

Abstract
Jun N-terminal kinase (JNK) signaling is a highly conserved pathway that controls both cytoskeletal remodeling and transcriptional regulation in response to a wide variety of signals. Despite the importance of JNK in the mammalian immune response, and various suggestions of its importance in Drosophila immunity, the actual contribution of JNK signaling in the Drosophila immune response has been unclear. Drosophila TAK1 has been implicated in the NF-κB/Relish-mediated activation of antimicrobial peptide genes. However, we demonstrate that Relish activation is intact in dTAK1 mutant animals, and that the immune response in these mutant animals was rescued by overexpression of a downstream JNKK. The expression of a JNK inhibitor and induction of JNK loss-of-function clones in immune responsive tissue revealed a general requirement for JNK signaling in the expression of antimicrobial peptides. Our data indicate that dTAK1 is not required for Relish activation, but instead is required in JNK signaling for antimicrobial peptide gene expression.
Keywords: Drosophila , innate immunity, JNK, NF-κB, Relish
Introduction
Innate immune responses are critical for a rapid host defense against pathogens. The signaling pathways that control these responses are present in all multicellular organisms, ranging from humans to flies, and are remarkably well conserved. Although the innate response lacks the antigen recognition capacity of vertebrate adaptive immunity, it is nevertheless complex and crucial for host survival (Medzhitov and Janeway, 1998; Dabbagh and Lewis, 2003; Takeda et al, 2003; Vercelli, 2003). Drosophila melanogaster is a proven genetic model organism for the study of innate immunity and has provided invaluable insights into the control of responses to infection.
Toll and Imd are the founding members of two principal innate immune response signaling pathways in Drosophila. Toll signals through two NF-κB/Rel family transcription factors, Dif and Dorsal, and is required for responses to fungal and Gram+ bacterial infections (Rutschmann et al, 2000a; De Gregorio et al, 2002). Imd signaling controls primarily Gram− bacteria-specific responses through the cleavage and activation of a third Rel family transcription factor, Relish, by the Drosophila caspase Dredd (Stöven et al, 2000, 2003). Relish activation also requires an IκB kinase (IKK) complex that is itself activated by Imd signaling (Rutschmann et al, 2000b; Silverman et al, 2000; Lu et al, 2001; Stöven et al, 2003). The transcriptional targets of Dif and Relish are not entirely distinct. For example, cecropinA expression requires either Relish or Dif, or both, depending on the type and strain of infecting microorganism (Hedengren-Olcott et al, 2004). More than 20 Drosophila genes have been implicated in these signaling pathways and nearly all of them have mammalian homologues with conserved immune functions (Brennan and Anderson, 2004).
Jun N-terminal kinase (JNK) signaling has been linked to stress responses, cell migration, apoptosis, and immune responses in both insects and mammals (Sluss et al, 1996; Leppèa and Bohmann, 1999; Stronach and Perrimon, 1999; Boutros et al, 2002; Dong et al, 2002). JNK activity can be induced by infection, lipopolysaccharide, and inflammatory cytokines such as tumor necrosis factor (TNF) in flies and mammals (Sluss et al, 1996; Boutros et al, 2002; Dong et al, 2002; Igaki et al, 2002; Moreno et al, 2002). Null mutations in JNK signaling components are typically embryonic lethal in flies and thus unlikely to appear as targets of mutagenesis screens designed to detect immune response genes in living animals. An exception to this rule is dTAK1. Overexpression and dominant-negative studies indicated that dTAK1 can act as a JNK kinase kinase (Mihaly et al, 2001; Igaki et al, 2002; Moreno et al, 2002).
Previously characterized dTAK1 mutations, however, showed no apparent JNK-like phenotype, but failed to express Relish-dependent antimicrobial peptides, suggesting a role in the Imd pathway (Vidal et al, 2001). Previous epistasis analysis using the UAS/GAL4 overexpression system (Brand and Perrimon, 1993) to ectopically express dTAK1 placed dTAK1 downstream of imd and upstream of the IKK complex in the Relish signaling pathway (Vidal et al, 2001). In vitro experiments implicated dTAK1 in the IKK-dependent phosphorylation of Relish in S2 cells (Silverman et al, 2003).
We uncovered evidence for a Relish-independent function of dTAK1 in the control of antimicrobial peptide gene expression. Several aspects of Relish activation appeared normal in infected dTAK1 mutant animals, including cleavage, nuclear localization, and promoter binding. We therefore tested if JNK pathway components mediated dTAK1 function in the immune response. We report here several lines of evidence for dTAK1 acting through the JNK cascade in the innate immune response. First, overexpression of Hemipterous, a JNKK, rescued attacin and diptericin expression in dTAK1 mutant animals, whereas overexpression of the downstream Imd component Dredd did not. Second, we found that expression of the Puckered (Puc) phosphatase, an inhibitor of JNK activity, suppressed the expression of antimicrobial peptide genes. To directly test for a JNK requirement in immune signaling, we induced JNK mutant clones in the fat body of larvae. Strikingly, diptericin, attacin, Metchnikowin, and drosomycin expression was lost in the mutant tissue.
We conclude that the JNK pathway is required to mediate dTAK1 signaling during the Drosophila immune response. Furthermore, we propose a model where the JNK and NF-κB signaling are both required to activate antimicrobial peptide gene expression during the immune response in the Drosophila fat body.
Results
Identification of a novel allele of dTAK1
We undertook an EMS mutagenesis screen to isolate adult viable mutations on the X-chromosome that impaired the expression of diptericin in response to bacterial challenge. In addition to immune response defects, one mutation, fb(x)179, exhibited a weakly penetrant maternal effect phenotype that was reminiscent of, and enhanced by, single maternal alleles of JNK pathway components. Recombination mapping, complementation testing, and sequencing indicated that this mutation fell within the Drosophila TGFβ-activated kinase 1 (dTAK1) gene (Figure 1, and not shown) revealing a glycine to aspartate missense mutation in the ATP binding motif in the kinase subdomain I region, which renders the protein an inactive kinase (Figure 1A). Based on these results, we henceforth will refer to fb(x)179 as dTAK1179.
Figure 1.
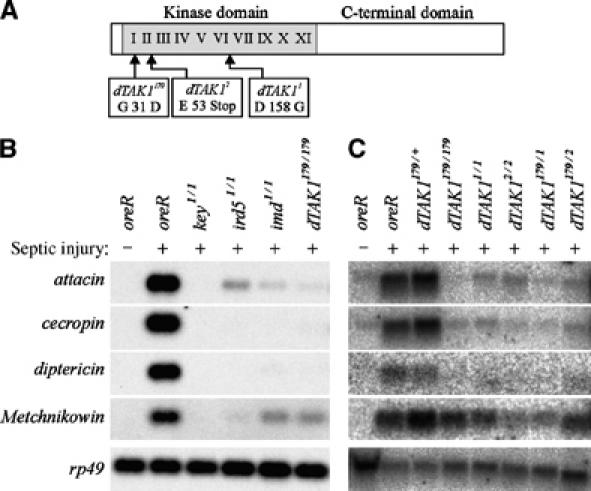
dTAK1 mutations block the expression of antibacterial peptide genes. (A) Diagram of genetic lesions in dTAK1 alleles used in this study. Roman numerals represent the kinase subdomains. (B) Northern blot comparison of dTAK1179 with mutations in Imd pathway genes. (C) Northern blot analysis of dTAK1 mutants and complementation test. Adult flies of the indicated genotypes were infected with E. coli and then incubated for 12 h (B) or overnight (C) at 25°C. RNA was then prepared and analyzed as described in Materials and methods.
dTAK1179 mutant animals failed to express Relish-dependent peptides in response to Escherichia coli infection (Figure 1B and C). We compared dTAK1179 with other imd pathway mutants. dTAK1179 mutants behaved like imd mutants and showed strongly reduced expression of Gram− antimicrobial genes like attacin, cecropin, and diptericin and a more modest reduction in Metchnikowin expression. We also observed reduced defensin, drosocin, and slightly reduced drosomycin expression (data not shown). These expression profiles are comparable to other dTAK1 alleles (Vidal et al, 2001) and complementation tests indicated that dTAK1179 behaves like a null (Figure 1C).
