin-recruiting signals in the cytoplasmic domain of the receptor. Mittler et al. also investigated the effect of domain truncations of ICAM-1 on the dynamin dependence of rhinovirus 14 (E.S. Mittler, J.M. Bergelson & J.M. Hogle, manuscript in preparation). They showed that while infection mediated by full-length (five domain) ICAM-1 was dynamin dependent, infection mediated by mutant receptors with deletions of domain 5 (membrane proximal), domains 4 and 5 or domain 3 were not. The results suggest that dynamin dependence is not an intrinsic property of the virus itself. Thus, the enteroviruses and rhinoviruses are not obligatorily dependent on dynamin-requiring pathways such as endocytosis for cell entry and may be promiscuous in their choice of pathways.
STRUCTURAL ALTERATIONS ASSOCIATED WITH CELL ENTRY For polioviruses, rhinoviruses, and most related enteroviruses, binding cells expressing the receptor at physiological temperatures result in the induction of conformational changes in the virus to produce a particle called the A particle or 135S particle (29, 33). In the course of a typical experimental infection, a significant fraction of the A particle subsequently elutes from cells in what is thought to represent an abortive infection. However, the A particle is also the predominant cell-associated form of the virus early in infection (within the first 20-30 min) (36). At later times (post infection) the levels of A particles begin to decrease. The timing of the disappearance of the A particle is correlated with the timing of the appearance of a second altered form of the virus that has lost its RNA and now sediments at 80S. The trigger for conversion to the 80S form is not known, but it does not require receptor.
The native-to-A particle conversion can also be induced by solubilized forms of the receptor (40, 55) and by the soluble ectodomain of the receptor in the absence of cells (3, 111). The A particle has altered sedimentation behavior (sedimenting at 135S versus 160S for the native virion) and altered antigenicity. In contrast to the virion (which is stable to the proteases and quite soluble), the A particle is sensitive to proteases and is hydrophobic (partitioning into detergent micelles). The A particle has externalized myristoyl-VP4 and the N-terminal extension of the capsid protein VP1 (36), both of which are in the interior of the native virion. The receptor-induced conversion to the A particle is apparently irreversible. Interestingly, transient and reversible exposure of VP4 and the N-terminal extension of VP1 occur when the virus is at physiological temperatures (but not room temperature) in a process that has been termed “breathing” (64). The breathing provides striking evidence for the dynamic nature of the poliovirus structure and suggests that the virus is literally primed to undergo larger, concerted, and irreversible changes associated with receptor binding. Breathing also occurs in other nonenveloped viruses such as Flock House virus (16) and rhinoviruses (63).
NEWLY EXPOSED SEQUENCES IN THE A PARTICLE FACILITATE MEMBRANE ATTACHMENT The exposed amino-terminal extension of VP1 (which is predicted to form an amphipathic helix in all entero- and rhinoviruses) enables the A particle to attach to liposomes (36). Insertion of the N terminus of VP1 (and perhaps the myristoyl group of VP4) may facilitate cell entry either by disrupting a membrane or by forming a pore in a membrane (36). The A particle (and the virus when incubated at 37°C where breathing is efficient) forms channels in planar bilayers (109). This observation, together with unpublished data that show that the channels become pores in the presence of receptor (M. Chow, personal communication), would support a model in which the inserted sequences formed a pore in the membrane through which the RNA could be extruded into the cytoplasm.
IS THE A PARTICLE A CELL ENTRY INTERMEDIATE? Although there is still considerable controversy concerning the role of the two altered particles, it is generally thought that the A particle may be an intermediate in the cell entry pathway and that the 80S empty particle is the final protein product that accumulates after the RNA is released into the cytoplasm to initiate translation and replication. There are several lines of evidence that the A particle is indeed an intermediate: (a) The antiviral activity of compounds that bind to the capsid correlates well with their ability to inhibit the formation of the A particle in vitro (110). (b) The kinetics of appearance and disappearance of the A particle in the cell during experimental infections at 37°C are consistent with it playing a role in both entry and RNA release (36). (c) The ability of the A particle to attach to liposomes provides a compelling model for membrane attachment during entry. (d) The ability of the A particle to form channels in membranes (109) could provide a mechanism for RNA translocation. (e) The A particle is infectious in a receptor-independent fashion (27). Although the efficiency of infection with the A particle is nearly four orders of magnitude lower than virions, this inefficiency is largely attributable to the lack of a receptor to bring the particle to high concentration at the cell surface. Indeed, the efficiency can be enhanced significantly (to within an order of magnitude of virus) by preincubating the A particles with non-neutralizing antibodies and using these virus/antibody complexes to infect cells expressing the Fc receptor (53).
However, the role of the A particle as a productive intermediate in the cell entry pathway has recently been questioned based on observations that it does not accumulate in cells when cold-adapted viruses are grown at 25°C (31). Although this observation raises an important caveat concerning the role of the A particle, the failure to observe an intermediate in a steady-state process in cell entry does not necessarily imply that the intermediate does not exist. In fact, in a steady-state process, an intermediate is expected to accumulate to appreciable levels only if a rate-limiting step in the pathway occurs downstream of the putative intermediate. Recent experiments using a neutral red assay to follow the kinetics of RNA release at 37°C and at 25°C for infections initiated by virions and by A particles demonstrate that RNA release is rate-limiting at 37°C, but that A particle formation is rate-limiting at 25°C (53). Thus, the A particle would not be expected to accumulate in infections at low temperature, and it (or perhaps a similar particle) remains a viable candidate as a cell entry intermediate.
IN VITRO PRODUCTION OF ALTERED PARTICLES The native-to-A particle and A particle-to-80S particle conversion can also be induced in the absence of receptor by warming the particle in hypotonic buffers in the presence of millimolar levels of calcium ions (27, 115). In the absence of calcium the reaction proceeds directly to the 80S particles, suggesting that calcium is required to stabilize the A particle and that depletion of calcium at some stage during the normal entry process may serve as a trigger for RNA release. Like the receptor-mediated native-to-A particle conversion, the thermal-mediated conversion is inhibited by capsid-binding antiviral agents (110, 116).
The ability to recapitulate the conformational alterations by simply warming the virion provides a convenient means for production of large amounts of the altered particles in vitro from purified virus. It also provides a convenient assay system for studying the kinetics of the virion-to-A particle conversion as a function of temperature. These kinetic studies have led to several observations that support the general model for cell entry discussed in the introduction and that provide additional insights into the role of the receptor in mediating the virion-to-A particle conversion.
THE VIRION IS KINETICALLY TRAPPED The dependence of the native-to-A particle conversion on elevated temperature suggests that there is a high activation energy barrier in the conversion pathway. This would be consistent with the model in which the virus is kinetically trapped in a metastable state. Analysis of the kinetics of both the thermal-induced and the receptor-induced virion-to-A particle conversion as a function of temperature supports this model (110). The rate of the thermal-induced conversion displays steep temperature dependence and is consistent with an activation energy barrier of 140 kcal/mole (110).
THE RECEPTOR FACILITATES CONVERSION BY LOWERING THE ACTIVATION BARRIER The dependence of the receptor-independent reaction on elevated temperature suggests that the receptor may facilitate the conversion at physiological temperature by lowering the activation barrier. Analysis of the kinetics of the receptor-mediated conversion as a function of temperature confirms that the receptor produces a significant enhancement of the rate, such that the rate becomes biologically relevant at physiological temperatures. The data also demonstrate that the receptor lowers the activation barrier for the reaction by nearly 50 kcal/mole. Thus, the receptor behaves like a classical transition-state catalyst (111).
THE CAPSID-BINDING ANTIVIRAL AGENTS STABILIZE VIRUS VIA ENTROPIC EFFECTS As a corollary to our original proposal that receptor would facilitate conversion by lowering the activation barrier, we had proposed that the capsid-binding antiviral agents that prevent receptor-mediated conversion at physiological temperatures would stabilize the virions by raising the activation barrier of the conversion. A model based on this prediction suggests that the antiviral agents act like a peg to rigidify the capsid. Surprisingly, kinetic studies of the thermal-mediated transition show that the capsid-binding antiviral agents have no effect on the activation barrier (an enthalpic term) and suggest that the drugs act via entropic stabilization of the virion (110). These experimental studies are consistent with computational studies of rhinovirus by Post and colleagues that show that binding the drugs increases rather than decreases the compressibility of the virus, and stabilizes the virus by providing a higher density of low-energy states to the virion (89, 90, 105).
THE RECEPTOR-CATALYZED REACTION PROCEEDS THROUGH AN ACTIVATED INTERMEDIATE Although the inhibition of receptor-independent conversion of virions to A particles by capsid-binding antivirals is mediated by strictly entropic effects, the inhibition of the receptor-mediated pathway includes both enthalpic and entropic contributions (111). These results can only be explained if the receptor-mediated pathway proceeds via an intermediate not present in the receptor-independent pathway. By analogy with classic enzyme kinetic models, we propose that this intermediate represents an activated virus-receptor complex in which the virus (and receptor?) undergoes conformational changes and switches from an initial complex to a “tight-binding” complex. This proposal is also consistent with the observation that soluble receptor has two affinities for the virus, with the low-affinity mode dominating at low temperature and the high-affinity mode becoming more prevalent as the temperature is increased (75). In classic models in enzymology, tight-binding complexes are “slow-binding” (the result of the enzyme closing down on the substrate), where a reduction in kon is more than compensated for by a large reduction in koff. In contrast, the tight-binding mode for the Pvr-poliovirus complex is characterized by a significant increase in kon, which more than compensates for a small increase in koff (75). This suggests that the tight-binding mode for the Pvr-poliovirus complex is the result of an opening of the receptor-binding site, making it more accessible to receptor binding.
SNAPSHOTS OF THE CELL ENTRY PATHWAY
In our studies of the poliovirus cell entry pathway we have attempted to obtain structural “snapshots” of stable intermediates and to couple the structural information with the results of genetic, biophysical, and biochemical observations to fill in the gaps. The structural snapshots begin with the virus structure, which has been determined at high resolution by X-ray crystallographic methods (52), and a high-resolution structure of the empty capsid assembly intermediate (which has not yet undergone the maturation cleavage of the immature capsid protein precursor VP0) (7). These snapshots also include structures of the virus-receptor complex (10, 46, 120), the A particle (9), and the 80S particle (9), which have been determined at approximately 22Å by image reconstruction analysis of electron micrographs of samples in vitreous ice.
