

Abstract
Members of the uncultivated bacterial division TM7 have been detected in the human mouth, but little information is available regarding their prevalence and diversity at this site. Human subgingival plaque samples from healthy sites and sites exhibiting various stages of periodontal disease were analyzed for the presence of TM7 bacteria. TM7 ribosomal DNA (rDNA) was found in 96% of the samples, and it accounted for approximately 0.3%, on average, of all bacterial rDNA in the samples as determined by real-time quantitative PCR. Two new phylotypes of this division were identified, and members of the division were found to exhibit filamentous morphology by fluorescence in situ hybridization. The abundance of TM7 rDNA relative to total bacterial rDNA was higher in sites with mild periodontitis (0.54% ± 0.1%) than in either healthy sites (0.21% ± 0.05%, P < 0.01) or sites with severe periodontitis (0.29% ± 0.06%, P < 0.05). One division subgroup, the I025 phylotype, was detected in 1 of 18 healthy samples and 38 of 58 disease samples. These data suggest that this phylotype, and the TM7 bacterial division in general, may play a role in the multifactorial process leading to periodontitis.
It has been well established that a view of the microbial world based on cultivable species vastly underestimates the diversity present in most environments. In 1998, Hugenholtz et al. constructed a tree that reflected the phylogeny of the domain Bacteria based on all 16S rRNA gene sequences in GenBank and identified 36 divisions or putative divisions (distinguished by ribosomal DNA [rDNA] sequence differences of approximately 20 to 25%) (8). One candidate division with no cultivated representatives is TM7. Although this division takes its name from the German peat bog from which the first sequence was obtained (Torf, mittlere Schicht, or peat, middle layer) (25), additional TM7 sequences deposited by several other investigators have demonstrated that members of this division are present in extremely diverse environments, including soil, freshwater, seawater, hot springs, mouse feces, and termite guts (9). The wide environmental range of this division suggested that it also might be human associated. Indeed, a survey of bacterial diversity in human subgingival plaque samples by broad-range PCR detected TM7 bacteria in several patients (24). Five TM7 phylotypes were identified, one of which, represented by clone I025, was suggested as a putative pathogen because it was found only in patients with various oral diseases.
While there is a general consensus that bacteria play a causative role in the development of periodontal disease, no single species is invariably detected at sites of disease. In almost all previous studies, the search for pathogens has been limited to cultivable species, a group that is estimated to comprise less than half of the oral flora (11, 24, 28). The initial detection of TM7 division members within the subgingival crevice by molecular methods suggested that further analysis of this major group of bacteria at this important human-associated site might shed light on its significance within the local complex microbial community. To explore the relationship of TM7 bacteria to the paradigm of successive colonization and to the development of local disease, we examined plaque from sites representing a continuum of periodontal disease. Our approaches included qualitative and quantitative molecular methods for bacterial community analysis. We chose real-time PCR (also known as 5′ nuclease assay) for its high sensitivity and adaptability to high-throughput operation and used fluorescence in situ hybridization (FISH) as a complementary and confirmatory approach. Using these techniques, we found that members of the TM7 division are widespread in the oral flora of both healthy and diseased sites and that the subgroup I025 is found primarily at diseased sites.
MATERIALS AND METHODS
Study population.
Forty-six volunteers were enrolled at the University of California, San Francisco (UCSF), School of Dentistry in the Center for Clinical Research (Division of Periodontology). The use of human subjects in this investigation was approved by the Stanford University Administrative Panel on Human Subjects in Medical Research and the UCSF Committee on Human Research. All volunteers read and signed a consent form approved by the UCSF committee. Participants were at least 21 years old and were missing no more than 14 teeth. Individuals were excluded if they were diabetic, human immunodeficiency virus positive, pregnant, lactating, or had taken antibiotics in the previous 3 months, since these factors have been implicated in altering bacterial composition (7, 14, 16, 21, 27). All volunteers completed a survey regarding smoking history (i.e., current smoker, former smoker, or never smoked), age, gender, and race. The average age of the patients was 47.3 years (standard deviation, 12.4 years; range, 23 to 74 years). The study population included 26 males (15 Caucasian subjects, 7 African American subjects, 1 Hispanic subject, 2 Asian subjects, and 1 Middle Eastern subject) and 20 females (10 Caucasian subjects, 5 African American subjects, and 5 Hispanic subjects). Twelve subjects were current smokers, 2 subjects were former smokers, and 32 subjects had never smoked.
Healthy volunteers (n = 4) had no probing depths (PD) greater than 4 mm and mean full-mouth clinical attachment loss (CAL) of ≤0.5 mm. Forty-two subjects had generalized chronic periodontitis (diagnosed by presence of periodontal pockets and bleeding on probing [BOP] of >30% of all teeth present). All subjects were free of other diseases of oral soft tissues. The periodontal status of each person was determined by measuring CAL to the nearest millimeter at the mesiobuccal, buccal, distobuccal, mesiolingual, lingual, and distolingual sites around each tooth with a calibrated periodontal probe.
Sample collection.
Six to 12 samples were collected from each subject, from healthy sites as well as sites exhibiting gingivitis or periodontitis. Clinical assessments at each site included the presence or absence of BOP, PD, and CAL. Clinical assessments and sample collections were performed by one of us (G. C. A.). Each site was classified as healthy (no BOP, CAL ≤ 1 mm, and PD ≤ 3 mm), exhibiting gingivitis (BOP, CAL ≤ 1 mm, and PD ≤ 4 mm), mild periodontitis (BOP, CAL ≥ 2 mm and ≤ 3 mm, and PD ≥ 4 mm), moderate periodontitis (BOP, CAL ≥ 4 mm and ≤ 5 mm, and PD ≥ 4 mm), or severe periodontitis (BOP, CAL ≥ 6 mm, and PD ≥ 4 mm). Supragingival plaque was removed from tooth surfaces before sampling, and subgingival plaque was collected by using Hartzell R-1 and R-2 curettes (a maximum of 2 curettes' worth of plaque was collected). A separate sterile curette was used for each plaque sample. A sample was taken from the dorsum of the tongue with a sterile plastic spatula. The samples were placed in 200 μl of sterile water (which had been subjected to filtration and γ-irradiated so as to destroy PCR-detectable DNA) and divided into two aliquots. One aliquot was prepared for FISH as described below. The other aliquot was frozen immediately and kept at −80°C before further processing.