Relish is activated normally in dTAK1 flies and larvae
Ectopic expression of dTAK1, as well as imd and dredd, constitutively activates diptericin expression (Vidal et al, 2001). Like the mammalian NF-κB proteins p100 and p105, Relish is a compound protein with an N-terminal DNA-binding Rel homology domain and a C-terminal inhibitory ankyrin-repeat domain. Signaling via the IKK complex results in Dredd-dependent cleavage of full-length Relish, REL-110, into a nuclear-active N-terminal fragment, REL-68, and a cytoplasmically stable C-terminal fragment, REL-49 (Stöven et al, 2000, 2003). In contrast to current models, Western blot analysis using an antibody specific for the C-terminal domain of Relish revealed that processing of endogenous Relish protein was intact in all three dTAK1 mutant strains including the protein null (Figure 2A). Processing did not occur in key1 mutant animals (Supplementary Figure 1; also see Stöven et al, 2003). We examined the intracellular localization of the REL-68 fragment in control and dTAK1 mutant fat body tissue using an antibody specific for the Rel homology domain (Stöven et al, 2000). Consistent with the above results, we detected an enrichment of REL-68 in the fat body nuclei of infected dTAK1 larvae just as in control animals (Figure 2B).
Figure 2.
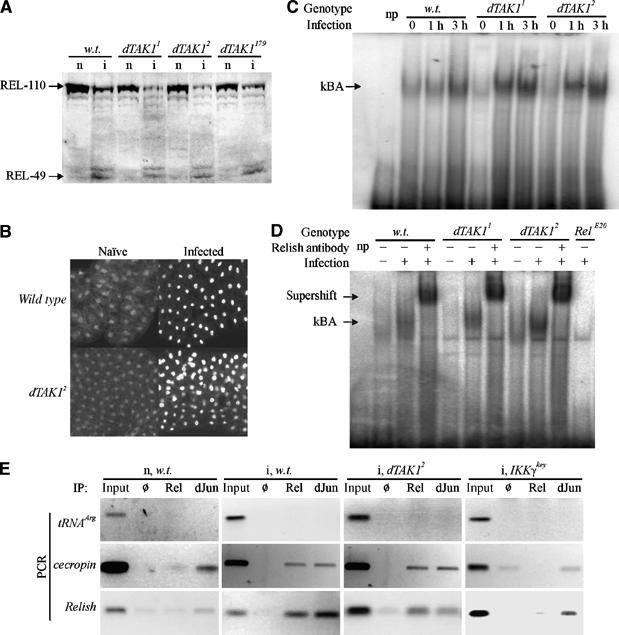
Relish activation is normal in dTAK1 mutant flies and larvae. (A) Relish is cleaved in dTAK1 larvae. Protein extracts were prepared from naïve (n) or infected (i) wandering third-instar larvae of the wild-type (w.t., Canton S), dTAK11, dTAK12, and dTAK179 backgrounds and analyzed by Western blotting with a monoclonal antibody specific for the C-terminal part of Relish, REL-49 (Stöven et al, 2000). (B) Relish is translocated to the nucleus in dTAK12 mutant larvae. Fat body from wild-type (Canton S) and dTAK12 third–instar larvae was fixed and the N-terminal part of Relish was visualized as described in Materials and methods. (C) κBA is present in dTAK1 mutant flies. Flies were challenged with a mixture of M. luteus and Enterobacter cloacae for the times indicated before preparing nuclear extracts and performing EMSA. A lane with no protein (np) is included. (D) Relish is a component of the κBA in TAK1 mutant flies. The κBA in both wild-type and dTAK1 mutant nuclear extracts is shifted by incubation with Relish-specific antibody and no κBA is observed in extracts from Relish mutant flies. The np lane contains no added protein extract. (E) Relish binds to target promoters in dTAK1 mutant fat body. Chromatin immunopurification was performed on extracts from fat bodies of naïve (n) w.t. (Oregon R) and infected (i) w.t., dTAK12, and IKKγkey animals using antibodies-specific for the Relish N-terminus (Rel) or dJun proteins or a blank (Ø) precipitation as indicated. Primers corresponding to sequences proximal to the promoters of tRNAArg (as negative control), cecropinA, and Relish genes were used to detect the presence of these sequences by PCR. Pre-IP input samples were used at 1000-fold dilution to provide a comparable positive control signal.
In further support of the finding that Relish cleavage and nuclear localization were normal in dTAK1 mutant animals, we tested the binding of Relish to the promoters of antimicrobial peptide genes. Drosophila κB binding motifs have been defined that are sufficient for Relish protein binding (Stöven et al, 2000). Using a cecropinA1 κB sequence as probe, we performed electromobility shift assays (EMSAs) to determine if binding activity persisted in dTAK1 mutant animals. We found κB binding activity (κBA) in protein extracts from dTAK1 mutant animals, just as in control extracts (Figure 2C). We confirmed that this κBA was Relish by supershift with Relish-specific antibody and the loss of κBA in extracts from Relish mutant animals (Figure 2D).
We sought to test the association of Relish with endogenous promoters in dTAK1 mutants. The analysis of Relish and association with endogenous promoter elements has been studied using the technique of chromatin immunoprecipitation (ChIP) in Drosophila S2 cells (Kim et al, 2005). We applied the ChIP technique to Drosophila larval fat body cells and compared samples from naïve and infected wild type and immune challenged dTAK1 and IKKγ mutants (Figure 2E). Antibodies specific for the N-terminal domain of Relish (Rel) or the Drosophila Jun (dJun) protein were used to IP the endogenous proteins and associated chromosomal sequences, and primers corresponding to promoters of the tRNAArg, CecropinA, and Relish genes were used to detect co-precipitated DNA by PCR. The tRNAArg promoter does not have any discernable NF-κB or dJun binding sites and thus served as a negative control (Figure 2E, row 1). In comparison, cecropinA and Relish sequences were detected in both Rel and dJun IP samples from infected wild-type larvae. Strikingly, cecropinA and Relish were also detected in infected dTAK1 mutant samples by both Rel and dJun IP (Figure 2E, column 3). Importantly, cecropinA and Relish were not readily detected in naïve, wild type or infected, IKKγkey samples by Rel IP, but were detected by dJun IP, reflecting the requirement for infection and IKKγkey function for Relish activation (Figure 2E, rows 1 and 2). We detected cecropinA and Relish in all dJun IP samples, regardless of experimental conditions, consistent with dJun being always nuclear and associated with promoters, even in the absence of signal (Weiss et al, 2003). In summary, these data indicate that the cleavage, nuclear translocation, and promoter binding activity of Relish persist in dTAK1 mutant animals, but are not sufficient for the expression of the antimicrobial peptide genes.