The Virion
The structures of the Mahoney strain of type 1 poliovirus (52) and of the closely related rhinovirus 14 (94) were first reported in 1985. Since then, structures of several other picornaviruses have been described (1, 34, 35, 41, 48, 56, 62, 65-67, 84, 85, 104, 112, 121, 122). The protein shell of the virion is composed of 60 copies of the 4 capsid proteins, VP1, VP2, VP3, and VP4, that are arranged on an icosahedral surface. The three large capsid proteins (VP1, VP2, and VP3) share a common fold (an eight-stranded beta-barrel) that is also seen in a number of other plant, insect, and animal viruses (Figure 3a). The beta-barrel cores of the capsid proteins are decorated with unique loops connecting the beta-strands, and with unique C-terminal and rather long N-terminal extensions (Figure 3b-d). The beta-barrel cores of the proteins make up the closed shell of the virus with the narrow end of VP1 packing around the fivefold axes and the narrow end of VP2 and VP3 alternating around the threefold axes (Figure 3e,f).
Figure 3.
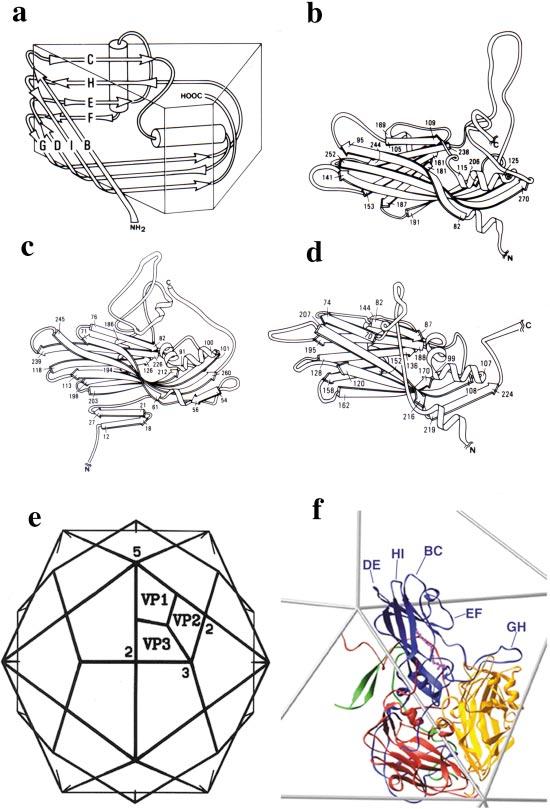
The structure of poliovirus. The virion is composed of 60 copies of 4 proteins, VP1, VP2, VP3, and VP4, arranged on an icosahedral surface. (a) A cartoon representation of the core structure eight-stranded beta-sandwich that is shared by VP1, VP2, and VP3 and the capsid proteins of a number of other icosahedral viruses. (b-d) Ribbon diagrams of VP1, VP2, and VP3 showing the common beta-sandwich core and the unique loops and terminal extensions of each of the subunits. The N-terminal extensions of VP1 and VP3 have been truncated for clarity. (e) An icosahedral framework showing the organization of VP1, VP2, and VP3 with respect to the symmetry axes of the particle. (f) A ribbon diagram representation of a single protomer arranged on a portion of the icosahedral framework, showing VP1 (blue), VP2 (yellow), VP3 (red), and VP4 (green). A fatty acid-like molecule, here modeled as a sphingosine (magenta), binds in the hydrophobic core of VP1.
THE OUTER SURFACE The shape of the structure formed by the cores alone is similar among all picornaviruses. The outer surface of the shell is decorated by the connecting loops and C-terminal extensions, which confer unique surface structure to each virus. The outer surface of the poliovirus and its closest relatives (rhinoviruses, Coxsackie A and B viruses, echoviruses, and enteroviruses) are dominated by star-shaped mesas at the fivefold axes and by a three-bladed propeller-like structure at the threefold axes (Figure 4). These prominent surface features are punctuated by depressions surrounding the fivefold axes and crossing the twofold axes. In polioviruses, related enteroviruses, and rhinoviruses the depressions surrounding the star-shaped mesa at the fivefold axes are joined to form a moat-like canyon, which has since been the site of receptor attachment for major group rhinoviruses (86), polioviruses (10, 46, 120), and Coxsackie A (119) and B viruses (47).
Figure 4.
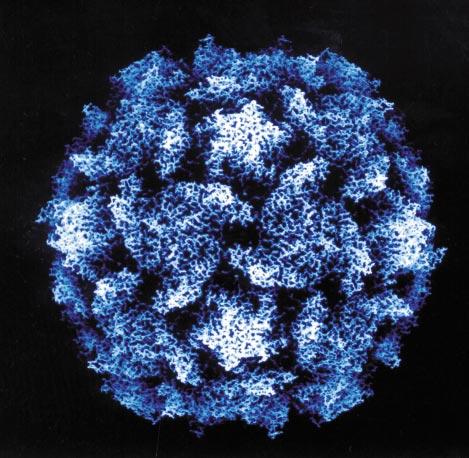
Radial depth cued view of the surface of poliovirus. The virus has been colored based on the distance of individual atoms from the center of the particle, with atoms closest to the center being dark and those furthest from the center being white. Note the star-shaped mesas at the fivefold axes, the three-bladed propellers at the threefold axes, and the deep canyon that separates the star-shaped mesas from the nearest blades of the propellers.
A HYDROPHOBIC POCKET IN VP1 At the base of the canyon there is an opening into a hydrophobic pocket in the hydrophobic core of the capsid protein VP1. In poliovirus and other enteroviruses, and in most rhinoviruses whose structures are known, the hydrophobic pocket is occupied by a fatty acid-like ligand or “pocket factor” (34) (Figure 3f). The pocket factor has been variously modeled as sphingosine, palmitate, or other shorter-chain fatty acids and is probably a mixture of fatty acid-like molecules. Genetic data suggest that the pocket factor may function to regulate the thermal stability of the virion (34, 68). The hydrophobic pocket is also the binding site for the capsid-binding antiviral agents whose antiviral activity stems from their ability to prevent the conformational changes required for cell entry (42, 102).
INTERNAL NETWORK The N-terminal extensions of VP1, VP2, and VP3 together with VP4 decorate the inner surface of the protein shell, forming an elaborate network that contributes substantially to the protein-protein interactions stabilizing the protein shell (Figures 5a and 6a,c). One particularly striking feature of this network is an interaction formed by five copies of the N terminus of VP3 as they intertwine around each of the fivefold axes to form a twisted parallel “beta-tube” (Figure 5b). This beta-tube is cradled by five copies of the myristoyl moiety at the N terminus of VP4 and is flanked on its inner surface side by five copies of a short three-stranded beta-sheet consisting of two strands from the N terminus of VP4 and one strand that is believed to represent residues from the extreme N terminus of VP1. The beta-tube forms a plug on the inner surface of the shell that blocks a solvent-filled channel (five copies of VP1 define the walls of the channel) between the five copies of VP1 as they pack around each of the fivefold axes.
Figure 5.
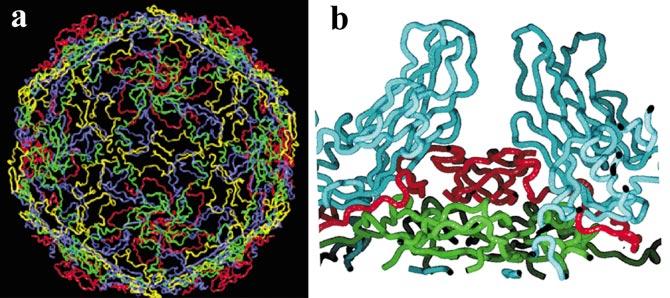
Network formed by the interaction of VP4 and the N-terminal extensions of VP1, VP2, and VP3 on the inner surface of the protein shell. (a) The network is viewed from the inside looking out. Only the VP4 (green) and N-terminal extensions of VP1 (blue), VP2 (yellow), and VP3 (red) are shown. Note the extensive interactions linking all portions of the structure. (b) A cutaway closeup of a plug formed by the N terminus of VP3 (red) and the N terminus of VP4 (green) that blocks otherwise open channels at the fivefold axes of the viral particle. In this view the inside of the virus is down and the outside is up. Two copies of VP1 flanking one such channel are shown in blue.
Figure 6.
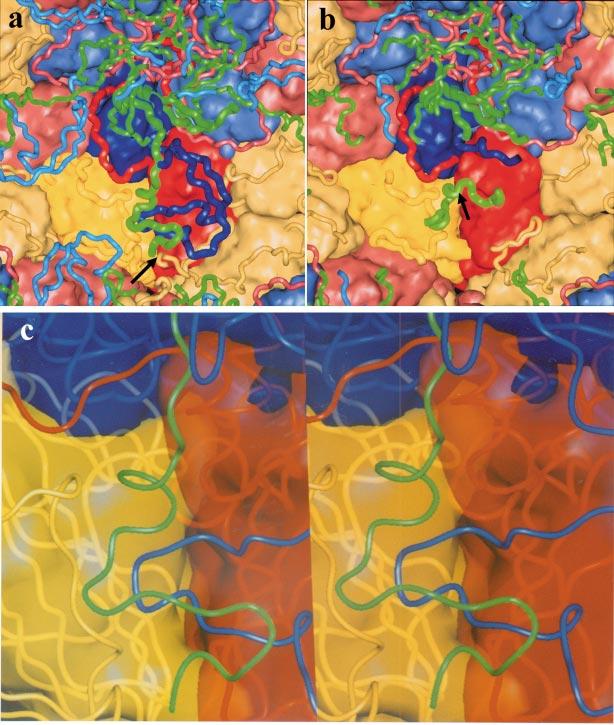
Changes on the inner network that occur upon cleavage of VP0. In these panels the view is from the inside of the particle. VP4 (green) and the N-terminal extensions of VP1 (blue), VP2 (yellow), and VP3 (red) are shown as tubes with the bodies of the subunits shown as either (a, b) opaque surfaces or (c) translucent surfaces. (a) The network in the mature virion. The N terminus of VP2 and the C terminus of VP4 (generated by the cleavage of VP0) are indicated by an arrow. (b) The network in the 75S particle prior to VP0 cleavage. The entire N-terminal extension of VP1 and the C terminus of VP4 are disordered, and the N-terminal extension of VP2 is either disordered or rearranged. Only the N-terminal extension of VP3 is intact. The peptide containing the scissile bond of VP0 (green) runs across the bottom center of the panel. The scissile bond is indicated by the arrow. Note that this peptide blocks access of the N-terminal extension of VP1 and the C terminus of VP4 from the site they occupy in the mature virion, and thus prevents the formation of the mature network. (c) A stereo view of the pocket in the inner surface where critical portions of the N terminus of VP1 and the C terminus of VP4 bind in the mature virus. Mutations in these residues suggest that the formation of this portion of the network (which extends otherwise tenuous interactions between VP2 and VP3 within a protomer) is important to viral stability.
