

Abstract
The overactivation of the HERs, a family of tyrosine kinase receptors, leads to the development of cancer. Although the canonical view contemplates HER receptors restricted to the secretory and endocytic pathways, full-length HER1, HER2 and HER3 have been detected in the nucleoplasm. Furthermore, limited proteolysis of HER4 generates nuclear C-terminal fragments (CTFs). Using cells expressing a panel of deletion and point mutants, here we show that HER2 CTFs are generated by alternative initiation of translation from methionines located near the transmembrane domain of the full-length molecule. In vitro and in vivo, HER2 CTFs are found in the cytoplasm and nucleus. Expression of HER2 CTFs to levels similar to those found in human tumors induces the growth of breast cancer xenografts in nude mice. Tumors dependent on CTFs are sensitive to inhibitors of the kinase activity but do not respond to therapeutic antibodies against HER2. Thus, the kinase domain seems necessary for the activity of HER2 CTFs and the presence of these HER2 fragments could account for the resistance to treatment with antibodies.
Keywords: alternative initiation of translation, breast cancer, HER2
Introduction
Exposure to epidermal growth factor (EGF) leads to modifications in several aspects of the cellular behavior related to the development of cancer (Yarden and Sliwkowski, 2001). EGF binds to HER1 (also known as EGF receptor or ErbB1), which is the prototype of a family that includes three additional members: HER2 (also known as neu or ErbB2), HER3 and HER4 (also known as ErbB3 and ErbB4, respectively) (Burgess et al, 2003). The generation of HER homo- or heterooligomers induces the activation of the intrinsic tyrosine kinase activity of the receptors. The subsequent phosphorylation of intracellular tyrosine residues leads to the recruitment of factors that transfer the signal from the plasma membrane to the nucleus. This induces changes in the expression of genes that coordinately regulate proliferation, migration, adhesion, differentiation and apoptosis (Yarden and Sliwkowski, 2001).
The involvement of HER receptors, and particularly HER2, in the development of a variety of cancers, including breast tumors, has led to the implementation of different therapeutic strategies (Yarden and Sliwkowski, 2001). These include monoclonal antibodies directed against the ectodomain of the receptors, such as Herceptin (also known as Trastuzumab), and small-molecule tyrosine kinase inhibitors (Baselga and Norton, 2002).
Although the canonical view contemplates HER receptors restricted to the secretory and endocytic pathways, accumulating evidence indicates the presence of full-length HER receptors or fragments of them within the cell nucleoplasm. HER1, HER2 and HER3 have been detected as full-length molecules in the nucleus of a variety of cells (Lin et al, 2001; Offterdinger et al, 2002; Wang et al, 2004). A C-terminal fragment (CTF) encompassing the cytoplasmic domain of HER4, generated by the sequential action of two types of proteolytic activities, is also transported to the cell nucleus. The mechanism of solubilization and transport to the nucleus of full-length receptors is still under debate (Oksvold et al, 2002; Wells and Marti, 2002; Carpenter, 2003; Johnson et al, 2004). In contrast, the generation and transport to the nucleus of fragments consisting of the cytoplasmic domain of certain type I transmembrane molecules, including HER4, seems to be a widespread mechanism of transducing signals directly from the plasma membrane to the nucleus (Carpenter, 2003). Upon ligand binding, the extracellular domain of HER4 is cleaved by the metalloprotease disintegrin TACE (tumor necrosis factor alpha converting enzyme) in a process known as ectodomain shedding. The resulting transmembrane-cytoplasmic domain is a substrate of presenilin-like protease(s) that releases the cytoplasmic domain (Ni et al, 2001), which bears a basic amino-acid-based nuclear localization signal (NLS) (Williams et al, 2004).
A series of CTFs, assumed to encompass the transmembrane and cytoplasmic domain of HER2, are frequently found in human mammary tumors (Molina et al, 2002, and references therein); in fact, these fragments are the main HER2 species in some tumors (Molina et al, 2002). Despite the functional potential and the clinical importance of these CTFs, which associate with nodal metastasis (Molina et al, 2002), the mechanism of generation is not understood. By analogy with HER4 and because ectodomain shedding is a frequent event that affects a numerous subset of cell surface proteins (reviewed in Arribas and Borroto, 2002), one possibility is that the HER2 CTFs arise through shedding of the extracellular domain of HER2. In agreement with this hypothesis, a discrete fragment of 95 kDa is generated after the shedding of HER2 by an unidentified metalloprotease (Codony-Servat et al, 1999). However, in contrast to the majority of shedding events, that of HER2 is very inefficient. It affects only a small percentage of the molecules, even when the cells are exposed to potent and nonspecific metalloprotease activators (Codony-Servat et al, 1999; Molina et al, 2001). Thus, the evidence available casts doubts on the hypothesis of ectodomain shedding as the main mechanism of generation of the HER2 CTFs in tumors.
Here, we show that CTFs similar to those observed in vivo are spontaneously expressed in cells stably transfected with the cDNA encoding full-length HER2. Biochemical evidence indicate that these fragments are not generated by proteolytic processing of full-length HER2; they are largely synthesized by alternative initiation of translation from methionines 611 and 687, located right before and after the transmembrane region, respectively. In vitro and in vivo, CTFs localize to the cytoplasm and nucleus of cells. In an effort to determine their possible biological activity and relevance in tumor development, we show that overexpression of HER2 CTFs induces the growth of breast cancer xenografts in nude mice. Tumors dependent on CTFs are sensitive to inhibitors of the kinase activity but do not respond to therapeutic antibodies against HER2. Thus, the kinase domain seems necessary for the activity of HER2 CTFs and the presence of these HER2 fragments could account for the resistance to treatment with antibodies.