JNK but not Relish signaling components mediate dTAK1 function
As dTAK1 has been implicated in JNK signaling during developmental patterning and apoptosis (Takatsu et al, 2000; Mihaly et al, 2001; Igaki et al, 2002), we tested downstream JNK signaling components in the context of the innate immune response. The Drosophila gene hemipterous (hep) encodes a JNK kinase and can act downstream of dTAK1 function. For example, null mutants in hep suppressed the planar cell polarity defects of ectopic dTAK1 expression in the Drosophila eye (Mihaly et al, 2001). If dTAK1 acts in JNK signaling rather than imd signaling during the immune response, then activated forms of downstream JNK pathway components might suppress the dTAK1 mutant phenotype. We expressed an activated form of Hep (Hep.CA) in dTAK1 mutant and control flies in the presence and absence of infection (Figure 3A). Unlike imd, dredd, and dTAK1, expression of Hep.CA itself caused no constitutive expression of diptericin. However, expression of activated Hep.CA in dTAK12 but not IKKγkey mutant flies resulted in diptericin expression, but only in response to infection (Figure 3A).
Figure 3.
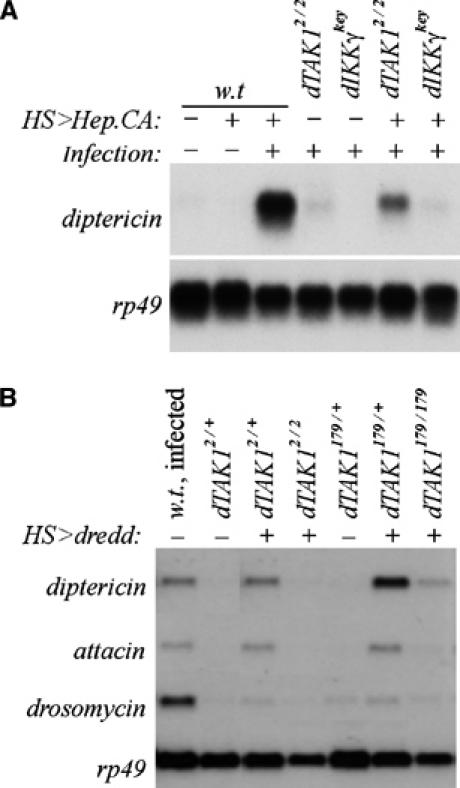
JNK but not Relish signaling components mediate dTAK1 function. (A) Flies of the indicated genotypes were either untreated (−) or subjected to heat shock (+) to active HS-GAL4-driven UAS-Hep.CA expression, and infected with a mixture of M. luteus and E. coli(+). (B) Wild-type Oregon R flies were infected overnight and RNA was prepared. HS-GAL4 and UAS-Dredd transgenes were crossed into dTAK12 and dTAK179 mutant backgrounds. Flies of the indicated genotypes were either untreated (−) or heat shocked (+) and analyzed by Northern blot (see Materials and methods).
As noted previously, overexpression of the Drosophila caspase Dredd can induce the expression of diptericin in the absence of infection (Vidal et al, 2001), presumably owing to ectopic cleavage of Relish. We confirmed that overexpression of Dredd could induce expression of diptericin and attacin in a wild-type background, but that this expression was suppressed in both dTAK2 and dTAK179 mutant animals (Figure 3B) consistent with a model that places dTAK1 downstream or parallel to Dredd. Overexpressed Dredd only weakly induced drosomycin expression. However, this low level of expression was still sensitive to loss of dTAK1 (Figure 3B). Given that Dredd has been shown to play a crucial role in Relish processing (Stöven et al, 2000, 2003) and that Relish cleavage is normal in dTAK1 mutant animals (Figure 2), dTAK1 does not act downstream of Dredd to activate Relish. An alternative role for Dredd has been proposed in the ubiquitin-mediated activation of dTAK1 and the dIKK complex (Zhou et al, 2005). Our data are consistent with this alternate Dredd function and do not exclude the possibility of either function. Nevertheless, we favor a model in which dTAK1 acts in the JNK signaling pathway in parallel to IKK signaling and Relish cleavage to control diptericin induction.
puc, an inhibitor of JNK signaling, suppresses antimicrobial peptide expression
As an independent test of the role of JNK signaling in the Drosophila immune response, we overexpressed the phosphatase Puc in the fat body. Puc is a negative feedback regulator of the JNK pathway that inactivates JNK function (Martin-Blanco et al, 1998). Strikingly, Puc overexpression in the fat body reduced antimicrobial gene expression upon infection by as much as 90% (Figure 4A). Interestingly, Puc suppressed more than strictly Relish-dependent peptides as both metchnikowin and drosomycin expression was also reduced in these animals (see also Discussion).
Figure 4.
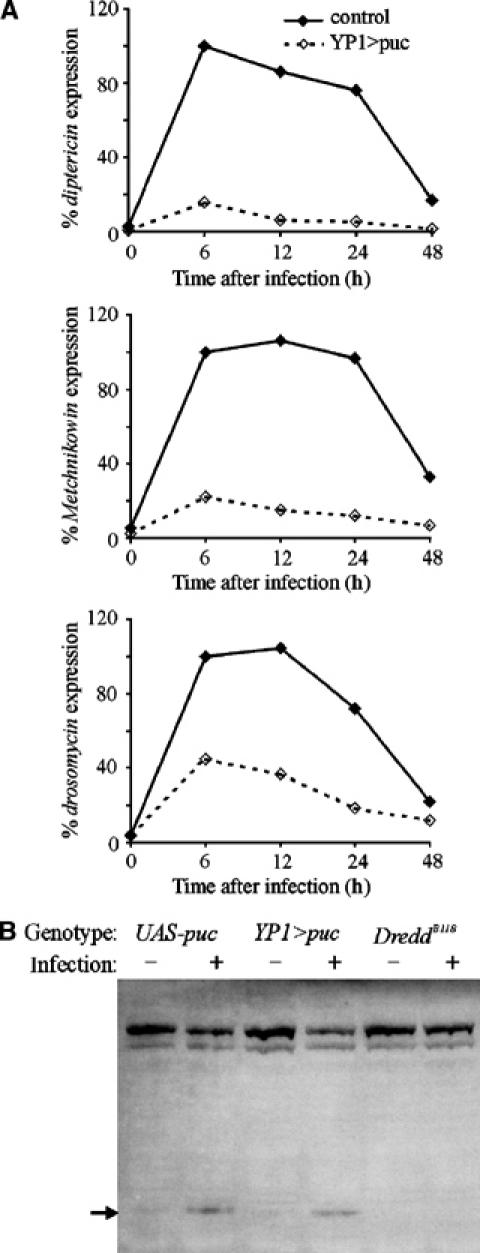
Overexpression of puc, an inhibitor of JNK signaling, suppresses antimicrobial peptide expression. (A) YP1-GAL4, an adult fat body driver, and UAS-puc transgenic lines were crossed. Control flies (solid lines) that carried only the YP1-GAL4 transgene or YP1>puc female flies that carried both transgenes (dashed lines) were analyzed for expression levels of antimicrobial peptide genes after bacterial infection using Northern hybridization. All data were normalized to rp49 signal and presented as percent signal intensity relative to the signal at 6 h, arbitrarily set to 100%. Almost complete elimination of diptericin was observed, and Metchnikowin and drosomycin were also reduced. (B) Overexpression of puc does not block Relish cleavage. Flies were crossed as in (A) and females that carried either the UAS-puc transgene alone or both the YP1-GAL4 and UAS-puc (YP1>puc) were infected as indicated. For comparison, extracts from naïve and infected DreddB118 flies were included that show no Relish cleavage.