The Empty Capsid Assembly Intermediate
THE INTERNAL NETWORK IS DISRUPTED IN THE EMPTY CAPSID An intermediate consisting of 60 copies of VP0, VP3, and VP1 and sedimenting at 75S accumulates to low levels in cells infected with some (but not all) picornaviruses. When properly isolated this particle is antigenically indistinguishable from virus and can be completely dissociated to assembly-competent 14S pentamers by treatment at slightly alkaline pH (pH 8.3). The 75S particles are unstable and are readily converted to a faster sedimenting, nondissociable particle with altered antigenicity by exposure to room temperature or extremes of ionic strength (7). Levels of the 75S empty capsid can be increased significantly by adding millimolar levels of guanidine to cells 2-3 h post infection. The guanidine is a specific inhibitor of RNA replication. The structure of the 75S empty capsid intermediate was reported in 1994 (7). As expected the structure of the outer surface and the protein shell are virtually identical to the structure of the protein shell of the virion. However, the network formed by the VP4 and the N-terminal extensions of the other capsid proteins is nearly completely disrupted (Figure 5a,b). Indeed, with the exception of the N-terminal extension of VP3 (including the beta-tube) and the N terminus of VP4, the peptide segments contributing to the internal network are either rearranged or completely disordered in the empty capsids. The failure to form the network disrupts interactions between pentamers, accounting for the ability to dissociate the capsids into pentamers. The disruption of the network also eliminates extensive contacts between VP1, VP2, and VP3 within a protomer (the copies derived from proteolysis of the capsid precursor P1) and between protomers in the pentamer. The loss of these critical interactions may explain the decreased stability of the empty capsid with respect to the virus.
A MODEL FOR AUTOCATALYTIC CLEAVAGE OF VP0 One portion of the network that is ordered in the empty capsids but in a position that is substantially different from that observed in the virus is the region spanning the scissile bond of VP0 (corresponding to the C terminus of VP4 and the N terminus of VP2 in the virion). In the virion the newly generated chain termini are located close together near the threefold axes (Figure 6a), and in both poliovirus and rhinovirus 14 there is an interaction between the hydroxyl oxygen of serine 10 of VP2 and one of the carboxylate oxygens of the C terminus of VP4. This led to the proposal that the autocatalytic cleavage of VP0 proceeds via a serine protease-like mechanism with hydroxyl of serine 10 serving as the nucleophile (4). However, mutagenesis studies revealed that the substitution of an alanine at position 10 did not affect cleavage (45). In the empty capsids the peptide segment spanning the scissile bond is located nearly 25Å away from the site occupied by the C terminus of VP4 and the N terminus of VP2 in the virion (Figure 6b). The scissile bond itself is located immediately above a segment of VP2 that contains a stretch of residues, including His 195 of VP2, that is highly conserved in all picornavirus sequences, leading to the proposal that the imidazole side chain served as a base that activates a water that then serves as the nucleophile to initiate peptide bond cleavage. Subsequent mutagenesis studies revealed that mutations of His 195 prevent VP0 cleavage and result in the production of highly unstable provirions (50).
HOW DOES VP0 CLEAVAGE TRAP THE VIRUS IN A METASTABLE STATE? The residues spanning the scissile bond pass over the top of a depression or pocket in the inner surface of the protein shell that produces a thin spot in the protein shell immediately below the interface between VP2 and VP3 from a single protomer. In this virion, this depression is filled with a loop of the N-terminal extension of VP1 [residues 44-56], which is covered on the inner surface by residues from the C terminus of VP4 (Figure 6c). Interactions of this loop of VP1 with the inner surface of VP2 and VP3 would be expected to contribute significantly to the stability of the capsid. Indeed, in the absence of the interactions involving this loop, the interactions between VP2 and VP3 from the protomer are restricted to a thin band of interactions at the outer edge of the interface joining their beta-barrel cores. Mutations in this loop of VP1, together with residues on the inner surface of the core of VP2 and residues from the C-terminal half of VP4 that contact it, cause a wide variety of phenotypes including alterations of thermal stability (34, 69), resistance to (and in some cases dependence on) capsid-binding drugs (83), mouse adaptation (25, 26, 81), and ability of the virus to grow in cells expressing mutant receptors (24). This suggests that these residues play an important role in regulating structural transitions of the virus and in cell entry. In the empty capsid structure the residues spanning the scissile bond block the N terminus of VP1 from accessing the site in the pocket that it occupies in the mature virion, suggesting that cleavage of VP0 is required for the completion of this portion of the network. This leads to a model in which the cleavage and subsequent rearrangement of the VP4 and the N-terminal extensions of VP1 and VP2 are responsible for locking the virus in the metastable state. The observation that some of the last segments of the network to be put in place (namely VP4 and the N terminus of VP1) are externalized reversibly when the virus “breathes” and irreversibly in receptor-mediated conformational rearrangements early in the entry process suggests that the cleavage and reorganization may also prime the virus for conformational changes required for cell entry. Indeed, analysis of thermal-induced conformational changes in the empty capsid demonstrates that exposure of sequences corresponding to VP4 cannot take place unless VP0 is cleaved (6). We have drawn a comparison of this process to the process of setting a mousetrap.
The Virus-Receptor Complex
THE CAM-LIKE RECEPTORS BIND IN THE CANYON The structures of picornavirus-receptor complexes have been solved at low resolution (15-25 Å) by image reconstruction analysis of cryo-electron micrographs. The complexes include complexes of rhinovirus 14 (59, 86), rhinovirus 16 (59), and Coxsackievirus A12 (119) with ICAM-1 (the receptor for the “major group” rhinoviruses), the complex of poliovirus 1 (Mahoney) with Pvr (10, 46, 120), the structure of the complex of Coxsackievirus B3 with CAR (Coxsackie adeno receptor) (47), and the structure of the complex of rhinovirus 2 with the VLDL receptor (the receptor for the “minor group” rhinoviruses) (49). The ectodomains of ICAM-1, Pvr, and CAR are CAM-like molecules with 5, 3, and 2 Ig-like domains in their ectodomains respectively. All three CAM-like receptors (Pvr, ICAM-1, and CAR) bind in the canyon of their cognate viruses, but the footprints of the receptors on the virus surface and the geometry of approach of the receptor to the virus surface differ greatly. Only the N-terminal (membrane distal) domain of the receptor contacts the virus surface. The area of the footprint of the Pvr on the poliovirus surface is considerably greater than the footprint of ICAM-1 on rhinovirus, which may account in part for the observation that the affinity of soluble Pvr for poliovirus is higher than the affinity of soluble ICAM-1 for rhinoviruses. Because there is clear density for all three domains of Pvr in the reconstruction of the Pvr-poliovirus complex, it has been possible to model how the complex would orient the virus in the context of the membrane (Figure 7c). If we assume that multiple receptors attach to the virus, the complex would result in the close approach of a particle fivefold axis to the membrane, which might facilitate insertion of multiple copies of VP4 and the N terminus of VP1 into the membrane upon conversion to the A particle.
Figure 7.
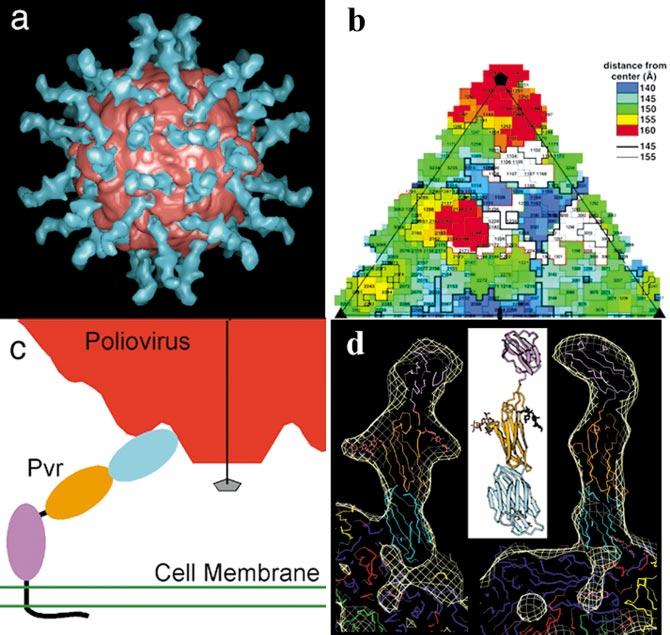
The virus-receptor complex. (a) Image reconstruction of the complex between poliovirus and its receptor Pvr. The portion of the reconstruction belonging to the virus is colored red, the remaining surface, which represents the receptor, is blue. There is compelling density for all three Ig-like domains of the receptor. Prominent arms on the density of the middle domain correspond to glycosylation sites. (b) A “roadmap” representation of the footprint of Pvr on the viral surface. A single icosahedral asymmetric unit is shown. The roadmap is color-coded by the distance of residues from the center of the particle with residues closest to the center in blue and those farthest from the center in red. The footprint is in white.(c) A cartoon showing how receptor binding would orient the virus with respect to the cell membrane. Note that the geometry of receptor binding observed in the complex would bring a particle fivefold axis (line with pentagon) in close proximity to the cell membrane. (d) A homology model for Pvr and the atomic model for the virus have been fit to the image reconstruction. Note the good fit to the density, including the fit of a model for glycosylation sites on the second domain to the prominent arms of the receptor reconstruction.
The reconstructions have been used to develop pseudoatomic models for the virus-receptor complexes by fitting the model of the virus derived from high-resolution crystallographic studies and crystallographic (ICAM-1 and CAR) or homology (Pvr) models of the receptor to the reconstruction density (Figure 7c). There is no evidence for significant conformational changes in the virus upon receptor attachment for any of these viruses.
NOT ALL RECEPTORS BIND IN THE CANYON The structure of the rhinovirus 2-LDL receptor complex differs significantly from the complexes with the CAM-like receptors. The LDL receptor does not bind in the canyon. Instead, it binds to sites at the top of the star-shaped mesas at the fivefold axes, interacting primarily with residues from the BC and the HI loop of VP1 (49). These loops are components of one of the major antigenic sites of rhinoviruses. Even prior to structural studies, the canyon had been predicted to be the most probable site of receptor attachment for rhinoviruses and poliovirus. The original rationale for the “Canyon Hypothesis” was that the receptor-binding site of the virus needed to be inaccessible to the antigen-binding sites of antibodies to prevent selection of mutants incapable of binding receptor under the pressure of a vigorous immune response (94). The underlying logic of the hypothesis has been questioned on theoretical grounds based on the premise that RNA viruses sample all possible mutations, but receptor-binding mutants are nonviable and could therefore never be fixed regardless of the selective pressure. The logic had also been challenged by two earlier experimental observations: (a) Smith and colleagues showed that the binding site of an antibody to one of the primary antigenic sites of rhinovirus 14 overlaps extensively with the ICAM-1-binding site (101, 103). (b) Stuart and colleagues showed that both the RGD sequence that is the presumed binding site for the integrin receptor for FMDV (1) and a site that is used by some tissue culture adapted strains of FMDV as a binding site for cell surface heparin sulfate (37) are highly exposed on the surface of the virus in areas that have been identified as antigenic sites.