Results
Transfection of HER2 into mammalian cells leads to the spontaneous generation of CTFs
As a tool to characterize the mechanism of generation of CTFs, we permanently transfected a cDNA encoding human HER2, tagged at the C-terminus with the HA and hexahistidine epitopes (Figure 1A), into CHO cells. This cell line has been frequently used as a model to study ectodomain shedding (Arribas et al, 1996). In order to select positive clones, we analyzed cell extracts by Western blot with antibodies against the cytoplasmic domain of HER2. In addition to the expected 185 kDa full-length form of the receptor, several clones expressed variable levels of ∼90–100 kDa species (data not shown), likely corresponding to fragments encompassing the intracellular domain of HER2. This conclusion was confirmed with the monoclonal antibodies L87, Herceptin, CB11and Ab3, directed against different regions of human HER2 (Figure 1A); while L87 and Herceptin recognized only full-length HER2 (data not shown), the antibodies against the cytoplasmic domain of HER2 readily detected the ∼95 kDa forms (Figure 1B). The apparent differences in the electrophoretic migration of the CTFs expressed by the three clones (Figure 1B) are attributable to the different levels of expression, because a similar analysis loading equivalent levels of CTFs showed indistinguishable electrophoretic patterns (Supplementary Figure 1A). To delimit further their length, we purified by Ni-chromatography, resolved by SDS–PAGE and trypsin digested the hexahistidine tagged CTFs. The tryptic fragments were analyzed by nano-LC-ESI ion trap mass spectrometry. We consistently identified several peptides, including those shown in Figure 1C. Thus, most of the cytoplasmic portion of HER2, likely containing the whole tyrosine kinase domain is contained in at least some of the CTFs.
Figure 1.

Spontaneous generation of CTFs in cells stably transfected with HER2. (A) Schematic of HER2 showing the N-terminus (N), the transmembrane domain (hatched box), the intracellular kinase domain (shaded box), the hemaglutinin antigen (HA) and hexahistidine (His) tags and the C-terminus (C). The localization of epitopes recognized by the monoclonal antibodies Herceptin® and Ab3 are shown. The monoclonal antibodies L87 and CB11 recognize undefined epitopes located in the extracellular and intracellular domain, respectively. (B) Analysis by Western blot with the indicated antibodies of parental CHO cells (C) and different clones of the same cells stably transfected with HER2 tagged with HA and His. (C) Schematic of peptides detected by mass spectrometry analysis in CTFs purified by Ni-chromatography. The sequence from lysine 676 to lysine 753 is shown. Methionines 687, 706 and 712 are shown in bold. The hatched and shaded boxes represent the transmembrane and tyrosine kinase domains, respectively. Peptides detected by mass spectrometry are indicated with thick lines. (D) Lysates from CHO cells stably transfected with untagged HER2 and from a human mammary tumor expressing CTFs were analyzed by Western blot with the antibody CB11.
To compare the electrophoretic migrations of the HER2 CTFs generated in transfected cells with that of those observed in human mammary tumors, untagged HER2 was stably transfected into CHO cells. Again, Western blot analysis with CB11 antibodies showed that a number of clones expressed variable levels of HER2 CTFs (data not shown). The electrophoretic migration of CTFs present in these clones partially coincide with those of CTFs expressed in human tumors (Figure 1D). Thus, part of the CTFs generated in CHO cells seem similar, if not identical, to those generated in tumors.
Ectodomain shedding is not the main mechanism of generation of HER2 CTFs
The shedding of HER2 produces a soluble fragment of 110 kDa, corresponding to the extracellular domain (HER2 ECD), and a ∼95 kDa cell-associated fragment encompassing the transmembrane and intracellular domains (Codony-Servat et al, 1999). The enzyme responsible is a metalloprotease sensitive to hydroxamic acid-derived compounds such as BB94 (Codony-Servat et al, 1999). To determine whether the HER2 CTFs observed in the stably transfected CHO cell lines arise through proteolytic shedding, we analyze the effect of different concentrations of BB94. As previously shown (Molina et al, 2001), 2 and 5 μM BB94 inhibited the shedding of HER2 in BT474 cells treated with metalloprotease activators (data not shown). However, these concentrations of BB94 do not have an effect on the levels of HER2 CTFs (Figure 2A), indicating that a metalloprotease sensitive to BB-94 is not required for the generation of the HER2 CTFs in CHO cells.
Figure 2.