Puc phosphatase activity is specific for JNKs and expression of Puc has no known effect on other kinases or pathways, and thus is not anticipated to have any inhibitory effect on IKK signaling (Martin-Blanco et al, 1998). To test this directly, we overexpressed Puc in the fat body of flies and examined Relish cleavage by Western analysis. Relish cleavage was normal in Puc-expressing flies as compared with siblings that lacked the YP1-Gal4 driver (Figure 4B). For comparison, no Relish cleavage was detected in flies mutant for the Dredd caspase (Figure 4B). This corroborates that the function of dTAK1 is independent of, and parallel to, Relish, and that together they have a combinatorial influence on downstream events. Together, these data suggest that JNK signaling is required in the fat body for the normal expression of antimicrobial genes and dTAK1 function.
Antimicrobial peptide gene expression is blocked in JNK mutant clones in vivo
JNKs as a kinase family are well conserved in both structure and in choice of phosphorylation targets. JNK signaling components are also expressed in most tissues. We detected by, RT–PCR, expression of bsk (dJNK) and hep in the larval fat body (data not shown). We also detected dJun and Bsk proteins in the larval fat body, confirming that JNK proponents are present in this tissue (data not shown). As JNK mutations are embryonic lethal, we examined the immune response in FLP/FRT-induced JNK mutant clones (using the null bsk2 and bsk170b alleles) in the fat body of infected larvae (Theodosiou and Xu, 1998; Manfruelli et al, 1999). We probed for the endogenous gene expression of antimicrobial peptides directly by in situ hybridization. Consistent with our hypothesis, we found that JNK−/− clones failed to express diptericin and attacin in response to infection (Figure 5A and data not shown). Consistent with the Puc expression data (Figure 4), JNK mutant tissue also showed reduced expression of metchnikowin and drosomycin (Figure 5B and C).
Figure 5.
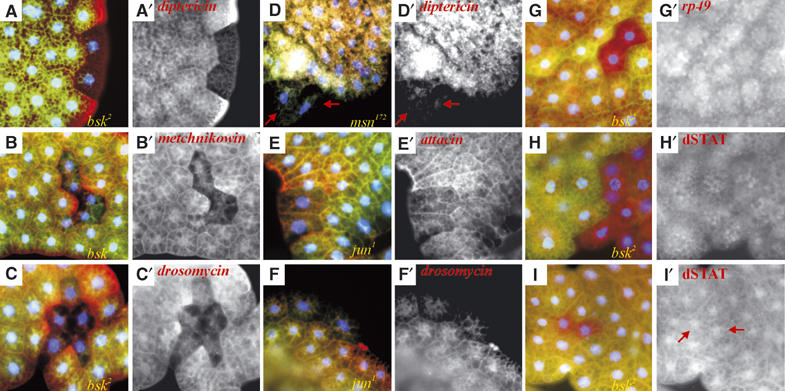
JNK pathway mutations block antimicrobial peptide gene expression in larval fat body tissue. Mosaic bsk2, msn172, and jun1 mutant larval fat body clones were each generated individually and analyzed as described (see Materials and methods). Nuclear Hoechst staining is in blue. Mutant clonal tissue is marked by the absence of GFP in green. Genotypes are noted in the lower right of three-color panels. (A) diptericin expression (red) is absent in bsk2 clones (all clones analyzed, >15, showed the same effect). (A′) diptericin expression shown in single channel. (B) Metchnikowin is lost in bsk2 mutant clones (16 clones analyzed, all showing the same effect) and (C) drosomycin expression (red) is lost in bsk2 mutant clones (again all out of 10 clones analyzed). (D) diptericin expression (red) is absent in msn172 clones (all of five clones analyzed, out of over 300 dissected larvae). Note in this example how the two mutant cells seem to be excluded from the fat body (red arrows). (E) attacin (red) and (F) drosomycin expression (red) is reduced in dJun1 mutant cells (two clones analyzed). (G) rp49 expression (red) is unimpaired in bsk2 mutant clones (seven clones analyzed). (H) dSTAT protein expression is normal in naïve bsk2 mutant clones. (I) dSTAT protein is nuclear in infected bsk2 mutant fat body cells (red arrows in (I′), five clones analyzed). Red channels are shown in (B′, C′, D′, E′, F′, G′, H′, I′).
We also performed similar clonal analyses with alleles of dJun and misshapen (msn, encoding a Drosophila MAPKKKK). Using either the msn102 or msn172 allele, we could only recover very few largely single cell clones. Although diptericin expression was absent in these cells, the mutant cells did not quite appear normal and in some cases were partially excluded from the surrounding tissue, rendering interpretation difficult (Figure 5D). Drosomycin expression was also reduced in mutant cells (not shown). Similarly for dJun, we could not recover mutant tissue for some alleles (dJun2 or dJun3, although we could identify GFP-bright twin spots). We recovered rare clones of the dJun1 allele and found that attacin and drosomycin expression was reduced (Figure 5E and F). In contrast to dTAK1 and bsk, we infer from these results that dJun and msn may be essential for viability of fat body tissue. Nevertheless, these results are consistent with a role for dJun and msn in the immune response in the fat body. In agreement with these data, RNA interference (RNAi) knockdown of kayak/dFos, msn, or hep in S2 cells can also block attacin and drosomycin expression (Kallio et al, 2005).
To control for the health and responsiveness of the mutant fat body cells, we looked at rp49 RNA expression levels and Drosophila STAT (dSTAT) protein levels and nuclear localization in response to infection (Agaisse et al, 2003). Unlike the antimicrobial genes, rp49 expression was not altered in bsk mutant tissue (Figure 5G). Basal dSTAT protein levels were unaltered across bsk mutant clone borders (Figure 5H). Upon infection, dSTAT protein localized normally to the nuclei in bsk mutant tissue (Figure 5I).
Based on the suppression of the immune response by puc overexpression and in bsk mutant clones, we conclude that JNK signaling is an essential component of the Drosophila immune response in the fat body (see also below). Taken together with the rescue of dTAK1 mutants by transgenic JNKK expression, our data indicate that dTAK1 signals through JNK in the immune response.
Discussion
The function of TAK1 in vertebrates has remained enigmatic. It was originally identified as a TGFβ-activated kinase, hence the name, in mammalian cell culture assays (Shibuya et al, 1996; Behrens, 2000). However, follow-up work in multicellular contexts and in vivo analyses in vertebrates, Caenorhabditis elegans, and Drosophila have shown no clear link to TGFβ signaling, but rather suggest a role for TAK family kinases in JNK activation or as upstream activators of Nemo-like kinases (Behrens, 2000). In mammalian systems, TAK1 is one of a number of kinases that can activate IKK complexes and, consequently, NF-κB signaling in vitro. In vitro studies of human cells have shown that targeting of TAK1 by RNAi reduces NF-κB activation by TNFα and IL-1 stimulation (Takaesu et al, 2003). Recent studies using fibroblasts derived from TAK1 mutant mouse embryos and mice with a B-cell-specific deletion of TAK1 showed that JNK activation was impaired in response to all stimuli tested in TAK1 mutant cells (Sato et al, 2005; Shim et al, 2005). Although NF-κB activation was impaired in response to stimulation by IL-1β, TNF, and TLR3 and TLR4 ligands, NF-κB activation by B-cell receptor or LT-β stimulation remained intact, suggesting a specific role for TAK1 upstream of IKKβ and JNK, but not IKKα (Sato et al, 2005; Shim et al, 2005). Interestingly, IKKα activation leads to the phosphorylation and processing of NF-κB2 from the p100 to the active p52 form (Hayden and Ghosh, 2004), reminiscent of Relish activation in Drosophila.