THE “CANYON HYPOTHESIS” REVISITED So why is it that the receptor-binding site for poliovirus and for major group rhinovirus is located in a deep invagination in the virus surface, whereas the receptor-binding sites for minor group rhinoviruses and for FMDV are located on highly exposed surfaces? The answer may rest in differences in the roles played by the receptors for these different viruses. Both rhinovirus 2 (91) and FMDV require acidification for productive cell entry, and the receptor for these viruses seems to play only one role, namely to increase the concentration of virus at the surface of the target cell. In contrast, in poliovirus, Coxsackie A and B viruses, and the major group rhinoviruses, the receptor must also trigger conformational changes that are necessary for subsequent events in entry. In order to induce the changes the receptor binding must provide sufficient energy to overcome the kinetic barrier that holds the virus in its native conformation. Thus, the receptor must donate some of its binding energy to facilitate structural rearrangements. To a first approximation the binding energy of a protein-protein interaction is proportional to the surface area that is buried in the interface between the proteins. Thus, one would anticipate that a virus whose receptor served the dual function of binding protein and “lever” to facilitate conformational change would have a larger area of contact with its receptor than a virus whose receptor served only as a binding protein. Locating the receptor-binding site in a deep invagination in the virus surface provides an efficient mechanism for increasing the surface area of the virus-receptor interaction and thus the binding energy available to do the work of initiating conformational changes required for subsequent steps in cell entry.
The A Particle and 80S Particle: Two Cell Entry Intermediates
STRUCTURES DERIVED FROM CRYO-ELECTRON MICROSCOPY Although we have obtained crystals of the A particle and the 80S empty particle, these crystals have not yet proved suitable for high-resolution studies. However, the structures have been solved at 22-23Å resolution by cryo-electron microscopy (9). The three-dimensional reconstructions for the A particle and 80S particle differ significantly from either a low-resolution surface generated from the high-resolution model of the virion or a reconstruction of the virion at comparable resolution. Models for the coat protein subunits have been docked into the resulting reconstructions and refined using methods that are analogous to the methods used to refine high-resolution crystallographic structures (Figure 8). Due to limited resolution, the modeling and refinement have assumed that the individual capsid proteins move as rigid bodies (a form of molecular tectonics). Despite this approximation, the models exhibit a remarkable fit to the reconstruction density, with the exception of several large loops (e.g., the GH loop of VP1), VP4, and the N-terminal extension of VP1, which are reorganized or externalized in the altered particles.
Figure 8.
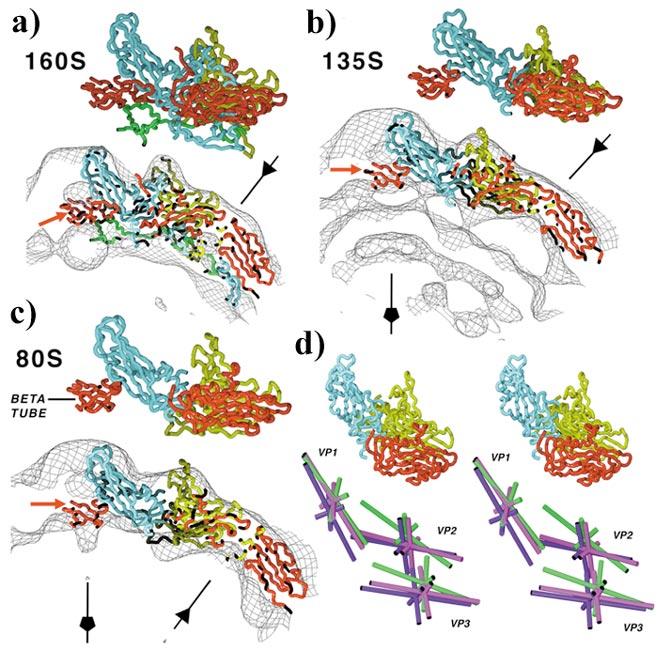
The structures of cell entry intermediates. (a) Models for the virus, (b) the A or 135S particle, and (c) the 80S empty particle have been fit to the reconstruction density, by treating the cores of VP1 (blue), VP2 (yellow), and VP3 (red) as rigid bodies. Note that all three reconstructions contain clear density for the plug formed by the VP3 beta-tube (red, indicated by red arrow). The particle fivefold axes (line with pentagon) and threefold axes (line with triangle) are indicated in each panel. (d) Stereo representations of simplified models for VP1, VP2, and VP3 in the virion (purple), the A particle (green), and the 80S particle (magenta), showing the movement of the proteins during the structural transitions. Umbrella-like motions of the subunits with VP1 pivoting about the fivefold axes and VP2 and VP3 pivoting about the threefold axes produce a flattened appearance of the model in the A or (b) 135S particle. This flattening results in a more angular appearance to the A particle than is readily apparent in reconstruction and in the original micrographs. Reproduced from (9) with permission.
THE STRUCTURAL CHANGES IN THE A PARTICLE Comparison of the models suggests that the virion-to-A particle transition is characterized by shifts in all three major capsid proteins (VP1, VP2, and VP3). In the model for the A particle, the subunits of VP1 have undergone a movement similar to the opening of an umbrella, with the tips of the subunits at the fivefold axes serving as a pivot and the wide end of the subunit (which forms the north wall and part of the base of the canyon) moving radially outward (Figure 8d). This pivoting about the residues near the fivefold axes may provide an explanation for previous observations that viruses with mutations in residues in the BC, GH, and DE loops of VP1 often have phenotypes that include alterations in their ability to undergo thermal- and receptor-mediated conformational changes (24, 73, 74, 116). VP2 and VP3 undergo a similar movement, with the narrow ends of the subunits pivoting about the threefold axes and the wide ends pivoting outward radially. VP1, VP2, and VP3 also undergo significant tangential reorientation (in the plane of the virus surface), with the tangential shifts of VP2 being largest. Because of the umbrella-like motions, the base of the canyon is moved outward appreciably, giving the A particle a more angular appearance, especially when viewed down the twofold axes. In this view the models, the reconstructions, and even the micrographs take on a distinctly hexagonal appearance. This is in sharp contrast to the virion, which appears roughly spherical in all views. The movement of VP1 also results in opening gaps at the base of the canyon between fivefold-related copies of VP1. The separation is also apparent in the reconstructions (the density is noticeably thinner) but is not sufficient to result in the appearance of “holes” in the density at 22Å resolution.
THE STRUCTURAL CHANGES ARE PARTIALLY REVERSED IN THE 80S PARTICLE In the models for the 80s particle the subunits have undergone a partial reversal of the structural rearrangements that characterized the virion-to-A particle transition (Figure 8c). Indeed the orientations of VP2 and VP3 in the 80S particle are similar to their orientations in the virion. In contrast the orientation of VP1 remains similar to that seen in the A particle.
WHERE DO VP4 AND THE N TERMINUS OF VP1 EXIT THE VIRION? Based on analogies to several other viruses, Rossmann and colleagues have proposed that during the N-to-A transition, VP4 and the N terminus of VP1 exit the virion via channels at the fivefold axes (44, 82, 95). The model is attractive because it would place five copies of the N terminus of VP1 (which is predicted to form an amphipathic helix) in a position where they could interact and insert into the membrane to form a channel. However, as discussed above, this channel is blocked at its base on the inside surface of the virion by a “plug” consisting of a beta-tube formed by five copies of the N terminus of VP3 interdigitating around the fivefold axis. In all three reconstructions (virion, A particle, and 80S particle) there is clear density for the VP3 plug occluding the channel (Figure 8a-c). More importantly, there is simply no room in the channel in either the A particle or the 80S particle reconstruction for five copies of the part N terminus of VP1, which must remain after the extreme N-terminal segment has been externalized (Figure 8b,c).
An alternative model for the egress of VP4 and the N terminus of VP1 has been proposed based on an analogy with the expansion of several structurally related plant viruses. These viruses undergo a significant (10%) expansion when exposed to slightly basic pH in the presence of chelators ˜ of divalent cations (54). The structure of the expanded state of TBSV has been solved at 7Å by X-ray crystallography (93), and the structure of the expanded state of CCMV has been solved at 25Å by cryo-electron microscopy (106). In both structures, expansion is characterized by a coordinated rotation and outward movement of the subunits analogous to the movements of VP2 and VP3 along the threefold axes, and by a rotation and outward movement of the subunits analogous to the movement VP1 along the fivefold axes. These coordinated movements result in the opening of large holes at the interfaces between the capsid protein subunits, at positions structurally analogous to the base of the canyon in the picornaviruses. In the case of TBSV, the N-terminal domain of two of the capsid protein subunits exits through this pore upon expansion (93). The extrusion of the N-terminal domain is reversible if the particles are “annealed” by gradually lowering the pH, but the N terminus is trapped outside if the particle is “quenched” by rapid acidification (39).
Mutations in the interfaces of poliovirus that are analogous to the interfaces disrupted during expansion of the plant viruses have been shown to affect viral stability (34, 69) and to alter the sensitivity of the virus to neutralization by soluble receptor (23). Although the capsid protein movements are less pronounced in the virion-to-A particle transition of poliovirus than in the expansion of the plant viruses, the umbrella-like movements of VP1, VP2, and VP3 do result in significant gaps between the subunits at the base of the canyon in positions. These gaps are in positions that are entirely analogous to the positions of larger openings seen in the plant viruses. Inspection of the reconstruction density near the gaps (which are located right where the N-terminal extension of VP1 leaves the beta-barrel core and enters the interior of the virion) suggests that the N-terminal extensions of VP1 may exit through these gaps and proceed up the outer surfaces of the mesas at the fivefold axes. Again this model is attractive, as it would locate five copies of the presumed amphipathic helix at the N terminus of VP1 close together such that they could form a fivefold helical bundle once inserted into the membrane.