One hundred thirty-seven samples were studied by one or more techniques. In total, 36 samples were taken from sites classified as healthy, 5 samples were taken from sites classified as gingivitis, 17 samples were taken from sites classified as mild periodontitis, 21 samples were taken from sites classified as moderate periodontitis, 53 samples were taken from sites classified as severe periodontitis, and 5 samples were taken from the tongue. The mean ± standard deviation of CAL and PD measurements for each study group were: healthy, CAL = 0.4 ± 0.5 mm and PD = 2.6 ± 0.6 mm; gingivitis, CAL = 1.0 ± 0 mm and PD = 3.4 ± 0.9 mm; mild periodontitis, CAL = 2.2 ± 0.4 mm and PD = 4.2 ± 0.9 mm; moderate periodontitis, CAL = 4.8 ± 0.4 mm and PD = 5.7 ± 0.7 mm; severe periodontitis, CAL = 7.9 ± 1.7 mm and PD = 7.9 ± 2.0 mm.
Samples from 16 patients were studied by qualitative PCR alone, samples from 7 patients were studied by FISH alone, samples from 27 patients were studied by real-time PCR alone, samples from 5 patients were studied by both qualitative PCR and FISH, a sample from 1 patient was studied by both qualitative and real-time PCR, and a sample from 1 patient was studied by all three methods. From all of these patients, 25 samples were studied by qualitative PCR alone, 11 samples were studied by FISH alone, 91 samples were studied by real-time PCR alone, 8 samples were studied by both qualitative PCR and FISH, 1 sample was studied by both qualitative and real-time PCR, and 1 sample was studied by all three methods.
As part of an initial analysis, we performed a qualitative PCR assay on 35 of these samples from 21 patients; this group of samples comprised 10 samples from healthy sites, 1 sample from a site exhibiting mild periodontitis, 3 samples from sites of moderate periodontitis, and 21 samples from sites of severe periodontitis. In later stages of this investigation, 20 samples from 12 patients were studied with FISH: 9 samples were from healthy sites and 11 samples were from severe-periodontitis sites. A further 93 samples from 25 patients were analyzed by real-time PCR. This set included 21 samples from healthy sites, 5 samples from gingivitis sites, 16 samples from mild-periodontitis sites, 18 samples from moderate-periodontitis sites, 28 samples from severe-periodontitis sites, and 5 samples from the tongue.
Sample preparation.
A rigorous cell lysis procedure was developed. One hundred microliters of cell lysis buffer (100 mM Tris-HCl [pH 7.4], 20 mM EDTA, 5 M guanidine isothiocyanate, 2% Triton X-100) and 50 μg of proteinase K were added to 100 μl of sample (modified from references 6 and 22). This mixture was incubated at 65°C for 30 min. After the addition of 0.1 g each of three sizes of baked zirconia beads (0.1, 0.5, and 1 mm) and 100 μl of water, the mixture was agitated in a FastPrep FP120 machine (Qbiogene, Carlsbad, Calif.) at 4.0 m/s for 30 s. The lysate was centrifuged at 16,000 × g for 1 min to pellet the beads, and the supernatant was removed to a fresh tube. As previously described, the supernatant was treated with 1.15% hexadecyltrimethylammonium bromide (Sigma, St. Louis, Mo.) to bind proteins and polysaccharides and facilitate their separation from the DNA, and then nucleic acids were isopropanol precipitated (33). After air drying, they were resuspended in 50 μl of water.
PCR and cloning.
A TM7-specific primer was designed by using the public database sequences belonging to that division and the probe design function of the ARB software package (31). Primer and probe sequences and positions are listed in Table 1. TM7-1177R was used in conjunction with the broad-range bacterial 16S rRNA primer Bac-8F to amplify TM7 16S rDNA from oral samples prepared as described above. The PCR mix contained 1 μl of the sample DNA solution, 10 pmol of each primer, 1× PCR buffer II (Applied Biosystems, Foster City, Calif.), 1.5 mM MgCl2, 0.6 mM concentrations of deoxynucleoside triphosphates, and 1.25 U of AmpliTaq DNA polymerase (other than the buffer, PCR reagents were from Perkin Elmer, Boston, Mass.). The reaction conditions were 96°C for 3 min; 35 cycles of 94°C for 1 min, 64°C for 1 min, and 72°C for 2 min; and a final extension at 72°C for 3 min. PCR products of the appropriate size were cloned by using the TOPO-TA kit (Invitrogen, Carlsbad, Calif.) following the manufacturer's instructions.
TABLE 1.
Primers and probes used for PCR, FISH, and real-time PCR
| Name | Use | E. coli 16S rRNA gene positions | Reference or sequence (5′ to 3′), if designed for this study |
|---|---|---|---|
| Bac-8F | Bacterial real-time PCR primer | 8-27 | 3 |
| Bac-515R | Bacterial real-time PCR primer | 515-533 | 12 |
| Bac-338R | Bacterial and I025 real-time PCR probe and FISH probe | 338-355 | 17 |
| TM7-910F | TM7 real-time PCR primer | 930-910 | CAT AAA GGA ATT GAC GGG GAC |
| TM7-1177R | TM7 PCR and real-time PCR primer | 1177-1200 | GAC CTG ACA TCA TCC CCT CCT TCC |
| TM7-1093F | TM7 real-time PCR probe | 1093-1113 | AGT CCA TCA ACG AGC GCA ACC |
| TM7-905 | TM7 FISH probe | 905-926 | 9 |
| I025-135F | I025 real-time PCR primer | 135-154 | CCC TGC AGT GAG GGA TAA GA |
| I025-590R | I025 real-time PCR primer | 590-611 | GTT TTC ATC GCT CGC TAA CTT G |
| I025-136 | I025 FISH probe | 136-155 | GTC TTA TCC CTC ACT GCA GG |
| Control probe | Negative control for FISH | 519-536 | 20 |
Sequencing and phylogenetic analysis.