Biochemical analyses in mammalian systems have demonstrated that TAK1 functions in multimeric protein complexes that can include TAB1, TAB2, and different TRAF proteins. The exact composition of these complexes seems to determine TAK1 responsiveness and downstream effects (Takaesu et al, 2003; Hayden and Ghosh, 2004, and references therein). In the fly, genetic studies found an interaction between dTRAF1 and dTAK1 in the activation of JNK signaling and apoptosis (Cha et al, 2003). Gain- and loss-of-function analyses indicate that dTRAF2, but not dTRAF1, is necessary for the activation of Relish-dependent gene expression; however, no interaction between dTRAF2 and dTAK1 in the activation of antimicrobial peptides was reported (Cha et al, 2003).
In vivo versus in vitro studies
Genome-wide analyses that examined in vivo responses in Drosophila identified dJun and puc as genes potentially regulated by Toll and Imd signaling, suggesting a cross-regulation between these pathways and the JNK signaling pathway (De Gregorio et al, 2002). A study recently reported that RNAi knockdown of kayak, msn, hep, or aop blocked E. coli-induced attacin and drosomycin expression in S2 cells (Kallio et al, 2005). Furthermore, in related studies, they also observed that, although dTAK1 RNAi-treated S2 cells failed to express an attacin reporter gene, Relish cleavage and nuclear localization remained intact in these cells (Kleino et al, 2005). Other RNAi analyses in S2 cells concluded that JNK signaling did not have a significant role in antimicrobial peptide gene expression (Boutros et al, 2002; Silverman et al, 2003; Park et al, 2004). However, RNAi against hep or bsk seemed to partially block antimicrobial peptide induction, especially of attacin and cecropinA (Silverman et al, 2003) and, accordingly, attacinD levels were lower in microarrays when the JNK pathway was blocked (Boutros et al, 2002). Our results confirm a positive role for JNK signaling in the antimicrobial peptide response in vivo.
Integration of JNK and NF-κB signaling
The placement of dTAK1 function upstream of JNK, rather than IKK, requires a remodeling of the signaling pathways that activate the antimicrobial peptide genes (Figure 6). Earlier models were based on studies that showed that dTAK1 mutations blocked the constitutive activation of diptericin by Imd overexpression (Vidal et al, 2001). In turn, IKK mutations blocked dTAK1-induced diptericin expression. One interpretation of these data places IKK directly downstream of dTAK1. However, if the activation of both JNK and IKK signaling pathways is required, then a disruption in either branch would be sufficient to suppress any upstream activation.
Figure 6.
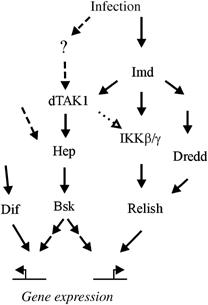
Integration of our data into current models, initially based on Brennan and Anderson (2004). Dotted arrows represent potentially pleiotropic signaling that may occur owing to overexpression. Dashed arrows represent hypothetical signaling events that could initially activate JNK signaling during infection or mediate signaling independently of dTAK1, such as during Dif-controlled drosomycin expression. The question mark (?) represents our speculation that other receptor signaling events, such as TNFR activation, also modulate the innate immune response in the Drosophila fat body.
Overexpression of dTAK1 is sufficient to induce antimicrobial peptide expression (Vidal et al, 2001). However, dTAK1 is an extremely potent activator of JNK signaling and apoptosis (Takatsu et al, 2000; Mihaly et al, 2001), and overexpression of dTAK1 could activate proteins that are not normal phosphorylation targets. Based on RNAi studies in S2 cells, dTAK1 is required for dIKK complex-dependent phosphorylation of Relish in vitro (Silverman et al, 2003). This could reflect a stringent requirement for dTAK1 in blood cell-derived S2 cells that is different in fat body tissue.
The new model (Figure 6) would predict that overexpression of the Dredd caspase would be insufficient to activate fully the antimicrobial peptides in dTAK1 mutant animals and this is indeed the case (Figure 3B). Overexpression of Dredd may be sufficient to induce antimicrobial peptide gene expression in a wild-type background because of inadvertent JNK pathway activation by ectopic caspase activity or by the heat-shock protocol itself (Gibson and Perrimon, 2005). Alternatively, an additional role for Dredd has been proposed in the ubiquitin-mediated activation of dTAK1 and the dIKK complex (Zhou et al, 2005). The suppression by dTAK1 mutants of ectopic Dredd expression is consistent with this model as well, and does not distinguish between the two potential functions of Dredd. Our data are consistent with a model that places dTAK1 activity in a pathway parallel to the functions of IKK and Relish and in which both these pathways are required for the activation of antibacterial peptide genes such as diptericin and attacin.
Promoter analyses of most antimicrobial peptide genes have not revealed any obvious binding sites for activator protein-1 (AP-1) complexes, the Jun/Fos heterodimer, and transcriptional mediator of JNK signaling (Kadalayil et al, 1997; Petersen et al, 1999; Senger et al, 2004). However, AP-1 binding sites can be quite diverse and are not always predictable directly from DNA sequence. Nevertheless, a recent study identified a functional AP-1 binding site in the attacinA promoter (Kim et al, 2005). Their data suggest that AP-1 binding represses attacinA transcription by recruiting histone deacetylase 1 (dHDAC1) to the promoter. In contrast, in mammalian studies, c-Jun function is itself repressed by association with HDAC3. This repression is relieved upon JNK signaling (Weiss et al, 2003). A similar mechanism may be employed in the Drosophila fat body. Accordingly, the sustained expression of attacin and other antimicrobial peptide genes in vivo would require an activation (or de-repression) of AP-1 function at the onset of the immune response. Such positive cooperation between AP-1 and NF-κB transcription factors was also seen in molecular studies of the human β-defensin-2 promoter (Wehkamp et al, 2004).
AP-1-dependent gene expression is normally rapid. Thus, if AP-1 activity is not directly required for diptericin expression, it could act indirectly through the activation of other genes. Alternatively, JNK could phosphorylate some targets other than the AP-1 complex proteins Jun and Fos. In mammalian studies, it has been shown that JNK can phosphorylate, and thereby inhibit, Insulin Receptor Substrate-1 (Lee et al, 2003). However, the recent finding that RNAi against kayak/dFos can block antimicrobial peptide expression and our dJun loss-of-function studies in vivo suggest that JNK does indeed signal through AP-1 to control expression of these genes (Kallio et al, 2005).
It is intriguing that overexpressed Puc not only blocked Relish-dependent antimicrobial peptide gene expression, but it also strongly blocked drosomycin expression, which is not true in dTAK1 mutants (Vidal et al, 2001). This suggests that JNK or JNK-related proteins, for example, p38a, p38b, and MPK2, may also be important for other aspects of the immune response, for example, the Toll/Dif-dependent antimicrobial genes (Sluss et al, 1996; Han et al, 1998). Our clonal analysis of JNK mutant tissue confirms that JNK is required not only for the expression of Gram−-specific peptides diptericin and attacin, but also for Metchnikowin (Gram+/fungal specific) (Levashina et al, 1995) and drosomycin (fungal specific) (Fehlbaum et al, 1994). Mutations in dTAK1 had less of an impact on Metchnikowin or drosomycin expression than on attacin, for example. Furthermore, reduced dJun activation occurred in dTAK1 mutant animals, indicating that other upstream kinases may be involved in the control of these genes. JNK is a member of a large family of mitogen-activated protein kinases (MAPKs). In the fly, there are at least five MAPKKKs, four MAPKKs, and five MAPKs, and so the potential redundancies are many. If these other proteins contribute to the immune response, how they do so has yet to be tested in genetic loss-of-function in vivo studies in the fat body.