ARE THERE ADDITIONAL INTERMEDIATES? One of the more notable features of the reconstructions of the A particle and the 80S particle is the lack of openings in the viral surface that would be sufficiently large to allow the facile extrusion of VP4 and the N-terminal extension of VP1 in the N-to-A particle transition or the RNA in the A particle-to-80S particle transition. Indeed, the relatively small extent of the expansion that occurs during the N-to-A particle transition was a surprise. Given the rather large shift in sedimentation coefficient (135S for the A particle versus 160S for the virion), we had anticipated a more significant expansion (on the order of 10%-15%). We therefore postulate that there are additional, as yet undetected, intermediates in the pathway located between the virion and the A particle and the A particle and the 80S particle. In these intermediates we propose that the particles would be sufficiently expanded to create openings for the release of the VP4 and the N terminus of VP1 in the N-to-A particle transition and of RNA in the A particle-to-80S particle transition. The failure to detect these intermediates to date may indicate that they are transient. The additional intermediate in the N-to-A pathway would then be analogous to the expanded form of the plant virus, and the A particle itself would correspond to the quenched form of the expanded plant virus in which the N-terminal arm is trapped outside the particle. The transient and reversible externalization of the N terminus of VP1 that occurs when the virus breathes may indicate a reversible equilibrium between the virion and the new intermediate that is analogous to the expansion and slow acidification transition that returns the N-terminal arm to the inside of the particle in the plant viruses. Efforts to identify conditions that would allow these putative additional intermediates to be isolated and characterized are in progress.
HOW DOES THE RNA EXIT THE VIRION? The nature of the intermediate linking the A particle and the 80S particle is less clear. Indeed, there are significant questions that remain to be answered concerning how the viral RNA is released from the particle. Nonetheless, it is clear that some factor must dictate that the RNA be released from a unique site in an otherwise icosahedrally symmetric particle, lest the RNA and protein shell be tied in an inextricable knot. In the course of a natural infection, one factor that could influence which site is chosen for RNA release could be the presence of membrane near one surface of the virus. However, the presence of membrane is not a necessary factor because the RNA is efficiently released in the absence of membrane in the in vitro (thermal-mediated) conversion. Other factors that could regulate which site is used, among the many otherwise equivalent sites, could include steric factors (e.g., the proximity of a specific structure in the viral RNA, including perhaps the genome-linked protein VPg) or kinetic factors (e.g., a slow initiation, followed by rapid release at the first initiation site that is established). Because the structures observed to date are symmetric, they can tell us little about the site of release. Indeed, the only clues from the structures that may indicate the site of release are differences in the density of the RNA in the reconstructions of the virion and the A particle. The density in the interior region of the virion provides a view of the icosahedrally averaged structure of the linear genome. The appearance of the average RNA structure differs significantly in the virion and the A particle, with the RNA making a significantly closer approach to the fivefold axes in the A particle.
A WORKING MODEL FOR POLIOVIRUS CELL ENTRY
Based on a combination of the structural, genetic, and biochemical evidence available to date we propose a working model for the cell entry of poliovirus, related enteroviruses, and major group rhinoviruses (Figure 9). The first step in the entry pathway is the formation of an initial binding complex with the receptor (Figure 9a). The formation would require no significant conformational alterations in either the virus or the receptor and would be the predominant form of the complex at low temperatures. We would propose that the published structures of the complexes represent this form of the complex. At more physiological temperatures the formation of this initial complex induces conformational changes in the virus (and perhaps the receptor) to form a tight-binding complex (Figure 9b).
Figure 9.
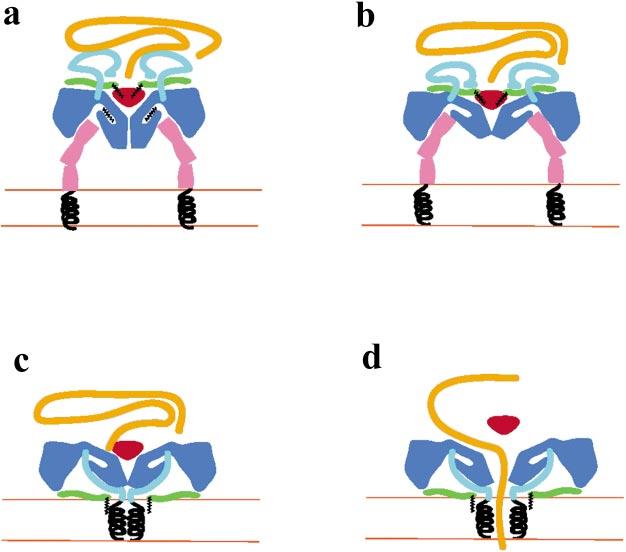
A cartoon representation of a working model for the cell entry mechanism of polio- and related viruses. (a) The virus attaches to the receptor on the membrane of a cell to form an initial complex. The three domains of the receptor are shown in pink with the transmembrane helices (black) anchored in the membrane. The body of the virus is in blue, with the pocket factor in black. The N-terminal extension of VP1 is cyan, the VP3 plug is red, VP4 is green, the myristate is black, and the viral RNA is orange. (b) At physiological temperatures the receptor induces a subtle conformational change that opens the receptor-binding site and presumably displaces the pocket factor to produce a tight-binding complex. (c) The virus undergoes conformational rearrangements that result in the insertion of the amphipathic helices of the N terminus of VP1 (black spirals) and the myristate group of VP4 into the membrane to form a channel. (d) A trigger (perhaps fluxes in Ca+2 concentration) results in the expansion of the particle, the expansion of the pore to form a channel, the temporary removal of the VP3 plug, and the release of the viral RNA into the cell.
Rossmann et al. (95) have proposed a model for the tight-binding complex in which the receptor is initially bound with most of the contacts between virus and receptor involving the “south wall” of the canyon. Subsequent structural changes in the virus cause the VP1 to move away from the fivefold axes, pinching the canyon such that the receptor makes bridging contacts with both the north and south walls of the canyon. In this model the pinching motion results in the opening of the channels at the fivefold axes. However, because the conformational alteration results in more extensive contacts with the receptor in a more “closed contact,” this model would predict that the tight-binding complex be characterized by a slower on-rate and a much slower off-rate for the receptor. This prediction is contradicted by kinetic data for both poliovirus/Pvr and rhinovirus/ICAM-1 binding, which show that the tight-binding complex is characterized instead by a faster off-rate, and a much faster on-rate (19, 75, 120). The Rossmann model also predicts that VP4 and the N terminus of VP1 are released through the newly opened fivefold channel, which is inconsistent with the structure of the A particle.
We propose an alternative model in which the transition from the initial binding complex to the tight-binding complex is characterized by movements of VP1, VP2, and VP3 that mimic the umbrella-like movements of the virion-to-A particle transition (Figure 9b). Consistent with the kinetic data, this would open the receptor-binding site, providing for a faster association rate for binding additional receptors. This tight-binding state is detectable at room temperature, but it may be transient at physiological temperature. At physiological temperatures binding of multiple receptors destabilizes the particles, resulting in the release of the pocket factor as the particle begins to expand to form the first transient state. This expansion permits the externalization of VP1 and the N terminus of VP4. Once these peptide segments are externalized, the particle undergoes a partial reversal of the expansion to form the A particle. At some time in this process, five copies of the N terminus of VP1 insert into the membrane by a mechanism that may be facilitated by the myristoyl group at the N terminus of VP4 (Figure 9c). The membrane-associated portions of VP1 then associate to form a channel composed of five amphipathic helices, each with its hydrophobic surface facing the membrane and its hydrophilic surface facing the interior of the channel (Figure 9c). At some point later in the entry process, some as yet unknown trigger results in a second round of expansion and RNA release. We would suggest that in this expansion the wide ends of VP1, VP2, and VP3 may serve as the pivot points for an umbrella-like movement of VP1 away from the fivefold axes. These movements would be coupled with a rotation of the five amphipathic helices, and a movement of the internal plug formed by the N termini of VP3 to form a large channel through which RNA is released (Figure 9d). Because the A particle does not have an appreciable affinity for the receptor, we feel that it is unlikely that the receptor plays any specific role in triggering this expansion. Although the trigger has not yet been identified, the observation that the 135S particle spontaneously converts to 80S particles in the absence of millimolar levels of calcium (115) suggests that changes in calcium concentration may play a role in triggering expansion and RNA release. After the RNA has been released, the VP3 plug is reinserted, the pore closes, and the particle shrinks to form the 80S structure.
ACKNOWLEDGMENTS
The author would like to acknowledge the contributions of the students and postdoctoral fellows whose works are cited in this review, the support of NIH (AI20566), and the collaborators who have contributed significantly to the ideas presented, including Dave Filman, Marie Chow, Jeff Bergelson, Vincent Racaniello, and Alasdair Steven. I would particularly like to acknowledge the contributions of Don Wiley, a teacher and colleague whose insights have shaped the way we all think about viruses and their interactions with cells.