CTFs are not generated by ectodomain shedding. (A) Stably transfected CHO cells expressing different HER2 species were incubated overnight with the indicated concentrations of the metalloprotease inhibitor BB-94 and analyzed by Western blot with the monoclonal anti-HER2 antibody CB11. (B) Stably transfected CHO cells expressing CTFs (clone #1) were treated for 24 h without (NA) or with 125 nM of γ-secretase inhibitor X, 10 μM of Calpain inhibitor or 10 μM of Caspase-3 inhibitor I and analyzed by Western blot with CB11 antibodies. (C) Parental CHO cells (C) or the same cells as in (A) were metabolically labeled with 35S-Translabel for 15 min, washed, lysed and immunoprecipitated with anti-HA antibodies. Immunoprecipitates were analyzed by SDS–PAGE and fluorography. (D) Top, CHO cells were transiently transfected with a control plasmid (C) or with plasmids encoding HER2 ΔNarI or HER2 as indicated. Cell lysates from transiently transfected cells were analyzed by Western blot with CB11 antiobodies. Bottom: Schematics showing the HER2 constructs transfected. The position of the Nar I sites used to make the HER2 ΔNar I construct are shown in wild-type HER2. The HER2 ΔNar I construct contains a stop codon after glycin 312 and generates a predicted protein of 32 kDa (solid line) the rest of the sequence in this construct is out of frame and is shown in dotted lines. The N-terminus (N), the transmembrane domain (hatched box), the intracellular kinase domain (shaded box) and the C-terminus (C) are shown. The position of the first in-frame methionine is indicated with the number 1. (E) T47D cells permanently transfected with wild-type HER2 mRNA were analyzed by Western blotting with antibodies against the intracellular domain of HER2 or by Northern blotting with probes specific for the extracellular (ECD) or intracellular (ICD) domains of HER2, respectively.
Certain proteolytic enzymes, such as γ-secretease, caspase-3 or calpain have been found to release the intracellular domain of transmembrane proteins (Marambaud et al, 2002; Kopan and Ilagan, 2004; Franco and Huttenlocher, 2005). To investigate the possible role of these proteases in the generation of CTFs from HER2, we treated permanently transfected cells with the corresponding inhibitors. None of the inhibitors prevented the production of CTFs (Figure 2B), arguing that the mentioned proteases do not participate in the generation of CTFs. Interestingly, the calpain inhibitor produced a slight but reproducible increase in the levels of the CTFs, indicating that a protease sensitive to this inhibitor participates in the degradation of CTFs.
Since the protease inhibitors tested did not have a detectable effect on the levels of CTFs, it is possible that CTFs are not the product of full-length HER2 cleavage. To examine this possibility, we analyzed the biosynthesis of CTFs by metabolic labeling and immunoprecipitation. Full-length HER2 was undetectable after short (15 min) metabolic pulses in clones expressing predominantly CTFs (Figure 2C), indicating that CTFs are not generated through proteolysis of the full-length receptor.
Despite the above results, it could be argued that the cleavage of HER2 in CHO cells is carried out by and unidentified protease that acts much faster and efficiently than expected and it is not inhibited by the compounds tested. To directly address whether CTFs are generated by proteolytic cleavage of full-length HER2, we made a HER2 cDNA deletion construct (HER2 ΔNar I). The deletion causes a frameshift after glycine 312 and a translation termination codon three amino acids downstream, preventing the formation of full-length HER2 (Figure 2D, bottom). Theoretically, this constructs generates a protein fragment with an estimated molecular weight of 32 kDa, obviously undetectable with antibodies against the cytoplasmic tail of HER2. As expected, the levels of full-length HER2 are similar in mock transfected cells and in cells transfected with the ΔNar I HER2 construct and dramatically increased in the same cells transfected with the full-length receptor (Figure 2D). However, CTFs are readily detected in cells transfected with the deletion construct (Figure 2D). This result directly shows that the generation of CTFs is independent of the presence of full-length HER2 and unveils the existence of a novel mechanism different from proteolytic processing. As shown in Figure 2D, CTFs are also generated in cells transiently transfected with the full-length cDNA, albeit with much lower efficiency.
The transcription of a short transcript from a cryptic promoter could explain the generation of CTFs. However, in transiently transfected cells such as those analyzed in Figure 2D, only single transcripts, with the size expected for full-length HER2 and the HER2 ΔNar I, respectively, can be detected by Northern blotting (data not shown). Furthermore, a single transcript is also apparent in different cells stably transfected with full-length cDNA encoding HER2 that express CTFs (Figure 2E and data not shown). Therefore, although we cannot rule out the existence of an undetectable mRNA species, alternative transcripts do not seem to explain the generation of CTFs.
CTFs arise from alternative initiation of translation
Considering the previous results, we analyzed the possibility that the HER2 CTFs arise by alternative initiation of translation. Full-length HER2 cDNA transcribed and translated in vitro in the presence of radiolabeled methionine gives rise to the full-length protein and to three main products, similar in size to those observed in transfected cells (Figure 3, WT). The specificity of these bands was assessed by Western blot with anti-HER2 monoclonal antibodies (data not shown). Northern blotting analysis shows the existence of a single transcript (Supplementary Figure 2), further supporting that alternative transcription is not the main mechanism of generation of CTFs. As expected, the deletion construct did not produce full-length HER2, but the generation of the CTFs remained unaltered (Figure 3, ΔNar I). The deduced sequence of the cDNA encoding HER2 bears several methionines (in positions 611, 687, 706, 712, respectively) surrounding the transmembrane domain (Figure 3, schematic). To analyze the possible role of these methionines in the synthesis of the CTFs, we mutated them to alanines, and the resulting constructs were transcribed and translated in vitro. Mutation of methionine 611 prevented the synthesis of the largest protein, indicating that this HER2 species starts before the transmembrane domain. Mutation of methionine 687 prevented the generation of the following protein and induced the synthesis of a slightly smaller species, indicating that methionine 687 acts as an initiator and that in its absence the next methionine (706) is used. Finally, mutation of methionines 706 and 712 did not have any effect, indicating that these methionines are not normally used.
Figure 3.