How JNK and NF-κB signals integrate to positively control gene expression is a critical question. Here, we have demonstrated that both are required for the expression of a particular set of immune responsive genes in vivo. Through the use of Drosophila genetics, we should be able to identify novel immune response genes that are controlled cooperatively by JNK and NF-κB signaling. From promoter analysis of these genes, we may be able to predict additional genes that are important for other biological processes. Both the JNK and NF-κB signaling pathways have been implicated many times in many different contexts. Continued analysis in Drosophila may lead to a general understanding of their roles in normal biological processes and developmental malignancies.
Materials and methods
Drosophila strains
Oregon R, Canton S, or diptericin-lacZ(X) (Reichhart et al, 1992), the parental strain of the EMS-induced dTAK1179 mutation, was used as a wild-type control. dTAK11 and dTAK12 were described previously (Vidal et al, 2001) as were ird51 (Lu et al, 2001), key1 (Silverman et al, 2000), imd1 (Lemaitre et al, 1995), DreddB118 (Leulier et al, 2000), bsk2, bsk170b (Sluss et al, 1996), msn102, msn172 (Treisman et al, 1997), dJun1, dJun3 (Kockel et al, 1997), and dJun2 (Nusslein-Volhard et al, 1984). For ectopic expression of hemipterous, puckered, and dredd, we employed the UAS/GAL4 system by crossing UAS-Hep.CA (Adachi-Yamada et al, 1999), UAS-puc (Martin-Blanco et al, 1998), and UAS-dredd (Vidal et al, 2001) to either YP1-GAL4, specific for female fat body (Vidal et al, 2001), or to Heat Shock-GAL4 and analyzed the offspring.
Northern analysis
To test antimicrobial gene expression, wandering third-instar larvae and adults were infected overnight with E. coli as described (Wu and Anderson, 1998) and total RNA extracted using Trizol Reagent (Invitrogen). A 5–10 μg portion of RNA was loaded per lane and then transferred to Hybond nylon membrane. Random prime labeled probes (Boehringer) were made for each of the antimicrobial peptides and rp49. For HS>dredd experiments, flies of the indicated genotypes were either untreated (−) or heat shocked (+) at 37°C for 1 h, rested overnight, heat shocked a second time, and rested 6 h. For HS>Hep.CA experiments, flies of the indicated genotypes were either untreated (−) or subjected to heat shock and/or infection with E. coli(+). Infection was just prior to a single 1 h heat shock at 37°C. RNA was prepared after an overnight incubation.
Western analysis and immunohistochemistry
Analysis of endogenous Relish protein was performed as described (Stöven et al, 2000). Preparation of larval fat body and immunohistochemical detection of Relish were carried out as previously described (Stöven et al, 2000) using an affinity-purified rabbit polyclonal antibody specific for the Rel homology domain and anti-rabbit IgG/Cy2 conjugate (Jackson ImmunoResearch). A rabbit polyclonal antibody specific for dSTAT was likewise used to detect dSTAT protein in larval fat body (Chen et al, 2002). Preparations of nuclear extracts were carried out as described (Uvell and Engström, 2003).
EMSA
EMSAs were carried out as described (Uvell and Engström, 2003) using the Cecropin A1 κB sequence as probe: tcgagacacGGGGATTTTTgcac. For supershift experiments, extracts were incubated with 1 μl of relish antiserum (Stöven et al, 2000) before the addition of probe.
ChIP
ChIP was carried largely as described for S2 cells (Kim et al, 2005). For a given experiment, 80–100 larvae were collected for direct processing or infected with a Micrococcus luteus/E. coli mixture for 10–15 min. Larvae were then dissected and fixed in 1% formaldehyde/PBS for 10 min, washed, and then fat body was dissected away from gut and integument and placed directly into lysis solution and then subjected to sonication. Immunopurification was performed with rabbit polyclonal antibodies specific for the Rel homology domain (Stöven et al, 2000) or dJun (Bohmann et al, 1994). The following primers were used for PCR detection of promoter sequences: tRNAArg-forward, 5′-CACAAGCAAACAGCAGAAGTAAAC-3′, tRNAArg-reverse, 5′-CATCGGTTTTATACCTCAAGATGC-3′, cecropinA-forward, 5′-GATTGTTCCCTAGATGTGCAG-3′, cecropinA-reverse, 5′-GCGACTGATGACTGCGATAC-3′, Relish-forward, GAACCGTAGTTTCCGTGAAAAGCT-3′, Relish-reverse, 5′-GCAGCGAATCGGGGAACTTTAGTG-3′.
In situ hybridization of mosaic clones
Mosaic clones were generated from the following crosses, bsk2: w, HS-FLP122; bsk2FRT40A/CyO X w, HS-FLP122; ubi-GFP FRT40A/CyO, msn172: w, HS-FLP122; msn172FRT80/TM6B X w, HS-FLP122; ubi-GFP FRT80, jun1: w, HS-FLP122; jun1FRT42/SM5a:TM6B X w, HS-FLP122; ubi-GFP FRT42. Embryos were collected 2–4 h after egg laying, incubated for 2 h, and then heat shocked for 1 h at 37°C and allowed to develop to third instar at 25°C. GFP+ larvae were sorted by fluorescence microscopy, infected with E. coli, and then allowed to rest for 3 h before dissection and overnight fixation in 8% formaldehyde/0.1% Tween 20/PBS at 4°C. In situ hybridization and subsequent visualizations with DIG-labeled RNA diptericin probe was carried out as described (Hauptmann, 2001; Agaisse et al, 2003). Sheep anti-DIG (Boehringer), biotinylated donkey anti-sheep (Jackson), and TSA TRITC (Perkin-Elmer) reagents were used to visualize in situ probes. FITC-labeled goat anti-rabbit (Jackson) and rabbit anti-GFP (Abcam) antibodies were used to visualize GFP. Nuclei were stained with Hoechst reagent. Analysis of diptericin expression induced by a mixture of M. luteus and Erwinia carotovora and overnight incubations post-infection yielded results similar to Figure 5A.
Supplementary Material
Supplementary Figure 1
Acknowledgments
We thank B Lemaitre and the Bloomington stock center for fly strains, YJ Kim for advice on the ChIP assays, D Bohmann for the dJun antibody and advice, and S Hou for the dSTAT antibody. We are grateful to K Brennan for invaluable discussions and close reading of the manuscript. SS would like to thank D Hultmark for financial support. Some of the experiments were carried out in his laboratory. JRD was partially supported by the Irvington Institute and T32-CA86796. This project was supported by grants from the NIH (RO1-EY13256 to MM) and the Swedish Research Council and the Swedish Cancer Society (to SS and YE).