LITERATURE CITED
- 1.Acharya R, Fry E, Stuart D, Fox G, Rowlands D, Brown F. The three-dimensional structure of foot-and-mouth disease virus at 2.9Å resolution. Nature. 1989;337:709–16. doi: 10.1038/337709a0. [DOI] [PubMed] [Google Scholar]
- 2.Aoki J, Koike S, Ise I, Sato-Yoshida Y, Nomoto A. Amino acid residues on human poliovirus receptor involved in interaction with poliovirus. J. Biol. Chem. 1994;269:8431–38. [PubMed] [Google Scholar]
- 3.Arita M, Koike S, Aoki J, Horie H, Nomoto A. Interaction of poliovirus with its purified receptor and conformational alteration in the virion. J. Virol. 1998;72:3578–86. doi: 10.1128/jvi.72.5.3578-3586.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Arnold E, Luo M, Vriend G, Rossmann MG, Palmenberg AC, et al. Implications of the picornavirus capsid structure for polyprotein processing. Proc. Natl. Acad. Sci. USA. 1987;84:21–25. doi: 10.1073/pnas.84.1.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Baker KA, Dutch RE, Lamb RA, Jardetzky TS. Structural basis for paramyxovirus-mediated membrane fusion. Mol. Cell. 1999;3:309–19. doi: 10.1016/s1097-2765(00)80458-x. [DOI] [PubMed] [Google Scholar]
- 6.Basavappa R, Gomez-Yafal A, Hogle JM. The poliovirus empty capsid specif!ically recognizes the poliovirus receptor and undergoes some, but not all, of the transitions associated with cell entry. J. Virol. 1998;72:7551–56. doi: 10.1128/jvi.72.9.7551-7556.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Basavappa R, Syed R, Flore O, Icenogle JP, Filman DJ, Hogle JM. Role and mechanism of the maturation cleavage of VP0 in poliovirus assembly: structure of the empty capsid assembly intermediate at 2.9 Å resolution. Protein Sci. 1994;3:1651–69. doi: 10.1002/pro.5560031005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bayer N, Prchla E, Schwab M, Blaas D, Fuchs R. Human rhinovirus HRV14 uncoats from early endosomes in the presence of bafilomycin. FEBS Lett. 1999;463:175–78. doi: 10.1016/s0014-5793(99)01610-5. [DOI] [PubMed] [Google Scholar]
- 9.Belnap DM, Filman DJ, Trus BL, Cheng N, Booy FP, et al. Molecular tectonic model of virus structural transitions: the putative cell entry states of poliovirus. J. Virol. 2000;74:1342–54. doi: 10.1128/jvi.74.3.1342-1354.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Belnap DM, McDermott BM, Jr, Filman DJ, Cheng N, Trus BL, et al. Three-dimensional structure of poliovirus receptor bound to poliovirus. Proc. Natl. Acad. Sci. USA. 2000;97:73–78. doi: 10.1073/pnas.97.1.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bergelson JM, Chan M, Solomon KR, St. John NF, Lin H, Finberg RW. Decay-accelerating factor (CD55), a glycosylphosphatidylinositol-anchored complement regulatory protein, is a receptor for several echoviruses. Proc. Natl. Acad. Sci. USA. 1994;91:6245–48. doi: 10.1073/pnas.91.13.6245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bergelson JM, Cunningham JA, Droguett G, Kurt-Jones EA, Krithivas A, et al. Isolation of a common receptor for coxsackie B viruses and adenoviruses 2 and 5. Science. 1997;275:1320–23. doi: 10.1126/science.275.5304.1320. [DOI] [PubMed] [Google Scholar]
- 13.Bergelson JM, Krithivas A, Celi L, Droguett G, Horwitz MS, et al. The murine CAR homolog is a receptor for coxsackie B viruses and adenoviruses. J. Virol. 1998;72:415–19. doi: 10.1128/jvi.72.1.415-419.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bergelson JM, Shepley MP, Chan BMC, Hemler ME, Finberg RW. Identification of the integrin VLA-2 as a receptor for echovirus 1. Science. 1995;255:1718–20. doi: 10.1126/science.1553561. [DOI] [PubMed] [Google Scholar]
- 15.Bernhardt G, Harber J, Zibert A, De-Crombrugghe M, Wimmer E. The poliovirus receptor: identification of domains and amino acid residues critical for virus binding. Virology. 1994;203:344–56. doi: 10.1006/viro.1994.1493. [DOI] [PubMed] [Google Scholar]
- 16.Bothner B, Dong XF, Bibbs L, Johnson JE, Siuzdak G. Evidence of viral capsid dynamics using limited proteolysis and mass spectrometry. J. Biol. Chem. 1998;273:673–76. doi: 10.1074/jbc.273.2.673. [DOI] [PubMed] [Google Scholar]
- 17.Bouchard MJ, Dong Y, McDermott BM, Jr, Lam DH, Brown KR, et al. Defects in nuclear and cytoskeletal morphology and mitochondrial localization in spermatozoa of mice lacking nectin-2, a component of cell-cell adherens junctions. Mol. Cell Biol. 2000;20:2865–73. doi: 10.1128/mcb.20.8.2865-2873.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bullough PA, Hughson FM, Skehel JJ, Wiley DC. Structure of influenza haemagglutinin at the pH of membrane fusion. Nature. 1994;371:37–43. doi: 10.1038/371037a0. [DOI] [PubMed] [Google Scholar]
- 19.Casasnovas JM, Springer TA. Kinetics and thermodynamics of virus binding to receptor. Studies with rhinovirus, intercellular adhesion molecule-1 (ICAM-1), and surface plasmon resonance. J. Biol. Chem. 1995;270:13216–24. doi: 10.1074/jbc.270.22.13216. [DOI] [PubMed] [Google Scholar]
- 20.Chan DC, Fass D, Berger JM, Kim PS. Core structure of gp41 from the HIV envelope glycoprotein. Cell. 1997;89:263–73. doi: 10.1016/s0092-8674(00)80205-6. [DOI] [PubMed] [Google Scholar]
- 21.Chen J, Lee KH, Steinhauer DA, Stevens DJ, Skehel JJ, Wiley DC. Structure of the hemagglutinin precursor cleavage site, a determinant of influenza pathogenicity and the origin of the labile conformation. Cell. 1998;95:409–17. doi: 10.1016/s0092-8674(00)81771-7. [DOI] [PubMed] [Google Scholar]
- 22.Chow M, Newman JFE, Filman D, Hogle JM, Rowlands DJ, Brown F. Myristylation of picornavirus capsid protein VP4 and its structural significance. Nature. 1987;327:482–86. doi: 10.1038/327482a0. [DOI] [PubMed] [Google Scholar]
- 23.Colston E, Racaniello VR. Soluble receptor-resistant poliovirus mutants identify surface and internal capsid residues that control interaction with the cell receptor. EMBO J. 1994;13:5855–62. doi: 10.1002/j.1460-2075.1994.tb06930.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Colston EM, Racaniello VR. Poliovirus variants selected on mutant receptor-expressing cells identify capsid residues that expand receptor recognition. J. Virol. 1995;69:4823–29. doi: 10.1128/jvi.69.8.4823-4829.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Couderc T, Guédo N, Calvez V, Pelletier I, Hogle J, et al. Substitutions in the capsids of poliovirus mutants selected in human neuroblastoma cells confer on the Mahoney type 1 strain a phenotype neurovirulent in mice. J. Virol. 1994;68:8386–91. doi: 10.1128/jvi.68.12.8386-8391.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Couderc T, Hogle J, Le Blay H, Horaud F, Blondel B. Molecular characterization of mouse-virulent poliovirus type 1 Mahoney mutants: involvement of residues of polypeptides VP1 and VP2 located on the inner surface of the capsid protein shell. J. Virol. 1993;67:3808–17. doi: 10.1128/jvi.67.7.3808-3817.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Curry S, Chow M, Hogle JM. The poliovirus 135S particle is infectious. J. Virol. 1996;70:7125–31. doi: 10.1128/jvi.70.10.7125-7131.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Damke H, Baba T, Warnock DE, Schmid SL. Induction of mutant dynamin specifically blocks endocytic coated vesicle formation. J. Cell Biol. 1994;127:915–34. doi: 10.1083/jcb.127.4.915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.De Sena J, Mandel B. Studies on the in vitro uncoating of poliovirus. II. Characteristics of the membrane-modified particle. Virology. 1977;78:554–66. doi: 10.1016/0042-6822(77)90130-1. [DOI] [PubMed] [Google Scholar]
- 30.DeTulleo L, Kirchhausen T. The clathrin endocytic pathway in viral infection. EMBO J. 1998;17:4585–93. doi: 10.1093/emboj/17.16.4585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dove AW, Racaniello VR. Cold-adapted poliovirus mutants bypass a post-entry replication block. J. Virol. 1997;71:4728–35. doi: 10.1128/jvi.71.6.4728-4735.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fass D, Harrison SC, Kim PS. Retrovirus envelope domain at 1.7 angstrom resolution. Nat. Struct. Biol. 1996;3:465–69. doi: 10.1038/nsb0596-465. [DOI] [PubMed] [Google Scholar]
- 33.Fenwick ML, Cooper PD. Early interactions between poliovirus and ERK cells. Some observations on the nature and significance of the rejected particles. Virology. 1962;18:212–23. doi: 10.1016/0042-6822(62)90007-7. [DOI] [PubMed] [Google Scholar]
- 34.Filman DJ, Syed R, Chow M, Macadam AJ, Minor PD, Hogle JM. Structural factors that control conformational transitions and serotype specificity in type 3 poliovirus. EMBO J. 1989;8:1567–79. doi: 10.1002/j.1460-2075.1989.tb03541.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Filman DJ, Wien MW, Cunningham JA, Bergelson JM, Hogle JM. Structure determination of echovirus 1. Acta Crystallogr. D. 1998;54:1261–72. doi: 10.1107/s0907444998002790. [DOI] [PubMed] [Google Scholar]
- 36.Fricks CE, Hogle JM. Cell-induced conformational change of poliovirus: Externalization of the amino terminus of VP1 is responsible for liposome binding. J. Virol. 1990;64:1934–45. doi: 10.1128/jvi.64.5.1934-1945.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fry EE, Lea SM, Jackson T, Newman JW, Ellard FM, et al. The structure and function of a foot-and-mouth disease virus-oligosaccharide receptor complex. EMBO J. 1999;18:543–54. doi: 10.1093/emboj/18.3.543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Geraghty RJ, Krummenacher C, Cohen GH, Eisenberg RJ, Spear PG. Entry of alphaherpesviruses mediated by poliovirus receptor-related protein 1 and poliovirus receptor. Science. 1998;280:1618–20. doi: 10.1126/science.280.5369.1618. [DOI] [PubMed] [Google Scholar]
- 39.Golden J, Harrison SC. Proteolytic dissection of turnip crinkle virus subunit in solution. Biochemistry. 1982;21:3862–66. doi: 10.1021/bi00259a022. [DOI] [PubMed] [Google Scholar]
- 40.Gomez Yafal A, Kaplan G, Racaniello VR, Hogle JM. Characterization of poliovirus conformational alteration mediated by soluble cell receptors. Virology. 1993;197:501–5. doi: 10.1006/viro.1993.1621. [DOI] [PubMed] [Google Scholar]