Eighty-five clones containing TM7 bacterial 16S rRNA gene fragments (from 7 patients) were subjected to one sequencing reaction with primer Bac-8F and the BigDye sequencing chemistry (Applied Biosystems). Six clone inserts were completely sequenced with an average of 3.5-fold coverage. The sequences were named with the prefix SBG (for subgingival).
Initial alignment of amplified sequences was performed with the automated 16S rDNA sequence aligner of the ARB software package (31) against a database of 12,569 complete and partial rDNA sequences. Ambiguously and incorrectly aligned positions were aligned manually on the basis of conserved primary sequence and secondary structure. Similarity matrices were generated from 180 masked (unambiguously aligned) positions. Following the proposition of Kroes et al., sequences with ≥99% identity were considered to be of a single phylotype (11). A single representative clone from each phylotype was chosen for further phylogenetic analysis. These clones were fully sequenced and aligned, and similarity matrices were generated from 1,076 masked positions. The phylogenetic associations of all representative sequences were determined by using a maximum-likelihood algorithm (18). The 16S rDNA sequences of Shewanella putrefaciens and Escherichia coli were used as out-groups to root the tree. These associations were confirmed using parsimony algorithms and a least-squares fit (4) of evolutionary distances (with Jukes-Cantor correction). The tree was bootstrapped with 100 replicate samples of the data set (5).
FISH.
Our protocol combined previously published procedures (1, 19), with modifications as follows. Fifty microliters of plaque sample collected in sterile water was mixed immediately with 50 μl of a solution of 2× phosphate-buffered saline-20% formalin, and cells were dispersed by sonication with a 3-mm-diameter microtip probe at maximum speed for 20 s in a calibrated XL2020 ultrasonic processor (Heat Systems, Inc., New York, N.Y.). Appropriate dilutions of cells were transferred to glass slides with wells etched with Teflon and coated with AddCell (to enhance cell adhesion) (Eric Scientific Company, Portsmouth, N.J.) and air dried. Cells were posttreated with 10 μl of methanol-formalin (90:10, vol/vol) per well for 15 min, rinsed with distilled water, and air dried.
Oligonucleotides (Table 1) labeled with Cy3 or Cy5 (Operon, Alameda, Calif.) were added at a 5-ng/μl concentration. Hybridization took place in 10 μl of hybridization buffer per well (19). The concentration of formamide in the hybridization buffer had been established previously for the probes Bac-338 (15) and TM7-905 (9). Because no cultivable strain contains fewer than three mismatches to the I025-136 probe, the 20% formamide used for this probe was determined based on its melting temperature as calculated with Lathe's equation (13). For each sample, hybridization to each probe was carried out in three separate wells.
Slides were incubated at 46°C for 2 h in the dark and then rinsed with 46°C water and placed in wash buffer (20 mM Tris-HCl [pH 7.2], 0.01% sodium dodecyl sulfate, and 0.225 M NaCl, as recommended for the probes in references 9 and 34) at 48°C for 15 min in the dark. Slides were quickly immersed in water and dried in a hybridization oven at 30°C for 5 min.
To visualize all cells, 10 μl of 2 μM YO-PRO-1 iodide (a general nucleic acid dye) (Molecular Probes, Eugene, Oreg.) was added to each well for 10 min. Slides were rinsed in water and air dried. Slides were mounted with the antifade solution Vectashield (Vector Laboratories, Inc., Burlingame, Calif.) under a 24- by 50-mm cover glass and sealed with nail polish.
FISH counts and micrographs were executed on a Nikon Eclipse TE300 laser scanning confocal microscope with a Nikon Plan Apo 100× objective lens and Bio-Rad LaserSharp MRC-1024 software with 1,024- by 1,024-pixel resolution. At least 30 YO-PRO-1-labeled cells in each of 10 randomly selected fields per well were observed, and the number of cells labeled with the specific probe was recorded. The results for the three replicate wells were averaged. Filaments were counted as one cell, but the number of cells in each filament was noted. No cells were fluorescent after hybridization with a control probe (the reverse complement of a broad-range 16S rDNA probe) or without any probe under nonstringent conditions. In addition, no cells became labeled with the I025-136 probe that were not also labeled with the TM7-905 probe.
Real-time PCR assays.
Three separate real-time PCR assays were developed and optimized to quantify total bacteria, relative abundance of TM7 division members, and relative abundance of TM7 subgroup I025 members. The reactions were carried out in an ABI Prism 7900HT sequence detection system (Applied Biosystems). Each reaction included 1× TaqMan Universal PCR mix with no AmpErase UNG (Applied Biosystems), 0.5 U of AmpliTaq Gold DNA polymerase (Applied Biosystems), and 1 μl of prepared DNA template in addition to the specific primers and probes described below. Characteristics of the final assays are listed in Table 2. All probes were conjugated to 5′ 6-carboxyfluorescein and 6-carboxy-tetramethylrhodamine.
TABLE 2.