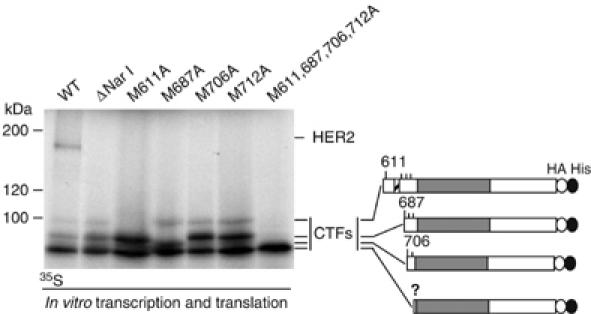
Biosynthesis of HER2 species from different mutant cDNAs in vitro. cDNAs encoding wild-type HER2, a Nar I deletion construct, or the indicated point mutants, also containing the Nar I deletion, were transcribed and translated in vitro in the presence of 35S-translabel and analyzed by SDS–PAGE and fluorography. Right: schematic showing the hypothetical HER2 CTFs generated, the numbers indicate the positions of different methionines (see also, Figure 1).
The smallest species is not affected by any mutation; a CTF starting in the next methionine, located at position 774, would be ∼8 kDa smaller than that initiated at methionine 706. Since the difference between the smaller CTFs corresponds to ∼1 kDa, the fastest migrating CTF likely represents the initiation at a noncanonical codon. However, we did not further investigate this possibility because this HER2 species cannot be detected in cells (see below).
To determine the relevance of these methionines in vivo, the different mutant HER2 constructs were transfected into MCF7 cells. Note that these cells express higher levels of endogenous HER2, compared to CHO cells (Figure 4A and data not shown). Mutation of methionine 611 prevented the formation of the two larger and less abundant CTFs (Figure 4A, CTFs 611), indicating that this methionine works as an initiator in vivo and suggesting that one of these CTFs is a post-transductional modification of the other. Mutation of methionine 687 did not affect the species generate from methionine 611 but resulted in a slight shift in the electrophoretic migration of the shorter and predominant ones (Figure 4A, CTFs 687), likely indicating that these forms arise from methionine 687. The electrophoretic shift corresponds to ∼2 kDa, indicating that, in the absence of methionine 687, CTFs start in methionine 706. Mutation of methionine 706 did not have any effect nor did mutation in methionine 712.
Figure 4.
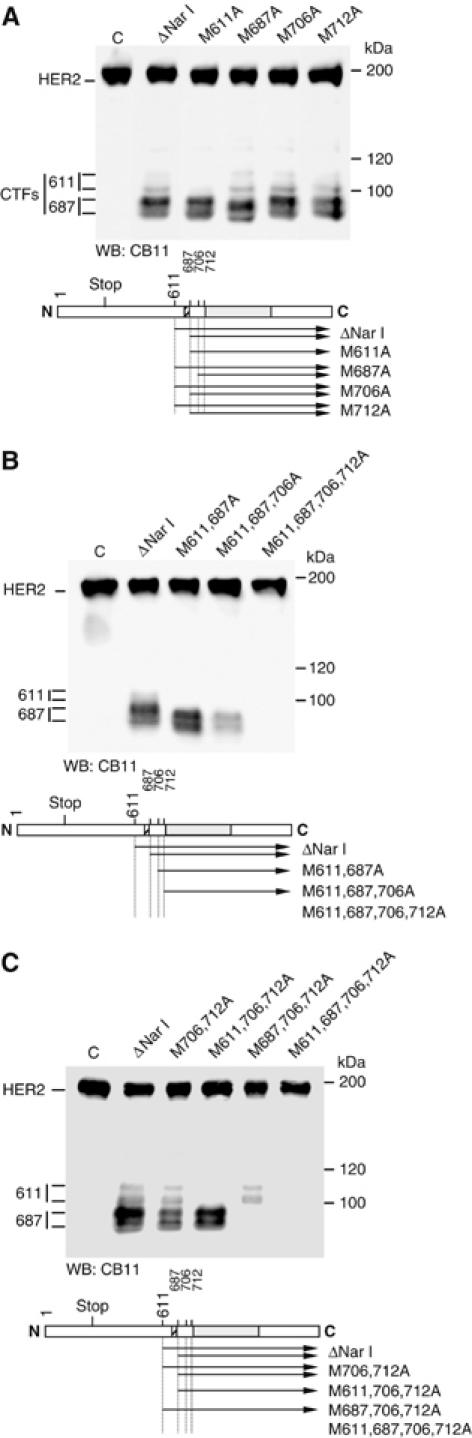
Biosynthesis of HER2 species from different mutant cDNAs in vivo. (A–C) MCF7 cells transiently transfected with a control plasmid (C) with the indicated single, double, triple or quadruple mutant constructs were lysed and cell lysates were analyzed by Western blot with CB11 antibodies. All the mutant constructs contain the ΔNar I deletion. Each panel contains the corresponding diagram showing the CTFs generated by the different constructs.
Next, we transfected double, triple and quadruple mutants. In agreement with the results shown in Figure 4A, mutation of methionines 611 and 687 prevented the generation of the two larger CTFs and shifted the mobility of the two smaller ones (Figure 4B). The 611, 687 and 706 triple mutant generated a product similar to that generated by the double mutant (Figure 4B), indicating that when the first three methionines are absent CTFs can be translated from methionine 712. Supporting this conclusion, the quadruple mutant did not produce any CTF (Figure 4B).