- 41.Grant RA, Filman DJ, Fujinami RS, Icenogle JP, Hogle JM. Three-dimensional structure of Theiler virus. Proc. Natl. Acad. Sci. USA. 1992;89:2061–65. doi: 10.1073/pnas.89.6.2061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Grant RA, Hiremath C, Filman DJ, Syed R, Andries K, Hogle JM. Structures of poliovirus complexes with antiviral drugs: implications for viral stability and drug design. Curr. Biol. 1994;4:784–97. doi: 10.1016/s0960-9822(00)00176-7. [DOI] [PubMed] [Google Scholar]
- 43.Greve JM, Davis G, Meyer AM, Forte CP, Yost SC, et al. The major human rhinovirus receptor is ICAM-1. Cell. 1989;56:839–47. doi: 10.1016/0092-8674(89)90688-0. [DOI] [PubMed] [Google Scholar]
- 44.Hadfield AT, Lee W-M, Zhao R, Oliveira MA, Minor I, et al. The refined structure of human rhinovirus 16 at 2.15Å resolution: implications for the viral life cycle. Structure. 1997;5:427–41. doi: 10.1016/s0969-2126(97)00199-8. [DOI] [PubMed] [Google Scholar]
- 45.Harber JJ, Bradley J, Anderson CW, Wimmer E. Catalysis of poliovirus VP0 maturation cleavage is not mediated by serine 10 of VP2. J. Virol. 1991;65:326–34. doi: 10.1128/jvi.65.1.326-334.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.He Y, Bowman VD, Mueller S, Bator CM, Bella J, et al. Interaction of the poliovirus receptor with poliovirus. Proc. Natl. Acad. Sci. USA. 2000;97:79–84. doi: 10.1073/pnas.97.1.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.He Y, Chipman PR, Howitt J, Bator CM, Whitt MA, et al. Interaction of coxsackievirus B3 with the full length coxsackievirus-adenovirus receptor. Nat. Struct. Biol. 2001;8:874–78. doi: 10.1038/nsb1001-874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hendry E, Hatanaka H, Fry E, Smyth M, Tate J, et al. The crystal structure of coxsackievirus A9: new insights into the uncoating mechanisms of enteroviruses. Struct. Fold Des. 1999;7:1527–38. doi: 10.1016/s0969-2126(00)88343-4. [DOI] [PubMed] [Google Scholar]
- 49.Hewat EA, Neumann E, Conway JF, Moser R, Ronacher B, et al. The cellular receptor to human rhinovirus 2 binds around the 5-fold axis and not in the canyon: a structural view. EMBO J. 2000;19:6317–25. doi: 10.1093/emboj/19.23.6317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hindiyeh M, Li QH, Basavappa R, Hogle JM, Chow M. Poliovirus mutants at histidine 195 of VP2 do not cleave VP0 into VP2 and VP4. J. Virol. 1999;73:9072–79. doi: 10.1128/jvi.73.11.9072-9079.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hofer F, Gruenberger M, Kowalski H, Machat H, Huettinger M, et al. Members of the low density lipoprotein receptor family mediate cell entry of a minor-group common cold virus. Proc. Natl. Acad. Sci. USA. 1994;91:1839–42. doi: 10.1073/pnas.91.5.1839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hogle JM, Chow M, Filman DJ. Three-dimensional structure of poliovirus at 2.9Å resolution. Science. 1985;229:1358–65. doi: 10.1126/science.2994218. [DOI] [PubMed] [Google Scholar]
- 53.Huang Y, Hogle JM, Chow M. Is the 135S poliovirus particle an intermediate during cell entry. J. Virol. 2000;74:8757–61. doi: 10.1128/jvi.74.18.8757-8761.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Incardona NL, Kaesberg P. A pH-induced structural change in bromegrass mosaic virus. Biophys. J. 1964;4:11–21. doi: 10.1016/s0006-3495(64)86766-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kaplan G, Freistadt MS, Racaniello VR. Neutralization of poliovirus by cell receptors expressed in insect cells. J. Virol. 1990;64:4697–702. doi: 10.1128/jvi.64.10.4697-4702.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kim S, Smith TJ, Chapman MS, Rossmann MG, Pevear DC, et al. Crystal structure of human rhinovirus serotype 1A (HRV1A) J. Mol. Biol. 1989;210:91–111. doi: 10.1016/0022-2836(89)90293-3. [DOI] [PubMed] [Google Scholar]
- 57.Kitamura N, Semler BL, Rothberg PG, Larsen GR, Adler CJ, et al. Primary structure, gene organization and polypeptide expression of poliovirus RNA. Nature. 1981;291:547–53. doi: 10.1038/291547a0. [DOI] [PubMed] [Google Scholar]
- 58.Koike S, Ise I, Nomoto A. Functional domains of the poliovirus receptor. Proc. Natl. Acad. Sci. USA. 1991;88:4104–8. doi: 10.1073/pnas.88.10.4104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kolatkar PR, Bella J, Olson NH, Bator CM, Baker TS, Rossmann MG. Structural studies of two rhinovirus serotypes complexed with fragments of their cellular receptor. EMBO J. 1999;18:6249–59. doi: 10.1093/emboj/18.22.6249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Deleted in proof.
- 61.Lamb RA, Joshi SB, Dutch RE. The paramyxovirus fusion protein forms an extremely stable core trimer: structural parallels to influenza virus haemagglutinin and HIV-1 gp41. Mol. Membr. Biol. 1999;16:11–19. doi: 10.1080/096876899294715. [DOI] [PubMed] [Google Scholar]
- 62.Lentz KN, Smith AD, Geisler SC, Cox S, Buontempo P, et al. Structure of poliovirus type 2 Lansing complexed with antiviral agent SCH48973: comparison of the structural and biological properties of three poliovirus serotypes. Structure. 1997;5:961–78. doi: 10.1016/s0969-2126(97)00249-9. [DOI] [PubMed] [Google Scholar]
- 63.Lewis JK, Bothner B, Smith TJ, Siuzdak G. Antiviral agent blocks breathing of the common cold virus. Proc. Natl. Acad. Sci. USA. 1998;95:6774–78. doi: 10.1073/pnas.95.12.6774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Li Q, Yafal AG, Lee YM-H, Hogle J, Chow M. Poliovirus neutralization by antibodies to internal epitopes of VP4 and VP1 results from reversible exposure of these sequences at physiological temperature. J. Virol. 1994;68:3965–70. doi: 10.1128/jvi.68.6.3965-3970.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Luo M, He C, Toth KS, Zhang CX, Lipton HL. Three-dimensional structure of Theiler murine encephalomyelitis virus (BeAn strain) Proc. Natl. Acad. Sci. USA. 1992;89:2409–13. doi: 10.1073/pnas.89.6.2409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Luo M, Toth KS, Zhou L, Pritchard A, Lipton HL. The structure of a highly virulent Theiler’s murine encephalomyelitis virus (GDVII) and implications for determinants of viral persistence. Virology. 1996;220:246–50. doi: 10.1006/viro.1996.0309. [DOI] [PubMed] [Google Scholar]
- 67.Luo M, Vriend G, Kamer G, Minor I, Arnold E, et al. The atomic structure of Mengo virus at 3.0 Å resolution. Science. 1987;235:182–91. doi: 10.1126/science.3026048. [DOI] [PubMed] [Google Scholar]
- 68.Macadam AJ, Arnold C, Howlett J, John A, Marsden S, et al. Reversion of the attenuated and temperature-sensitive phenotypes of the Sabin type 3 strain of poliovirus in vaccinees. Virology. 1989;172:408–14. doi: 10.1016/0042-6822(89)90183-9. [DOI] [PubMed] [Google Scholar]
- 69.Macadam AJ, Ferguson M, Arnold C, Minor PD. Anassembly defect as a result of an attenuating mutation in the capsid proteins of the poliovirus type 3 vaccine strain. J. Virol. 1991;65:5225–31. doi: 10.1128/jvi.65.10.5225-5231.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Madshus IH, Olsnes S, Sandvig K. Requirements for entry of poliovirus into cells at low pH. EMBO J. 1984;3:1945–50. doi: 10.1002/j.1460-2075.1984.tb02074.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Malashkevich VN, Chan DC, Chutkowski CT, Kim PS. Crystal structure of the simian immunodeficiency virus (SIV) gp41 core: Conserved helical interactions underlie the broad inhibitory activity of gp41 peptides. Proc. Natl. Acad. Sci. USA. 1998;95:9134–39. doi: 10.1073/pnas.95.16.9134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Malashkevich VN, Schneider BJ, Mc-Nally ML, Milhollen MA, Pang JX, Kim PS. Core structure of the envelope glycoprotein GP2 from Ebola virus at 1.9Å resolution. Proc. Natl. Acad. Sci. USA. 1999;96:2662–67. doi: 10.1073/pnas.96.6.2662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Martin A, Benichou D, Couderc T, Hogle JM, Wychowski C, et al. Use of type 1/type 2 chimaeric polioviruses to study determinants of poliovirus type 1 neurovirulence in a mouse model. Virology. 1991;180:648–58. doi: 10.1016/0042-6822(91)90078-p. [DOI] [PubMed] [Google Scholar]
- 74.Martin A, Wychowski C, Couderc T, Crainic R, Hogle J, Girard M. Engineering a poliovirus type 2 antigenic site on a type 1 capsid results in a chimaeric virus which is neurovirulent for mice. EMBO J. 1988;7:2839–47. doi: 10.1002/j.1460-2075.1988.tb03140.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.McDermott BM, Rux AH, Eisenberg RJ, Cohen GH, Racaniello VR. Two distinct binding affinities of poliovirus for its cellular receptor. J. Biol. Chem. 2000;275:23089–96. doi: 10.1074/jbc.M002146200. [DOI] [PubMed] [Google Scholar]
- 76.Mendelsohn CL, Wimmer E, Racaniello VR. Cellular receptor for poliovirus: molecular cloning, nucleotide sequence, and expression of a new member of the immunoglobulin superfamily. Cell. 1989;56:855–65. doi: 10.1016/0092-8674(89)90690-9. [DOI] [PubMed] [Google Scholar]
- 77.Molla A, Paul AV, Wimmer E. Cell-free, de novo synthesis of poliovirus. Science. 1991;254:1647–51. doi: 10.1126/science.1661029. [DOI] [PubMed] [Google Scholar]
- 78.Molla A, Paul AV, Wimmer E. In vitro synthesis of poliovirus. Dev. Biol. Stand. 1993;78:39–53. [PubMed] [Google Scholar]