Characteristics of the three real-time PCR assays developed in this study
| Assay target | Approx amplicon length (bp) | Sensitivity (no. of rDNA target copies) | Maximum of the dynamic range (no. of rDNA target copies) |
|---|---|---|---|
| All bacteria | 525 | <10 | ≥107 |
| TM7 division | 276 | <10 | ≥106 |
| I025 subgroup | 441 | <1,000 | ≥106 |
Each sample was assayed in duplicate on two separate days, and the 4 measurements were averaged. The intraexperimental coefficient of variation (CV) was calculated as:
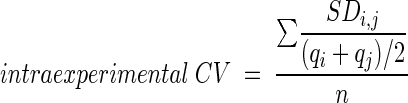 |
(1) |
where qi and qj are the quantities calculated from two duplicate reactions with the same sample, SDi,j is the standard deviation of qi and qj, and n is the number of samples. To determine interexperimental coefficient of variation, qi and qj for each sample in each experiment were averaged to produce a single quantity (Q). These values were used to calculate the interexperimental coefficient of variation with the equation:
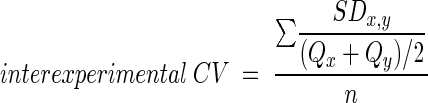 |
(2) |
where Qx and Qy are the quantities calculated in two independent experiments with the same sample, SDx,y is the standard deviation of Qx and Qy, and n is the number of samples.
The total-bacteria assay used the primers Bac-8F and Bac-515R (9 pmol of each per reaction mixture), and probe Bac-338R (2 pmol per reaction mixture). The cycling conditions were 95° for 10 min followed by 50 cycles of 95°C for 30 s, 55°C for 30 s, 60°C for 45 s, 65°C for 15 s, and 72°C for 15 s. The threshold was set at 0.01, with the baseline measured from cycles 3 to 15. The standards for this assay were dilutions of a plasmid containing the E. coli 16S rRNA gene. The specificity of the assay was tested by adding either no DNA or 1.8 ng of human DNA, neither of which produced a signal. The sensitivity and specificity of the assay were not altered when a plasmid containing a cloned 16S rRNA gene from a TM7 division member was used for the standards.
Three TM7 division-specific primer/probe sequences were designed by using the complete set of TM7 sequences deposited in GenBank as well as sequences obtained during the course of this study. The TM7-division real-time PCR mix included 10 pmol of primer TM7-910F, 1 pmol of primer TM7-1177R, and 1.2 pmol of probe TM7-1093F. The cycling conditions were 95°C for 10 min followed by 40 cycles of 95°C for 30 s and 61°C for 1 min. The threshold was set at 0.15, with the baseline measured from cycles 3 to 16. This assay was tested for specificity by using the cloned 16S rRNA gene from Bordetella holmesii, which has the fewest mismatches to the primers and probes of any known non-TM7 16S rRNA gene (one mismatch to primer TM7-910F, 3 mismatches to primer TM7-1177R, and 2 mismatches to the probe TM7-1093F). This template was not amplified when present in up to 108 copies. Human DNA also was not amplified, nor was product detected in reactions containing no DNA. The standards were dilutions of a plasmid containing SBG6 (Fig. 1). No systematic bias was evident in the sensitivity or specificity of the assay with cloned 16S rRNA sequences from two widely divergent oral members of the TM7 division (SBG1 and SBG6) (Fig. 1). To ensure that the processed plaque specimens did not contain real-time PCR inhibitors, 1 μl of samples that tested negative for TM7 rDNA by real-time PCR was added to the standard reactions. No effect on the quantification of the standards was observed.
FIG. 1.

Phylogeny of representative 16S rDNA sequences of oral TM7 phylotypes. The tree was created by using a maximum-likelihood algorithm and 1,076 positions. The 16S rDNA sequences of S. putrefaciens and E. coli were used as out-groups to root the tree. Phylotypes detected in this study are named with the prefix SBG (for subgingival). The two sequences highlighted in gray are novel phylotypes detected in this study. Bootstrap values greater than 50 are shown. The bar represents evolutionary distance. Accession numbers are given in parentheses.
Two primers were designed for the I025 group of the TM7 division by using the sequences of the published I025 clone (accession number AF125206) and three I025-related clones obtained in the course of this study. The I025 real-time PCR assay used 0.5 pmol of primer I025-135F, 2.0 pmol of primer I025-590R, and 1.0 pmol of probe Bac-338R. The cycling conditions were 95°C for 10 min followed by 40 cycles of 95°C for 30 s, 50°C for 30 s, 55°C for 15 s, and 60°C for 45 s. The threshold was set at 0.08, with the baseline measured from cycles 8 to 19. Sensitivity was tested by including the cloned 16S rRNA gene from a non-I025 TM7 member (SBG2) which contains 4 mismatches to primer I025-135F, 11 mismatches to primer I025-590R, and no mismatches to the probe Bac-338R. This fragment did not generate detectable product when present in up to 107 copies, nor was product generated in reactions with human DNA or no added DNA. The standards for this assay were dilutions of a plasmid containing the insert SBG1, the fully sequenced 16S rRNA gene closely related to that of sequence I025 (Fig. 1). Adding 1 μl of I025-negative samples to the standard reactions did not affect the quantification of the standards.
Limitations of real-time PCR for analysis of uncultivated species in bacterial communities.
In all of the real-time PCR assays developed for this study, two standard curves generated independently with the same samples gave slightly different best-fit lines, resulting in as much as a 10-fold difference in the estimation of DNA quantity in the experimental samples. This may have been due to small variations in template quantity during creation of the standard dilution series, whose effect was magnified in the log-scale conversion in the calculations. Since a separate standard curve was used in each set of reactions, this produced average interexperimental coefficients of variation above 0.5. To decrease the impact of this variability on the calculated quantities of target in unknown samples, an average standard curve was created for each reaction which could be utilized to quantify every sample. To do this, the standard dilutions were assayed in triplicate in two (for the TM7 assay) or three (for the bacteria assay) separate reactions, and the average of the three CT values (the cycle at which the signal passes a predetermined threshold value) was used for each standard dilution on each day to create a single standard curve. When this average standard curve was used to estimate DNA quantity in plaque samples, the coefficients of variation for the assays were significantly lower than when separate standard curves were used (0.39 versus 0.54 for the bacterial assay and 0.21 versus 0.61 for the TM7-division assay).