Thus, in agreement with the results of the in vitro transcription and translation, expression of mutant cDNAs in cells indicate that CTFs are translated from methionines 611 and 687; however, the predominant products arise from the latter. To confirm this conclusion we mutated methionines 611 or 687 in cDNAs containing mutations in methionines 706 and 712 (Figure 4C). In this background, mutation of methionine 611 prevented the synthesis of the largest CTFs and mutation of methionine 687 prevented that of the major HER2 species. Collectively, these results strongly support that CTFs arise from alternative initiation of translation from methionines 611 and 687.
At least two products arise from methionine 611 indicating that one of them is a post-translational modification or a proteolytic product of the other. Similarly, more than one product arises from methionine 687. To analyze the possible involvement of proteolytic activities on the generation of different CTFs from the same methionine, we treated cells permanently transfected with the HER2 ΔNar I construct with different inhibitors. In agreement with the results obtained in Figure 2, inhibitors of γ-secretease, caspase-3 or calpain did not affect the generation of the different CTFs, arguing that these proteolytic activities do not participate in their generation (Figure 5). Serine protease inhibitors (leupeptin and aprotinin) or EDTA were also without effect (Figure 5). Interestingly, inhibitors of the proteasome and, to less extent, the calpain inhibitor (which also inhibits the proteasome) increased the levels of the CTFs, particularly those starting in methionine 611, suggesting that the proteasome participates in the degradation of CTFs. Furthermore, high molecular weight species of ∼120 kDa are detected in cells treated with proteasome inhibitors and EGTA (Figure 5). This result indicates that the different species observed are not proteolytic products from larger CTFs and opens the possibility that high molecular weight species of short half-life, normally degraded by the proteasome or a proteolytic activity sensitive to EGTA, are generated from methionines 611 and/or 687.
Figure 5.
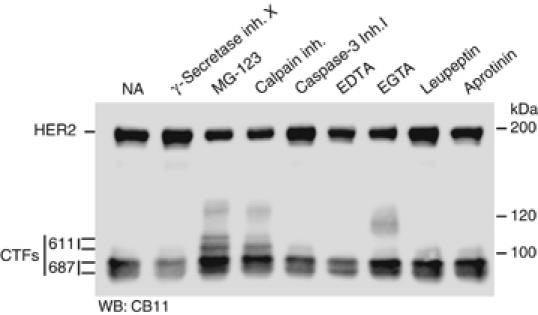
Effect of different protease inhibitors on the expression of CTFs. T47D cells permanently transfected with the ΔNar I construct were treated for 24 h without (NA) or with 125 nM of γ-secretase inhibitor X, 5 μM of the proteasome inhibitor MG132, 10 μM of Calpain inhibitor, 10 μM of Caspase-3 inhibitor I, 10 mM of EDTA, 10 mM of EGTA, 100 μM of leupeptin or 10 μg/ml of aprotinin. Treated cells were lysed and cell lysated analyzed with CB11 antibodies.
Subcellular localization of HER2 CTFs
To analyze the subcellular location of HER2 CTFs, we stained cells stably expressing HER2 CTFs or full-length HER2, respectively, with antibodies against the intracellular domain of HER2. The pattern observed in cells expressing full-length HER2 is that expected for cell surface molecules and, in addition, intracellular vesicles (Figure 6A), likely representing HER2 in transit through the secretory and/or endocytic pathways. In contrast, in CTFs-expressing CHO cells diffuse cytoplasmic and a nuclear staining is apparent (Figure 6A). The same results were obtained using T47D or MCF7 cells transfected with the ΔNar I HER2 construct (data not shown), confirming that the CTFs can be transported to the nucleus of a variety of cell lines. To complement these results, we analyzed the localization of full-length HER2 and CTFs through a simple biochemical fractionation. As expected, full-length HER2 was almost exclusively detectable in the membrane fraction of CHO cells (Figure 6B). In contrast, all detectable CTFs were present in the soluble fraction (Figure 6B).
Figure 6.

Subcellular localization of different HER2 species. (A) Stably transfected CHO cells expressing predominantly HA-tagged full-length HER2 (#3) and HER2 CTFs (#1), respectively, were fixed, permeabilized and stained with anti-HA and FITC-labeled anti-mouse antibodies. (B) The same cells were homogenized and fractionated by ultracentrifugation. Aliquots from soluble and membrane fractions were analyzed by Western blotting and probed with CB11 antibody or, as a control, with anti-β-tubulin antibodies. (C) MDA-MB-468 cells stably expressing CTFs WT or KD versions were fixed, permeabilized and stained with anti-HER2 (CB11) and FITC-labeled anti-mouse antibodies. (D) Homogenates from tumor samples were analyzed by Western blot with the anti-HER2 monoclonal antibody CB11. (E) Immunohistochemical staining with CB11 antibodies of the same tumor samples. In the magnification, nuclear staining is marked with arrows. (F) Tumor 48 was homogenized as in B and the different fractions analyzed by Western with the monoclonal antibody CB11.
Treatment of cells with tyrosine kinase inhibitors that target HER2 prevents the accumulation of the full-length receptor in the nucleus (Wang et al, 2004). Thus, we analyzed the possible role of the tyrosine kinase activity on the subcellular localization of the CTFs. To avoid interference with full-length HER2, we used MDA-MB-468 cells, which do not express detectable levels of the receptor. In agreement with the results shown in Figure 6A, analysis of MDA-MB-468 expressing CTFs cells showed nuclear staining (Figure 6C). In contrast, CTFs bearing a point mutation that disrupts the kinase activity of HER2 show a cytoplasmic distribution and accumulate in cell–cell contacts (Figure 6C). Thus, the tyrosine kinase activity seems to be required for the accumulation of CTFs in the cell nucleus.