References
- Adachi-Yamada T, Fujimura-Kamada K, Nishida Y, Matsumoto K (1999) Distortion of proximodistal information causes JNK-dependent apoptosis in Drosophila wing. Nature 400: 166–169 [DOI] [PubMed] [Google Scholar]
- Agaisse H, Petersen UM, Boutros M, Mathey-Prevot B, Perrimon N (2003) Signaling role of hemocytes in Drosophila JAK/STAT-dependent response to septic injury. Dev Cell 5: 441–450 [DOI] [PubMed] [Google Scholar]
- Behrens J (2000) Cross-regulation of the Wnt signalling pathway: a role of MAP kinases. J Cell Sci 113: 911–919 [DOI] [PubMed] [Google Scholar]
- Bohmann D, Ellis MC, Staszewski LM, Mlodzik M (1994) Drosophila Jun mediates Ras-dependent photoreceptor determination. Cell 78: 973–986 [DOI] [PubMed] [Google Scholar]
- Boutros M, Agaisse H, Perrimon N (2002) Sequential activation of signaling pathways during innate immune responses in Drosophila. Dev Cell 3: 711–722 [DOI] [PubMed] [Google Scholar]
- Brand AH, Perrimon N (1993) Targeted gene expression as a means of altering cell fates and generating dominant phenotypes. Development 118: 401–415 [DOI] [PubMed] [Google Scholar]
- Brennan CA, Anderson KV (2004) Drosophila: the genetics of innate immune recognition and response. Annu Rev Immunol 22: 457–483 [DOI] [PubMed] [Google Scholar]
- Cha G-H, Cho KS, Lee JH, Kim M, Kim E, Park J, Lee SB, Chung J (2003) Discrete functions of TRAF1 and TRAF2 in Drosophila melanogaster mediated by c-Jun N-terminal kinase and NF-κB-dependent signaling pathways. Mol Cell Biol 23: 7982–7991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H-W, Chen X, Oh S-W, Marinissen MJ, Gutkind JS, Hou SX (2002) mom identifies a receptor for the Drosophila JAK/STAT signal transduction pathway and encodes a protein distantly related to the mammalian cytokine receptor family. Genes Dev 16: 388–398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dabbagh K, Lewis DB (2003) Toll-like receptors and T-helper-1/T-helper-2 responses. Curr Opin Infect Dis 16: 199–204 [DOI] [PubMed] [Google Scholar]
- De Gregorio E, Spellman PT, Tzou P, Rubin GM, Lemaitre B (2002) The Toll and Imd pathways are the major regulators of the immune response in Drosophila. EMBO J 21: 2568–2579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong C, Davis RJ, Flavell RA (2002) MAP kinases in the immune response. Annu Rev Immunol 20: 55–72 [DOI] [PubMed] [Google Scholar]
- Fehlbaum P, Bulet P, Michaut L, Lagueux M, Broekaert W, Hetru C, Hoffmann J (1994) Insect immunity. Septic injury of Drosophila induces the synthesis of a potent antifungal peptide with sequence homology to plant antifungal peptides. J Biol Chem 269: 33159–33163 [PubMed] [Google Scholar]
- Gibson MC, Perrimon N (2005) Extrusion and death of DPP/BMP-compromised epithelial cells in the developing Drosophila wing. Science 307: 1785–1789 [DOI] [PubMed] [Google Scholar]
- Han ZS, Enslen H, Hu X, Meng X, Wu IH, Barrett T, Davis RJ, Ip YT (1998) A conserved p38 mitogen-activated protein kinase pathway regulates Drosophila immunity gene expression. Mol Cell Biol 18: 3527–3539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauptmann G (2001) One-, two-, and three-color whole-mount in situ hybridization to Drosophila embryos. Methods 23: 359–372 [DOI] [PubMed] [Google Scholar]
- Hayden MS, Ghosh S (2004) Signaling to NF-κB. Genes Dev 18: 2195–2224 [DOI] [PubMed] [Google Scholar]
- Hedengren-Olcott M, Olcott MC, Mooney DT, Ekengren S, Geller BL, Taylor BJ (2004) Differential activation of the NF-κB-like factors Relish and Dif in Drosophila melanogaster by fungi and Gram-positive bacteria. J Biol Chem 279: 21121–21127 [DOI] [PubMed] [Google Scholar]
- Igaki T, Kanda H, Yamamoto-Goto Y, Kanuka H, Kuranaga E, Aigaki T, Miura M (2002) Eiger, a TNF superfamily ligand that triggers the Drosophila JNK pathway. EMBO J 21: 3009–3018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadalayil L, Petersen UM, Engström Y (1997) Adjacent GATA and kappa B-like motifs regulate the expression of a Drosophila immune gene. Nucleic Acids Res 25: 1233–1239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kallio J, Leinonen A, Ulvila J, Valanne S, Ezekowitz RA, Rämet M (2005) Functional analysis of immune response genes in Drosophila identifies JNK pathway as a regulator of antimicrobial peptide gene expression in S2 cells. Microb Infect 7: 811–819 [DOI] [PubMed] [Google Scholar]
- Kim T, Yoon J, Cho H, Lee W-B, Kim J, Song Y-H, Kim SN, Yoon JH, Kim-Ha J, Kim Y-J (2005) Downregulation of lipopolysaccharide response in Drosophila by negative crosstalk between the AP1 and NF-κB signaling modules. Nat Immunol 6: 211–218 [DOI] [PubMed] [Google Scholar]
- Kleino A, Valanne S, Ulvila J, Kallio J, Myllymäki H, Enwald H, Stöven S, Poidevin M, Ueda R, Hultmark D, Lemaitre B, Rämet M (2005) Inhibitor of apoptosis 2 and TAK1-binding protein are components of the Drosophila Imd pathway. EMBO J 24: 3423–3434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kockel L, Zeitlinger J, Staszewski L, Mlodzik M, Bohmann D (1997) Jun in Drosophila development: redundant and nonredundant functions and regulation by two MAPK signal transduction pathways [published erratum appears in Genes Dev 1998 Feb 1;12(3):447]. Genes Dev 11: 1748–1758 [DOI] [PubMed] [Google Scholar]
- Lee YH, Giraud J, Davis RJ, White MF (2003) c-Jun N-terminal Kinase (JNK) mediates feedback inhibition of the insulin signaling cascade. J Biol Chem 278: 2896–2902 [DOI] [PubMed] [Google Scholar]
- Lemaitre B, Kromer-Metzger E, Michaut L, Nicolas E, Meister M, Georgel P, Reichhart JM, Hoffmann JA (1995) A recessive mutation, immune deficiency (imd), defines two distinct control pathways in the Drosophila host defense. Proc Natl Acad Sci USA 92: 9465–9469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leppèa S, Bohmann D (1999) Diverse functions of JNK signaling and c-Jun in stress response and apoptosis. Oncogene 18: 6158–6162 [DOI] [PubMed] [Google Scholar]
- Leulier F, Rodriguez A, Khush RS, Abrams JM, Lemaitre B (2000) The Drosophila caspase Dredd is required to resist gram-negative bacterial infection. EMBO Rep 1: 353–358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levashina EA, Ohresser S, Bulet P, Reichhart J-M, Hetru C, Hoffmann JA (1995) Metchnikowin, a novel immune-inducible proline-rich peptide from Drosophila with antibacterial and antifungal properties. Eur J Biochem 233: 694–700 [DOI] [PubMed] [Google Scholar]
- Lu Y, Wu LP, Anderson KV (2001) The antibacterial arm of the Drosophila innate immune response requires an IkappaB kinase. Genes Dev 15: 104–110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manfruelli P, Reichhart JM, Steward R, Hoffmann JA, Lemaitre B (1999) A mosaic analysis in Drosophila fat body cells of the control of antimicrobial peptide genes by the Rel proteins Dorsal and DIF. EMBO J 18: 3380–3391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin-Blanco E, Gampel A, Ring J, Virdee K, Kirov N, Tolkovsky AM, Martinez-Arias A (1998) puckered encodes a phosphatase that mediates a feedback loop regulating JNK activity during dorsal closure in Drosophila. Genes Dev 12: 557–570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medzhitov R, Janeway CA Jr (1998) An ancient system of host defense. Curr Opin Immunol 10: 12–15 [DOI] [PubMed] [Google Scholar]