RESULTS
TM7 prevalence and diversity in subgingival samples.
In an initial assessment of TM7 prevalence in subgingival plaque samples, a qualitative PCR assay with a TM7 division-specific primer produced bands of the expected size in 30 of 35 samples. The detection limit of this reaction was approximately 105 rDNA target copies. The PCR products from four positive samples from healthy sites and three positive samples from periodontitis sites were cloned and sequenced with a single reaction from the 5′ end to provide an overview of TM7 division diversity in the subgingival crevice. Of the 85 TM7 bacteria 16S rDNA sequences obtained, 62 were phylogenetically most similar to clone AH040 and all 7 samples contained sequences of this type. One disease sample also contained sequences that most closely matched sequence I025, which was previously found in 4 out of 24 patients with certain forms of periodontal disease (2 with refractory periodontitis and 2 with acute necrotizing ulcerative gingivitis) and 0 out of 6 healthy patients (24). A more sensitive and quantitative assessment of TM7 bacteria and subgroup I025 prevalence was provided by using real-time PCR (see below).
Phylogeny of representative oral TM7 clones.
Six representative amplified 16S rRNA genes located in different branches of the TM7 division were chosen from the 85 screened clones for complete sequencing. Four of these sequences were ≥99% identical to previously published sequences. The other two sequences, SBG2 and SBG3, represent two new phylotypes of the TM7 division. Both are most similar to the previously published sequence BE109 (92.6 and 93.4% similar, respectively). The phylogeny inferred from these sequences is displayed in Fig. 1. The topology of this tree is congruent with previously published trees of TM7 division members (9, 24).
Morphology of oral TM7 and I025 bacteria.
We used FISH to explore the morphological diversity of TM7 division members in the subgingival crevice. The TM7 division probe labeled both short and long filaments from 4 to 30 μm long (12 μm on average, 52 filaments measured) and 1 to 1.5 μm thick (Fig. 2B). Long filaments were composed of multiple cells (visible on the microscope, although difficult to distinguish in reproductions), each ranging from 3 to 4 μm long. The I025 phylotype probe labeled only long filaments, averaging 20 μm long (range, 10 to 30 μm; 21 filaments measured) and 1 μm thick (Fig. 2C).
FIG. 2.

FISH images of TM7 and I025 bacteria. Panel A shows cells staining with YO-PRO-1 iodide (nonspecific nucleic acid dye), panel B shows cells in the same field that hybridize with the TM7 bacterial division probe (labeled with Cy5), and panel C shows a cell that hybridizes with the I025 subdivision-specific probe (labeled with Cy3).
Quantification of TM7 in subgingival specimens.
Because we expected that the total amount of subgingival plaque and the quantity of bacteria per sample would differ, we developed real-time quantitative PCR assays to measure the quantity of total bacterial rDNA as well as the amount of TM7 rDNA (Fig. 3A). This allowed us to compare the relative abundance of TM7 across many samples. Almost all samples (86 of 90, 96%) contained detectable TM7 rDNA, including all 7 samples from periodontally healthy subjects. Figure 3B depicts the percentages of bacterial rDNA that were detected in the TM7-specific assay for samples in the five disease categories. Although the proportion of TM7 rDNA in healthy samples was not different from that in severe-periodontitis samples, the percentage of TM7 rDNA in mild-periodontitis samples (0.54% ± 0.10% [standard error {SE}]) was significantly higher than in both healthy (0.21% ± 0.05%, P < 0.01 [Student's t test]) and severe-periodontitis samples (0.29% ± 0.06%, P < 0.05). Tongue samples were included for comparison.
Samples from Caucasian patients had lower proportions of TM7 rDNA (0.22% ± 0.03% [SE], n = 33) than those from Asian patients (0.49% ± 0.15%, n = 8, P < 0.008) and black patients (0.37% ± 0.05%, n = 37, P < 0.05). Although this analysis did not include data on socioeconomic status, which has been correlated with both ethnicity and periodontal health, the finding that race is associated with subgingival microflora composition in adult periodontitis is consistent with previous studies (27). There was no correlation between the relative abundance of TM7 rDNA and either gender (proportion of TM7 rDNA in samples from males = 0.32% ± 0.045% [SE], n = 42; proportion of TM7 rDNA in samples from females = 0.33% ± 0.052%, n = 38; P = 0.94), smoking status (proportion of TM7 rDNA in samples from current smokers = 0.372% ± 0.061%, n = 25; proportion of TM7 rDNA in samples from patients who never smoked = 0.323% ± 0.046%, n = 48; P = 0.52), or age (point biserial correlation coefficient = −0.041).
About 9% of the reactions had an intraexperimental coefficient of variation (between duplicate reactions in the same plate) of greater than 1. These reactions were repeated, and the samples were excluded from the analyses if the coefficient of variation was still greater than 1. The interexperimental coefficient of variation (between reactions performed on separate dates) of 7% of the samples was greater than 1, and these were excluded from further analyses. The average intra- and interexperimental coefficients of variation were both approximately 0.4 for the bacterial assay and 0.2 for the TM7-division assay. The I025 subgroup assay, which was performed only once, had an intraexperimental coefficient of variation of 0.2.
We used FISH as an independent method to corroborate the real-time PCR findings. TM7 cells were counted in 20 samples (9 from healthy sites and 11 from sites with severe periodontitis) by using FISH. The range of TM7 relative abundance (as a percentage of total bacteria labeling with the broad-range bacterial probe Bac-338) in these samples was 0% to 6.3%. The average percentage of TM7 bacteria was not significantly different in samples from healthy sites (2.1% ± 0.76% [SE]) and those from sites with severe periodontitis (1.5 ± 0.43%, P = 0.47). TM7 bacteria in samples from sites with mild and moderate periodontitis cannot be compared because of low numbers of samples of these types. No differences in arrangement or morphology of TM7 bacteria were observed between healthy and disease samples.
Quantification of subgroup I025.