To analyze the subcellular localization of the CTFs in vivo, we analyzed by immunohistochemistry six samples from human mammary tumors: three expressing predominantly full-length HER2 and three samples expressing detectable levels of CTFs as judged by Western blot (Figure 6D and data not shown). As expected, the signal corresponding to samples expressing full-length HER2 stained with antibodies against the intracellular domain of HER2 was largely associated to membranes (Figure 6E, sample 52 and Supplementary Figure 3B). In samples expressing CTFs, in addition to the membrane pattern, a clear cytoplasmic signal and occasional nuclear staining is apparent (Figure 6E, sample 48 and Supplementary Figure 3B). In both samples, predominant surface staining was apparent with antibodies against the extracellular domain of HER2 that do not detect CTFs (supplementary Figure 3B). Biochemical fractionation of tumor samples confirmed that full-length HER2 distribute largely to the membrane fraction (Figure 6F). The high molecular weight HER2 CTFs observed in tumors co-fractionated with membranes, indicating that part of the HER2 CTFs observed in tumors may contain the transmembrane domain and, therefore, could be generated by alternative initiation of translation from methionine 611 or ectodomain shedding (Figure 6F). In contrast, HER2 CTFs of low molecular weight are soluble proteins. Thus, in contrast to the full-length receptor, some HER2 CTFs are soluble proteins that are expressed in the cytoplasm and nucleus of cells.
Biological activity of HER2 CTFs
Oligomerization of HER2 with other HER receptors leads to the activation of their tyrosine kinase activity (Yarden and Sliwkowski, 2001). As a first approximation to characterize the biological activity of the CTFs, we analyzed their phosphotyrosine content. Western blot analysis with anti-phosphotyrosine antibodies showed that the CTFs of higher molecular weight contain phosphotyrosines (Figure 7A), indicating that, similarly to the full-length receptor, they can be activated by phosporylation.
Figure 7.
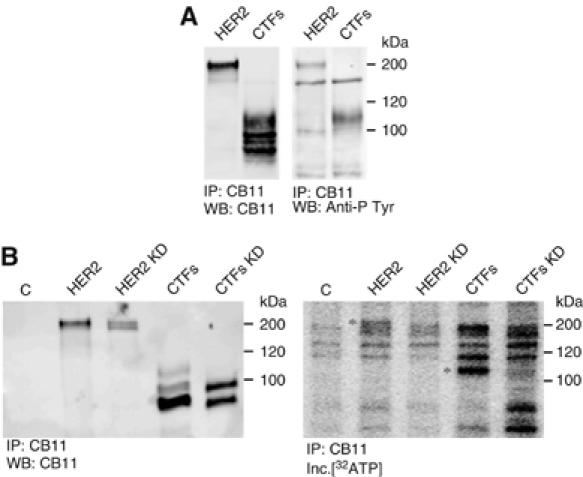
Phosphorylation of CTFs. (A) MDA-MB-468 cells transfected with wild-type HER2 or a deletion construct that encodes CTFs were lysed. Cell lysates were immunoprecipitated with CB11 antibodies and analyzed by Western blot with the same antibodies or anti-phosphotyrosine antibodies as indicated. (B) Cell lysates from parental cells MDA-MB-468 (C) or the same cells transfected with constructs expressing HER2 or CTFs (WT or KD versions) were lysed and immunoprecipitated as in (A). Immunoprecipitates were incubated with [γ32ATP] and analyzed by SDS–PAGE and autoradiography.
Next, we analyzed the possible kinase activity of the CTFs. Incubation of enolase, a substrate frequently used for in vitro kinase asssays, with immunoprecipitated CTFs, did not result in the phosphorylation of the former (data not shown). However, in this assay we could consistently detect specific phosphorylation of the CTFs (see Figure 7B), opening the possibility that the kinase domain of the CTFs is active and that can autophosphorylate. The introduction of a point mutation that abolishes the kinase activity of HER2, also abolishes the phosphorylation of CTFs (Figure 7B), indicating that phosphorylation observed is carried out by the CTFs and not by a unidentified co-precipitating kinase. These results strongly support that CTFs are active kinases.
One classical biological assay for HER2 is the growth of xenografts in nude mice (see, for example, Baselga et al, 1998). In agreement with previous reports, parental T47D exhibited low tumorigenic potential in this system (Bagatell et al, 2001). To analyze the effect of HER2 overexpression, we used clones expressing the full-length protein (Figure 8A). As expected, overexpression of HER2 lead to the vigorous growth of tumors (Figure 8B). To analyze specifically the effect of HER2 CTFs, we transfected T47D cells with the ΔNar I deletion construct. Interestingly, even levels of HER2 CTFs lower than those observed in certain tumors, as judged by immunohistochemistry and Western blot (Supplementary Figure 1 and Supplementary Table I), clearly augmented the tumorigenic potential of T47D cells, further supporting the biological activity of this HER2 isoform and indicating its involvement in the development of tumors. As expected, treatment with Herceptin reduced the size of tumors induced by T47D/HER2 cells. In contrast, the tumors induced by overexpression of the CTFs were insensitive to this treatment indicating that HER2 CTFs could be involved in the resistance to treatment with Herceptin.
Figure 8.