- 79.Morrison ME, He YJ, Wien MW, Hogle JM, Racaniello VR. Homolog-scanning mutagenesis reveals poliovirus receptor residues important for virus binding and replication. J. Virol. 1994;68:2578–88. doi: 10.1128/jvi.68.4.2578-2588.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Morrison ME, Racaniello VR. Molecular cloning and expression of a murine homolog of the human poliovirus receptor gene. J. Virol. 1992;66:2807–13. doi: 10.1128/jvi.66.5.2807-2813.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Moss EG, Racaniello VR. Host range determinants located on the interior of the poliovirus capsid. EMBO J. 1991;10:1067–74. doi: 10.1002/j.1460-2075.1991.tb08046.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Mosser AG, Rueckert RR. WIN 51711-dependent mutants of poliovirus type 3: evidence that virions decay after release from cells unless drug is present. J. Virol. 1993;67:1246–54. doi: 10.1128/jvi.67.3.1246-1254.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Mosser AG, Sgro JY, Rueckert RR. Distribution of drug resistance mutations in type 3 poliovirus identifies three regions involved in uncoating functions. J. Virol. 1994;68:8193–201. doi: 10.1128/jvi.68.12.8193-8201.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Muckelbauer JK, Kremer M, Minor I, Diana G, Dutko FJ, et al. The structure of coxsackievirus B3 at 3.5Å resolution. Structure. 1995;3:653–67. doi: 10.1016/s0969-2126(01)00201-5. [DOI] [PubMed] [Google Scholar]
- 85.Oliveira MA, Zhao R, Lee WM, Kremer MJ, Minor I, et al. The structure of human rhinovirus 16. Structure. 1993;1:51–68. doi: 10.1016/0969-2126(93)90008-5. [DOI] [PubMed] [Google Scholar]
- 86.Olson NH, Kolatkar PR, Oliveira MA, Cheng RH, Greve JM, et al. Structure of a human rhinovirus complexed with its receptor molecule. Proc. Natl. Acad. Sci. USA. 1993;90:507–11. doi: 10.1073/pnas.90.2.507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Pallansch MA, Kew O, Semler BL, Omilianowski DR, Anderson CW, et al. Protein processing map of poliovirus. J. Virol. 1984;49:873–80. doi: 10.1128/jvi.49.3.873-880.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Perez L, Carrasco L. Entry of poliovirus into cells does not require a low-pH step. J. Virol. 1993;67:4543–48. doi: 10.1128/jvi.67.8.4543-4548.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Phelps D, Post C. Molecular dynamics investigation of the effect of an antiviral compound on human rhinovirus. Protein Sci. 1999;8:2281–89. doi: 10.1110/ps.8.11.2281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Phelps DK, Speelman B, Post CB. Theoretical studies of viral capsid proteins. Curr. Opin. Struct. Biol. 2000;10:170–73. doi: 10.1016/s0959-440x(00)00064-6. [DOI] [PubMed] [Google Scholar]
- 91.Prchla E, Kuechler E, Blaas D, Fuchs R. Uncoating of human rhinovirus serotype 2 from late endosomes. J. Virol. 1994;68:3713–23. doi: 10.1128/jvi.68.6.3713-3723.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Racaniello VR. Picornaviridae: the viruses and their replication. In: Knipe D, Howley P, editors. Fields Virology. Lippincott Williams & Wilkens; New York: 2001. pp. 685–722. [Google Scholar]
- 93.Robinson IK, Harrison SC. Structure of the expanded state of tomato bushy stunt virus. Nature. 1982;297:563–68. [Google Scholar]
- 94.Rossmann MG, Arnold E, Erickson JW, Frankenberger EA, Griffith JP, et al. Structure of a human common cold virus and functional relationship to other picornaviruses. Nature. 1985;317:145–53. doi: 10.1038/317145a0. [DOI] [PubMed] [Google Scholar]
- 95.Rossmann MG, Bella J, Kolatkar PR, He Y, Wimmer E, et al. Cell recognition and entry by rhino- and enteroviruses. Virology. 2000;269:239–47. doi: 10.1006/viro.2000.0258. [DOI] [PubMed] [Google Scholar]
- 96.Selinka H-C, Zibert A, Wimmer E. Poliovirus can enter and infect mammalian cells by way of an intercellular adhesion molecule 1 pathway. Proc. Natl. Acad. Sci. USA. 1991;88:3598–602. doi: 10.1073/pnas.88.9.3598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Selinka H-C, Zibert A, Wimmer E. A chimeric poliovirus/CD4 receptor confers susceptibility to poliovirus on mouse cells. J. Virol. 1992;66:2523–26. doi: 10.1128/jvi.66.4.2523-2526.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Shafren DR, Bates RC, Agrez MV, Herd RL, Burns GF, Barry RD. Coxsackieviruses B1, B3, and B5 use decay accelerating factor as a receptor for cell attachment. J. Virol. 1995;69:3873–77. doi: 10.1128/jvi.69.6.3873-3877.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Shukla D, Rowe CL, Dong Y, Racaniello VR, Spear PG. The murine homolog (Mph) of human herpesvirus entry protein B (HveB) mediates entry of pseudorabies virus but not herpes simplex virus types 1 and 2. J. Virol. 1999;73:4493–97. doi: 10.1128/jvi.73.5.4493-4497.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Skehel JJ, Wiley DC. Receptor binding and membrane fusion in virus entry: the influenza hemagglutinin. Annu. Rev. Biochem. 2000;69:531–69. doi: 10.1146/annurev.biochem.69.1.531. [DOI] [PubMed] [Google Scholar]
- 101.Smith TJ, Chase ES, Schmidt TJ, Olson NH, Baker TS. Neutralizing antibody to human rhinovirus 14 penetrates the receptor-binding canyon. Nature. 1996;383:350–54. doi: 10.1038/383350a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Smith TJ, Kremer MJ, Luo M, Vriend G, Arnold E, et al. The site of attachment in human rhinovirus 14 for antiviral agents that inhibit uncoating. Science. 1986;233:1286–93. doi: 10.1126/science.3018924. [DOI] [PubMed] [Google Scholar]
- 103.Smith TJ, Olson NH, Cheng RH, Liu H, Chase ES, et al. Structure of human rhinovirus complexed with Fab fragments from a neutralizing antibody. J. Virol. 1993;67:1148–58. doi: 10.1128/jvi.67.3.1148-1158.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Smyth M, Tate J, Hoey E, Lyons C, Martin S, Stuart D. Implications for viral uncoating from the structure of bovine enterovirus. Struct. Biol. 1995;2:224–31. doi: 10.1038/nsb0395-224. [DOI] [PubMed] [Google Scholar]
- 105.Speelman B, Brooks BR, Post CB. Molecular dynamics simulations of human rhinovirus and an antiviral compound. Biophys. J. 2001;80:121–29. doi: 10.1016/S0006-3495(01)75999-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Speir JA, Munshi S, Wang G, Baker TS, Johnson JE. Structures of the native and swollen forms of cowpea chlorotic mottle virus determined by X-ray crystallography and cryo-electron microscopy. Structure. 1995;3:63–78. doi: 10.1016/s0969-2126(01)00135-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Staunton DE, Merluzzi VJ, Rothlein R, Barton R, Marlin SD, Springer TA. A cell adhesion molecule, ICAM-1, is the major surface receptor for rhinoviruses. Cell. 1989;56:849–53. doi: 10.1016/0092-8674(89)90689-2. [DOI] [PubMed] [Google Scholar]
- 108.Takahashi K, Nakanishi H, Miyahara M, Mandai K, Satoh K, et al. Nectin/PRR: An immunoglobulin-like cell adhesion molecule recruited to cadherin-based adherens junctions through interaction with Afadin, a PDZ domain-containing protein. J. Cell Biol. 1999;145:539–49. doi: 10.1083/jcb.145.3.539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Tosteson MT, Chow M. Characterization of the ion channels formed by poliovirus in planar lipid membranes. J. Virol. 1997;71:507–11. doi: 10.1128/jvi.71.1.507-511.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Tsang SK, Danthi P, Chow M, Hogle JM. Stabilization of poliovirus by capsid-binding antiviral drugs is due to entropic effects. J. Mol. Biol. 2000;296:335–40. doi: 10.1006/jmbi.1999.3483. [DOI] [PubMed] [Google Scholar]
- 111.Tsang SK, McDermott BM, Racaniello VR, Hogle JM. A kinetic analysis of the effect of poliovirus receptor on viral uncoating: the receptor as a catalyst. J. Virol. 2001;75:4984–89. doi: 10.1128/JVI.75.11.4984-4989.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Verdaguer N, Blaas D, Fita I. Structure of human rhinovirus serotype 2 (HRV2) J. Mol. Biol. 2000;300:1179–94. doi: 10.1006/jmbi.2000.3943. [DOI] [PubMed] [Google Scholar]
- 113.Weissenhorn W, Calder LJ, Wharton SA, Skehel JJ, Wiley DC. The central structural feature of the membrane fusion protein subunit from the Ebola virus glycoprotein is a long triple-stranded coiled coil. Proc. Natl. Acad. Sci. USA. 1998;95:6032–36. doi: 10.1073/pnas.95.11.6032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Weissenhorn W, Dessen A, Harrison SC, Skehel JJ, Wiley DC. Atomic structure of the ectodomain from HIV-1 gp41. Nature. 1997;387:426–30. doi: 10.1038/387426a0. [DOI] [PubMed] [Google Scholar]
- 115.Wetz K, Kucinski T. Influence of different ionic and pH environments on structural alterations of poliovirus and their possible relation to virus uncoating. J. Gen. Virol. 1991;72:2541–44. doi: 10.1099/0022-1317-72-10-2541. [DOI] [PubMed] [Google Scholar]
- 116.Wien MW, Curry S, Filman DJ, Hogle JM. Structural studies of poliovirus mutants that overcome receptor defects. Nat. Struct. Biol. 1997;4:666–74. doi: 10.1038/nsb0897-666. [DOI] [PubMed] [Google Scholar]
- 117.Wiley DC, Skehel JJ. The structure and function of the hemagglutinin membrane glycoprotein of influenza virus. Annu. Rev. Biochem. 1987;56:365–94. doi: 10.1146/annurev.bi.56.070187.002053. [DOI] [PubMed] [Google Scholar]
- 118.Wilson IA, Skehel JJ, Wiley DC. Structure of the haemagglutinin membrane glycoprotein of influenza virus at 3Å resolution. Nature. 1981;289:366–73. doi: 10.1038/289366a0. [DOI] [PubMed] [Google Scholar]
- 119.Xiao C, Bator CM, Bowman VD, Rieder E, He Y, et al. Interaction of coxsackievirus A21 with its cellular receptor, ICAM-1. J. Virol. 2001;75:2444–51. doi: 10.1128/JVI.75.5.2444-2451.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Xing L, Tjarnlund K, Lindqvist B, Kaplan GG, Feigelstock D, et al. Distinct cellular receptor interactions in poliovirus and rhinoviruses. EMBO J. 2000;19:1207–16. doi: 10.1093/emboj/19.6.1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Yeates TO, Jacobson DH, Martin A, Wychowski C, Girard M, et al. Three-dimensional structure of a mouse-adapted type 2/type 1 poliovirus chimera. EMBO J. 1991;10:2331–41. doi: 10.1002/j.1460-2075.1991.tb07772.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Zhao R, Pevear DC, Kremer MJ, Giranda VL, Kofron JA, et al. Human rhinovirus 3 at 3.0Å resolution. Structure. 1996;4:1205–20. doi: 10.1016/s0969-2126(96)00128-1. [DOI] [PubMed] [Google Scholar]
- 123.Zhao X, Singh M, Malashkevich VN, Kim PS. Structural characterization of the human respiratory syncytial virus fusion protein core. Proc. Natl. Acad. Sci. USA. 2000;97:14172–77. doi: 10.1073/pnas.260499197. [DOI] [PMC free article] [PubMed] [Google Scholar]