The I025 subgroup of the TM7 division, which was previously found only in subgingival samples from sites with disease (24), was also examined by real-time PCR. Overall, I025 rDNA was detected in 51% (39 of 76) of the samples (with a sensitivity of detection of 1,000 rDNA target copies/μl). Healthy samples were significantly less likely to be positive (1 of 18) for I025 rDNA than samples from diseased sites (38 of 58, P < 0.001 [Fisher's exact test]) (Table 3). Among the samples which were positive for the I025 phylotype, the amount of I025 as a percentage of total bacterial rDNA was highly variable (from 0.002 to 1.25%), but in all cases, it was well above the detection threshold for the I025 real-time PCR assay (at least 10-fold higher than the threshold, and on average about 500-fold higher). Therefore, the lack of detection of I025 bacteria in healthy samples was not due to the fact that these samples had fewer total bacteria than the disease samples.
TABLE 3.
Detection of I025-subgroup DNA by real-time PCR in relation to disease category
| Disease status | No. of samples | No. of samples with detectable I025 rDNA | Percentage of samples with detectable I025 rDNA |
|---|---|---|---|
| Healthy | 18 | 1 | 6 |
| Gingivitis | 5 | 4 | 80 |
| Mild periodontitis | 17 | 11 | 65 |
| Moderate periodontitis | 15 | 10 | 67 |
| Severe periodontitis | 21 | 13 | 62 |
| Total | 76 | 39 | 51 |
Because of the relatively low numbers of patients with I025, the mean percent I025 in different disease categories was significantly different only in mild periodontitis (0.099% ± 0.036% [SE]) compared to moderate periodontitis (0.297% ± 0.086%, P < 0.05), but there appeared to be a trend towards a greater relative abundance of I025 bacteria in more-advanced stages of disease (Fig. 4).
FIG. 4.

Abundance of I025 rDNA relative to total bacterial rDNA in subgingival plaque at sites with various states of disease. Only sites with detectable quantities of I025 are included. SEs are indicated. The difference between relative abundance at sites with mild and moderate periodontitis was significant at P < 0.04.
DISCUSSION
TM7 is a cosmopolitan and highly divergent division of bacteria. The human oral TM7 bacteria appear to form a monophyletic group within the division, along with a putative member whose sequence was isolated from mouse feces, suggesting that the ability to colonize mammals may be a conserved feature of this group of TM7 phylotypes (Fig. 1). Oral TM7 cells visualized by FISH were larger than the TM7 bacteria previously visualized by transmission electron microscopy in laboratory-scale bioreactor sludge samples (9). This difference may reflect true variation in the morphotypes of TM7 bacteria found in the mouth versus reactor sludge, or it may be an artifact caused by the different visualization methods employed (electron microscopy may cause contraction while measurements taken by epifluorescence, as performed in this study, may be slight overestimates due to light refraction). In addition, bacteria in the nutrient-rich milieu of the subgingival crevice may grow to larger sizes than those found in nutrient-poor environments. We did not observe any coccoid cells labeled with the TM7 division probe. Cocci detected in the sludge samples may represent phylotypes of TM7 bacteria which are not present in the subgingival crevice, or they may have been bacilli which appeared coccoid in the sludge samples because of different nutrient availability.
Oral TM7 phylotypes, like other members of the division, are not restricted to a small geographic area or a single environmental source, based on their identification in the human mouth in both this study and a previous study (24). Although previous investigators detected TM7 bacteria in only a small fraction of samples, we were able to detect members of this division much more frequently, in nearly every sample. Negative controls were interspersed throughout the assays, ensuring that the detection of TM7 bacteria was not due to contamination. We used a technique with high sensitivity (real-time PCR) and primers designed to specifically amplify the TM7 division, which allowed us to gain a clearer picture of its prevalence and abundance in the subgingival crevice.
We found that TM7 bacteria made up 0.3% ± 0.03% (SE) of total bacteria in the human subgingival crevice by real-time PCR and 1.8% ± 0.4% by FISH, on average. These estimates are reasonably consistent with the identification of TM7 bacteria in 1.3% of clones in a broad-range study of diversity in the subgingival crevice (24). The presence and prevalence of TM7 in most of the samples analyzed in our study suggest that this division is a common, and perhaps permanent, component of the oral flora, with the capacity to maintain its growth under the disparate conditions of health and severe disease. The known diversity of this bacterial division is expanded by our identification of two new phylotypes of TM7, indicating that there are at least seven genus-level groups of this division found in the human subgingival crevice. Considering that TM7 is a division with potentially hundreds or thousands of species, the observed increase in the proportion of TM7 in mild-periodontitis samples may be due to an expansion of the TM7 bacteria already present at the site or to the replacement of one TM7 species by another which is better adapted to the environment of the mild-periodontitis subgingival crevice.
There are several possible explanations for the preferential association of TM7 with the early stages of periodontal disease. One explanation is that members of this division require an environmental factor which is present at optimal levels only in the early stages of disease. Another explanation is that the further progression of disease creates conditions in which other organisms compete more favorably with TM7. While it is not possible to assign a causal role to TM7 in the development of periodontal disease, the significant increase in relative abundance of this division at sites with mild periodontitis indicates its close link with the changing conditions present in the subgingival crevice as periodontal health deteriorates.
The I025 subgroup was previously associated with some forms of periodontal disease (24) and therefore seemed to be an attractive choice for a more-detailed examination. Fifty-one percent of the samples in the present study contained a detectable quantity of I025 bacteria. I025 rDNA was detected at only 1 of 18 healthy sites but at 38 of 58 diseased sites. It should be noted that the I025 phylotype may itself be a large group containing some members which are more closely disease associated and others which are not.