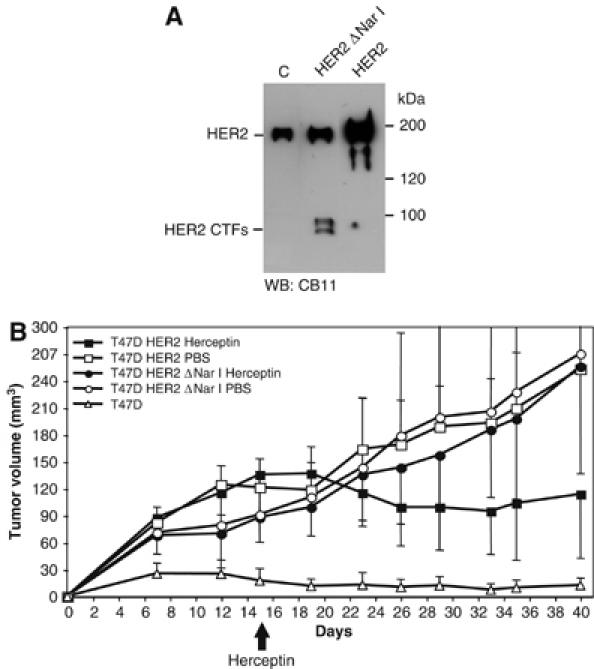
Analysis of the tumors induced by parental T47D cells and the same cells expressing full-length HER2 or HER2 CTFs. (A) Parental T47D cells (C) or the same cells stably transfected with cDNAs encoding HER2 ΔNar I or HER2 were lysed and the cell lysates analyzed by Western blot with antibodies against the cytoplasmic domain of HER2 (CB11). (B) The same cells were subcutaneously injected into nude mice. Tumor volumes were measured at the indicated days after injection. Herceptin was given (i.p.) from day 15 (arrow) twice a week at a dose of 30 mg/kg. The control group was treated with PBS. Each point represents the mean of six individual determination±s.d.
We next determined whether the tyrosine kinase activity of the CTFs contributes to the generation of tumors. Lapatinib, an inhibitor that targets the kinase domain of HER1 and HER2 readily prevented the phosphorylation of the CTFs (Figure 9A), indicating that it is a useful reagent to analyze the effect of the kinase activity on the development of tumors. As shown in Figure 9B, lapatinib clearly reduced tumor growth, indicating that the kinase activity of CTFs does contribute to tumor growth and that, tyrosine kinase inhibitors could be a good approach to treat tumors expressing CTFs.
Figure 9.

Effect of Lapatinib on the phosphorylation of CTFs and on tumour growth. (A) Cells stably transfected with cDNAs encoding HER2 ΔNar I were treated with lapatinib (10 μM) during 8 h. Cell lysates were immunoprecipitated with anti-HA antibodies and analyzed by Western blot with the same antibody or anti-phosphotyrosine antibodies as indicated. Note that the T47D clone used in (A) express higher levels of CTFs than that used in Figure 8B. (B) The same cells T47D cells expressing CTFs used in Figure 8A and B were subcutaneously injected into nude mice. Tumor volumes were measured at the indicated days after injection. Lapatinib was given orally from day 22th (arrow) twice a day at a dose of 75 mg/kg. The vehicle was administered to the control group.
Discussion
A novel mechanism to generate nuclear fragments of HER2 receptors
It is becoming increasingly clear that, in addition to the canonical signaling mode, HER receptors can function in a more direct way by becoming signal transducers that are transported to the cell nucleus where they probably modulate gene expression (reviewed in Carpenter, 2003). On the one hand, full-length HER1, HER2 and HER3 have been detected in the nucleoplasm under certain circumstances (reviewed in Wells and Marti, 2002). On the other hand, a proteolytic fragment of HER4, encompassing the cytoplasmic domain, has been shown to contain a functional NLS (Williams et al, 2004). Our results unveil a novel mechanism to generate soluble HER2 fragments (CTFs) with the ability to accumulate in the cell nucleus: alternative initiation of translation. The major fragments produced encompass the cytoplasmic domain and start immediately after the transmembrane domain. Consistent with this prediction, CTFs are expressed as soluble proteins in the cytoplasm and nucleus of cells, and the same distribution can be seen in samples from mammary tumors expressing CTFs. Thus, HER2 joins the growing list of proteins containing apparent internal translation initiation sites (reviewed in Komar and Hatzoglou, 2005).
A recent report indicates that a juxtamembrane intracellular region rich in basic aminoacids (aa 676-KRRQQKIRKYTMRR-689) is required for the nuclear accumulation of full-length HER2 (Giri et al, 2005). This region is absent in the most abundant CTFs analyzed in this report, which start at methioning 687. Thus, the mechanism of nuclear localization of full-length HER2 is likely different from that of the CTFs.
CTFs are not generated exclusively by ectodomain shedding
Previous reports indicated that HER2 CTFs were likely generated through shedding of the full-length molecule by metalloproteases (Codony-Servat et al, 1999; Molina et al, 2002). However, the basal shedding of HER2 affects a small proportion of the molecules expressed by the cell lines analyzed (∼5%, see Codony-Servat et al, 1999) and it reaches only 20% of the molecules, when cells are exposed to potent and nonspecific metalloprotease activators (Molina et al, 2001). The results with tumors indicate that ectodomain shedding, and/or alternative initiation from methionine 611, can account for the high molecular weight CTFs. The CTFs of lower molecular size are soluble proteins similar, if not identical to those observed in transfected cell lines, showing that in cells and, very likely, in vivo, alternative initiation of translation from methionine 687 is a relevant mechanism of generation of CTFs.