- Mihaly J, Kockel L, Gaengel K, Weber U, Bohmann D, Mlodzik M (2001) The role of the Drosophila TAK homologue dTAK during development. Mech Dev 102: 67–79 [DOI] [PubMed] [Google Scholar]
- Moreno E, Yan M, Basler K (2002) Evolution of TNF signaling mechanisms. JNK-dependent apoptosis triggered by Eiger, the Drosophila homolog of the TNF superfamily. Curr Biol 12: 1263. [DOI] [PubMed] [Google Scholar]
- Nusslein-Volhard C, Wieschaus E, Kluding H (1984) Mutations affecting the pattern of the larval cuticle in Drosophila melanogaster. Roux's Arch Dev Biol 193: 267–282 [DOI] [PubMed] [Google Scholar]
- Park JM, Brady H, Ruocco MG, Sun H, Williams D, Lee SJ, Kato T Jr, Richards N, Chan K, Mercurio F, Karin M, Wasserman SA (2004) Targeting of TAK1 by the NF-κB protein Relish regulates the JNK-mediated immune response in Drosophila. Genes Dev 18: 584–594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen UM, Kadalayil L, Rehorn KP, Hoshizaki DK, Reuter R, Engström Y (1999) Serpent regulates Drosophila immunity genes in the larval fat body through an essential GATA motif. EMBO J 18: 4013–4022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reichhart JM, Meister M, Dimarcq JL, Zachary D, Hoffmann D, Ruiz C, Richards G, Hoffmann JA (1992) Insect immunity: developmental and inducible activity of the Drosophila Diptericin promoter. EMBO J 11: 1469–1477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutschmann S, Jung AC, Hetru C, Reichhart JM, Hoffmann JA, Ferrandon D (2000a) The Rel protein DIF mediates the antifungal but not the antibacterial host defense in Drosophila. Immunity 12: 569–580 [DOI] [PubMed] [Google Scholar]
- Rutschmann S, Jung AC, Zhou R, Silverman N, Hoffmann JA, Ferrandon D (2000b) Role of Drosophila IKK gamma in a toll-independent antibacterial immune response. Nat Immunol 1: 342–347 [DOI] [PubMed] [Google Scholar]
- Sato S, Sanjo H, Takeda K, Ninomiya-Tsuji J, Yamamoto M, Kawai T, Matsumoto K, Takeuchi O, Akira S (2005) Essential function for the kinase TAK1 in innate and adaptive immune responses. 6: 1087–1095 [DOI] [PubMed]
- Senger K, Armstrong GW, Rowell WJ, Kwan JM, Markstein M, Levine M (2004) Immunity regulatory DNAs share common organizational features in Drosophila. Mol Cell 13: 19–32 [DOI] [PubMed] [Google Scholar]
- Shibuya H, Yamaguchi K, Shirakabe K, Tonegawa A, Gotoh Y, Ueno N, Irie K, Nishida E, Matsumoto K (1996) TAB1: an activator of the TAK1 MAPKKK in TGF-beta signal transduction. Science 272: 1179–1182 [DOI] [PubMed] [Google Scholar]
- Shim J-H, Xiao C, Paschal AE, Bailey ST, Rao P, Hayden MS, Lee K-Y, Bussey C, Steckel M, Tanaka N, Yamada G, Akira S, Matsumoto K, Ghosh S (2005) TAK1, but not TAB1 or TAB2, plays an essential role in multiple signaling pathways in vivo. Genes Dev 19: 2668–2681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silverman N, Zhou R, Erlich RL, Hunter M, Bernstein E, Schneider D, Maniatis T (2003) Immune activation of NF-kappaB and JNK requires Drosophila TAK1. J Biol Chem 278: 48928–48934 [DOI] [PubMed] [Google Scholar]
- Silverman N, Zhou R, Stoven S, Pandey N, Hultmark D, Maniatis T (2000) A Drosophila Ikappa B kinase complex required for Relish cleavage and antibacterial immunity. Genes Dev 14: 2461–2471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sluss HK, Han Z, Barrett T, Davis RJ, Ip YT (1996) A JNK signal transduction pathway that mediates morphogenesis and an immune response in Drosophila. Genes Dev 10: 2745–2758 [DOI] [PubMed] [Google Scholar]
- Stöven S, Ando I, Kadalayil L, Engström Y, Hultmark D (2000) Activation of the Drosophila NF-kappaB factor Relish by rapid endoproteolytic cleavage. EMBO Rep 1: 347–352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stöven S, Silverman N, Junell A, Hedengren-Olcott M, Erturk D, Engström Y, Maniatis T, Hultmark D (2003) Caspase-mediated processing of the Drosophila NF-kappa B factor Relish. Proc Natl Acad Sci USA 100: 5991–5996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stronach BE, Perrimon N (1999) Stress signaling in Drosophila. Oncogene 18: 6172–6182 [DOI] [PubMed] [Google Scholar]
- Takaesu G, Surabhi RM, Park K-J, Ninomiya-Tsuji J, Matsumoto K, Gaynor RB (2003) TAK1 is critical for IκB kinase-mediated activation of the NF-κB pathway. J Mol Biol 326: 105–115 [DOI] [PubMed] [Google Scholar]
- Takatsu Y, Nakamura M, Stapleton M, Danos MC, Matsumoto K, O'Connor MB, Shibuya H, Ueno N (2000) TAK1 participates in c-Jun N-terminal kinase signaling during Drosophila development. Mol Cell Biol 20: 3015–3026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeda K, Kaisho T, Akira S (2003) Toll-like receptors. Annu Rev Immunol 21: 335–376 [DOI] [PubMed] [Google Scholar]
- Theodosiou NA, Xu T (1998) Use of FLP/FRT system to study Drosophila development. Methods 14: 355–365 [DOI] [PubMed] [Google Scholar]
- Treisman JE, Ito N, Rubin GM (1997) misshapen encodes a protein kinase involved in cell shape control in Drosophila. Gene 186: 119–125 [DOI] [PubMed] [Google Scholar]
- Uvell H, Engström Y (2003) Functional characterization of a novel promoter element required for an innate immune response in Drosophila. Mol Cell Biol 23: 8272–8281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vercelli D (2003) Innate immunity: sensing the environment and regulating the regulators. Curr Opin Allergy Clin Immunol 3: 343–346 [DOI] [PubMed] [Google Scholar]
- Vidal S, Khush RS, Leulier F, Tzou P, Nakamura M, Lemaitre B (2001) Mutations in the Drosophila dTAK1 gene reveal a conserved function for MAPKKKs in the control of rel/NF-kappaB-dependent innate immune responses. Genes Dev 15: 1900–1912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wehkamp J, Harder J, Wehkamp K, Meissner BW-v, Schlee M, Enders C, Sonnenborn U, Nuding S, Bengmark S, Fellermann K, Schroder JM, Stange EF (2004) NF-κB- and AP-1-mediated induction of human beta defensin-2 in intestinal epithelial cells by Escherichia coli Nissle 1917: a novel effect of a probiotic bacterium. Infect Immun 72: 5750–5758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss C, Schneider S, Wagner EF, Zhang X, Seto E, Bohmann D (2003) JNK phosphorylation relieves HDAC3-dependent suppression of the transcriptional activity of c-Jun. EMBO J 22: 3686–3695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu LP, Anderson KV (1998) Regulated nuclear import of Rel proteins in the Drosophila immune response. Nature 392: 93–97 [DOI] [PubMed] [Google Scholar]
- Zhou R, Silverman N, Hong M, Liao DS, Chung Y, Chen ZJ, Maniatis T (2005) The role of ubiquitination in Drosophila innate immunity. J Biol Chem 280: 34048–34055 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1