Real-time PCR is an attractive approach for microbial community analysis because of its large dynamic range and high sensitivity. We were able to decrease the level of interexperimental variability in our real-time PCR assays by creating average standard curves by using standards from several experiments. However, we found that the variability inherent in this type of assay does not allow for reliable detection of differences of less than two- to threefold between groups given the degree of intersample variation we observed. Other researchers have reported similar levels of variability in real-time PCR assay results (2, 10, 23, 32). Although it is much more laborious, FISH provides an independent approach for estimating phylotype abundance.
One factor that may have biased our results was the use of multiple samples from sites in the same patient displaying various degrees of disease and the possibility of intraoral site-to-site seeding. The extent of this phenomenon is largely unknown, but its effect would be predicted to diminish the differences in community composition that might occur solely as a function of disease state. Indeed, Riviere et al. (26), by using immunocytochemical detection methods, found that spirochetes and Porphyromonas gingivalis were detected more frequently at healthy sites of diseased subjects than at healthy sites in periodontally healthy subjects. However, incorporation of bacteria into dental plaque biofilms is governed by highly specific host-microbe and microbe-microbe interactions (29, 30). Since colonization of tooth surfaces and existing biofilms are not random events, it is unlikely that the presence of microflora at diseased sites is a dominant ecological determinant of the types of bacteria that colonize healthy sites. If the latter were true, one would expect to find that most sites in a given patient have the same amount of periodontal disease, which is clearly not the case. Therefore, in this study we presumed that each site provides a unique environment for bacteria and supports its own microbiota, relatively independent of other sites in the mouth. In support of this assumption, we frequently observed patients with detectable I025 rDNA at some sites while other sites within the same patient had no I025.
This study explored the disease associations of an uncultivated division of bacteria. As molecular detection methods are increasingly applied to problems of human health and disease, it is useful to consider the criteria that should be used to evaluate whether a species which is detected solely by its genetic material is pathogenic. Importantly, the detection and quantity of the genetic material of a pathogen should be temporally associated with disease. The presence of the genetic material should reliably predict the development of disease, and the amount of genetic material should decrease with resolution of disease. Another criterion for evaluating pathogenicity is that the genetic material should be spatially distributed in or on tissue in a disease-specific manner, that is, it should be found in higher quantities at sites of disease than at healthy sites from the same patient. Paired samples from individual subjects are useful in this regard. Accurate quantification is also necessary in order to address these issues, especially given the sensitivity and nonquantitative features of PCR when used in a qualitative format. We have been able to determine that the I025 phylotype is rarely present in healthy samples, but data from a greater number of samples will be required before a more complete understanding of the role of these and other TM7 division members in oral disease can be achieved.
Acknowledgments
This work was supported in part by grants from the NIH (NIDCR) to D.A.R. (R01-DE13541), the Ellison Medical Foundation to D.A.R. (ID-SS-0103-01), Procter & Gamble to P.W.L., the NIH (NIAID) to C.C.O. (5-T32 AI07328), and the NIH (NIDCR) to G.C.A. (P60-DE13058) and by the NIH Digestive Disease Research Center at Stanford (DK56339).
We thank Kristofer Jennings of the Stanford University Department of Statistics for help with data analysis, Muna Khan at the UCSF School of Dentistry for help with the collection of plaque samples, and Danielle Perry for assistance with sample preparation and FISH.
REFERENCES
- 1.Bond, P. L., R. Erhart, M. Wagner, J. Keller, and L. L. Blackall. 1999. Identification of some of the major groups of bacteria in efficient and nonefficient biological phosphorus removal activated sludge systems. Appl. Environ. Microbiol. 65:4077-4084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chen, R. W., H. Piiparinen, M. Seppänen, P. Koskela, S. Sarna, and M. Lappalainen. 2001. Real-time PCR for detection and quantitation of hepatitis B virus DNA. J. Med. Virol. 65:250-256. [DOI] [PubMed] [Google Scholar]
- 3.DeLong, E. F. 1992. Archaea in coastal marine environments. Proc. Natl. Acad. Sci. USA 89:5685-5689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.DeSoete, G. 1983. A least squares algorithm for fitting additive trees to proximity data. Psychometrika 48:621-626. [Google Scholar]
- 5.Felsenstein, J. 1985. Confidence limits on phylogenies: an approach using the bootstrap. Evolution 39:783-791. [DOI] [PubMed] [Google Scholar]
- 6.Fredricks, D. N., and D. A. Relman. 1998. Improved amplification of microbial DNA from blood cultures by removal of the PCR inhibitor sodium polyanetholesulfonate. J. Clin. Microbiol. 36:2810-2816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Genco, R. J. 1996. Current view of risk factors for periodontal diseases. J. Periodontol. 67:1041-1049. [DOI] [PubMed] [Google Scholar]
- 8.Hugenholtz, P., B. M. Goebel, and N. R. Pace. 1998. Impact of culture-independent studies on the emerging phylogenetic view of bacterial diversity. J. Bacteriol. 180:4765-4774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hugenholtz, P., G. W. Tyson, R. I. Webb, A. M. Wagner, and L. L. Blackall. 2001. Investigation of candidate division TM7, a recently recognized major lineage of the domain Bacteria with no known pure-culture representatives. Appl. Environ. Microbiol. 67:411-419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kleiber, J., T. Walter, G. Haberhausen, S. Tsang, R. Babiel, and M. Rosenstraus. 2000. Performance characteristics of a quantitative, homogeneous TaqMan RT-PCR test for HCV RNA. J. Mol. Diagn. 2:158-166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kroes, I., P. W. Lepp, and D. A. Relman. 1999. Bacterial diversity within the human subgingival crevice. Proc. Natl. Acad. Sci. USA 96:14547-14552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lane, D. J., B. Pace, G. J. Olsen, D. A. Stahl, M. L. Sogin, and N. R. Pace. 1985. Rapid determination of 16S ribosomal RNA sequences for phylogenetic analyses. Proc. Natl. Acad. Sci. USA 82:6955-6959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lathe, R. 1985. Synthetic oligonucleotide probes deduced from amino acid sequence data. Theoretical and practical considerations. J. Mol. Biol. 183:1-12. [DOI] [PubMed] [Google Scholar]
- 14.Löe, H., and J. Silness. 1963. Periodontal disease in pregnancy. I. Prevalence and severity. Acta Odontol. Scand. 21:533-551. [DOI] [PubMed] [Google Scholar]
- 15.Manz, W., R. Amann, W. Ludwig, M. Wagner, and K.-H. Schleifer. 1992. Phylogenetic oligodeoxynucleotide probes for the major subclasses of proteobacteria: problems and solutions. Syst. Appl. Microbiol. 15:593-600. [Google Scholar]
- 16.Muramatsu, Y., and Y. Takaesu. 1994. Oral health status related to subgingival bacterial flora and sex hormones in saliva during pregnancy. Bull. Tokyo Dent. Coll. 35:139-151. [PubMed] [Google Scholar]
- 17.Murray, A. E., C. M. Preston, R. Massana, L. T. Taylor, A. Blakis, K. Wu, and E. F. DeLong. 1998. Seasonal and spatial variability of bacterial and archaeal assemblages in the coastal waters near Anvers Island, Antarctica. Appl. Environ. Microbiol. 64:2585-2595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Olsen, G. J., H. Matsuda, R. Hagstrom, and R. Overbeek. 1994. fastDNAmL: a tool for construction of phylogenetic trees of DNA sequences using maximum likelihood. Comput. Appl. Biosci. 10:41-48. [DOI] [PubMed] [Google Scholar]
- 19.Ouverney, C., and J. A. Fuhrman. 1997. Increase in fluorescence intensity of 16S rRNA in situ hybridization in natural samples treated with chloramphenicol. Appl. Environ. Microbiol. 63:2735-2740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ouverney, C. C., and J. A. Fuhrman. 1999. Combined microautoradiography-16S rRNA probe technique for determination of radioisotope uptake by specific microbial cell types in situ. Appl. Environ. Microbiol. 65:1746-1752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Page, R. C., and J. D. Beck. 1997. Risk assessment for periodontal diseases. Int. Dent. J. 47:61-87. [DOI] [PubMed] [Google Scholar]
- 22.Parrish, K. D., and E. P. Greenberg. 1995. A rapid method for extraction and purification of DNA from dental plaque. Appl. Environ. Microbiol. 61:4120-4123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pas, S. D., E. Fries, R. A. De Man, A. D. M. E. Osterhaus, and H. G. M. Niesters. 2000. Development of a quantitative real-time detection assay for hepatitis B virus DNA and comparison with two commercial assays. J. Clin. Microbiol. 38:2897-2901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Paster, B. J., S. K. Boches, J. L. Galvin, R. E. Ericson, C. N. Lau, V. A. Levanos, A. Sahasrabudhe, and F. E. Dewhirst. 2001. Bacterial diversity in human subgingival plaque. J. Bacteriol. 183:3770-3783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rheims, H., C. Sproer, F. A. Rainey, and E. Stackebrandt. 1996. Molecular biological evidence for the occurrence of uncultured members of the actinomycete line of descent in different environments and geographical locations. Microbiology 142:2863-2870. [DOI] [PubMed] [Google Scholar]
- 26.Riviere, G. R., K. S. Smith, E. Tzagaroulaki, S. L. Kay, X. Zhu, T. A. DeRouen, and D. F. Adams. 1996. Periodontal status and detection frequency of bacteria at sites of periodontal health and gingivitis. J. Periodontol. 67:109-115. [DOI] [PubMed] [Google Scholar]
- 27.Schenkein, H. A., J. A. Burmeister, T. E. Koertge, C. N. Brooks, A. M. Best, L. V. Moore, and W. E. Moore. 1993. The influence of race and gender on periodontal microflora. J. Periodontol. 64:292-296. [DOI] [PubMed] [Google Scholar]
- 28.Socransky, S. S., R. J. Gibbons, A. C. Dale, L. Bortnick, E. Rosenthal, and J. B. Macdonald. 1963. The microbiota of the gingival crevice area of man. I. Total microscopic and viable counts and counts of specific organisms. Arch. Oral Biol. 8:275-280. [DOI] [PubMed] [Google Scholar]
- 29.Socransky, S. S., and A. D. Haffajee. 2000. Dental biofilms: difficult therapeutic targets. Periodontology 28:12-55. [DOI] [PubMed] [Google Scholar]
- 30.Socransky, S. S., A. D. Haffajee, L. A. Ximenez-Fyvie, M. Feres, and D. Mager. 1999. Ecological considerations in the treatment of Actinobacillus actinomycetemcomitans and Porphyromonas gingivalis periodontal infections. Periodontol. 2000 20:341-362. [DOI] [PubMed] [Google Scholar]
- 31.Strunk, O., O. Gross, B. Reichel, M. May, N. Herman, N. Stuckmann, B. Nonhoff, T. Ginhart, A. Vilbig, M. Lenke, T. Ludvig, A. Bode, K.-H. Schleifer, and W. Ludwig. 1999. ARB: a software environment for sequence data. Department of Microbiology, Tenische Universität München, Munich, Germany.
- 32.Suzuki, M. T., L. T. Taylor, and E. F. DeLong. 2000. Quantitative analysis of small-subunit rRNA genes in mixed microbial populations via 5′-nuclease assays. Appl. Environ. Microbiol. 66:4605-4614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.van Soolingen, D., P. E. de Haas, P. W. Hermans, and J. D. van Embden. 1994. DNA fingerprinting of Mycobacterium tuberculosis. Methods Enzymol. 235:196-205. [DOI] [PubMed] [Google Scholar]
- 34.Zarda, B., D. Hahn, A. Chazinotas, W. Schönhuber, A. Neef, R. I. Amann, and J. Zeyer. 1997. Analysis of bacterial community structure in bulk soil by in situ hybridization. Arch. Microbiol. 168:185-192. [Google Scholar]