In vitro transcription and translation experiments with the cDNA of HER2 show that the methionines located in positions 611 and 687 (before and after the transmembrane domain, respectively) can act as initiators. In agreement with these results, in vivo, methionine 687 and, to a lesser extent, methionine 611 seem to be the main initiators of translation. Apparently, the several CTFs observed arise only from two methionines in cells. Thus, the different species observed in SDS–PAGE gels are likely due to post-translational modifications of two polypeptides. One of these modifications is, as expected, phosphorylation as judged by immunoreactivity with anti-phosphotyrosine antibodies, but we cannot exclude others.
Alternative usage of different start codons
A question immediately arises from these results. Why in some transfected cells are methionines 611 and 687 used instead of methionine 1? This question is relevant to explain the great variability in the levels of CTFs expressed in tumor samples (Molina et al, 2002). A similar situation has been observed for Notch2 or Hairless (Lauring and Overbaugh, 2000; Maier et al, 2002). It has been shown that the coding region of both proteins contains an internal ribosome entry site (IRES) that allows 5′cap-independent recruitment of ribosomal subunits to mRNAs (Martinez-Salas et al, 2004). An obvious possibility is the existence of an IRES within the coding sequence of HER2 and we are currently addressing it. IRES are tightly regulated under situations not completely understood (Komar and Hatzoglou, 2005). The characterization of the regulation of a putative IRES within HER2 would help to address the variability of the expression of these HER2 species in cells and in tumors.
CTFs promote the formation of tumors
Despite the fact that CTFs were described some time ago, it has been difficult to assay its possible role in the development of tumors. The identification of the mechanism of generation allows directly addressing this point. The transfection of cells with deletion cDNA mutants lacking the initial methionine allows the specific overexpression of CTFs. The vigorous growth of different cells types overexpressing HER2 CTFs in nude mice clearly show the tumorogenic potential of the CTFs. In view of the early reports showing that the cytoplasmic domain of HER2 induces potent transactivation of transcription (Xie and Hung, 1994), a possible scenario would be the modification of transcription of certain genes by the CTFs that, in turn, would lead to tumor growth.
Materials and methods
Reagents and antibodies
Herceptin was kindly provided by Genentech Inc. (South San Francisco, CA). Lapatinib was kindly provided by GlaxoSmithKline (Research Triangle Park, NC). The following monoclonal antibodies were purchased: CB11 (Biogenex, San Ramon, CA), L87 (NeoMarkers, Fremont, CA), anti-Phosphotyrosine, clone 4G10 (Upstate, Lake Placid, NY), c-neu (Ab-3), anti-actin (ab-1) (both from Oncogene research Products, Boston, MA), anti-β-tubulin (Clone 2-28-33) (Sigma, Saint Louis, MI), anti-HA hybridome (Babco, Richmon, CA). Secondary antibodies conjugated to horseradish peroxidase or FITC were from Amersham Biosciences (Little Chafont, UK).
BB-94 was provided by British Biotech Pharmaceuticals, Ltd (Oxford, UK). Caspase-3 inhibitor I, MG132, γ-secretase inhibitor X, EDTA and EGTA were from Calbiochem (Dramstatd, Germany). Calpain Inhibitor I, Leupeptin and Aprotinin were purchased from Sigma (Saint Louis, MI).
Cell culture and tissue samples
CHO, MCF7, MDA-MB-468 and T47D cells were obtained from the American Type Culture Collection (Rockville, MD) and grown as monolayers at 37°C and 5% CO2/air in DMEM/F12 supplemented with 10% fetal bovine serum and 2 mM of glutamine (all from Life Technologies, Inc. Ltd, Paisley, UK).
Breast tissues used in this study were surgical resections specimens obtained at Vall d'Hebron University Hospital (Spain) following Institutional Guidelines. All samples shown in this report were positive by FISH.
Plasmids and transfections
All the cDNA constructs were generated using standard techniques. The final constructs were confirmed by sequencing, subcloned in the pcDNA3.1 Zeo(+) and cotransfected in CHO, MDA-MB-468 and T47D cells with the selectable plasmid pREP4 at a DNA ratio of 1:10 using the calcium phosphate precipitation method in CHO cells or the FuGENE 6 Transfection Reagent (Roche, Indianapolis, IN) following the instructions of the manufacturer in T47D and MDA-MB-468 cells. Stable transfectants were selected and subcloned in 500 μg/ml hygromycin (lifetechnologies, Inc.) for CHO cells or in 150 μg/ml hygromycin for T47D and MDA-MB-468 cells. The expression of HER2 was analyzed by Western blot.
Transient transfections in MCF7 cells were performed using the FuGENE 6 Transfection Reagent following the instructions of the manufacturer.
Western blot
Lysates from breast tissues and cultured cells were prepared and analyzed by Western blot as previously described (Molina et al, 2001).
LC MS/MS analysis of CTFs
Exponentially growing CHO cells expressing CTFs were lysed in lysis buffer without EDTA. After removal of cell debris by centrifugation lysates were incubated with Ni-NTA Agarose (Qiagen) during 2 h at 4°C. Agarose beads were washed twice during 10 min with lysis buffer with 20 mM imidazole and eluted in 40 μl with lysis buffer with 400 mM imidazole. The eluted sample was separated by 1D SDS–PAGE (8% gel) and bands were detected by silver staining. An aliquot of the sample was analyzed in the same conditions by Western blot with CB-11 antibody. Bands corresponding to the electrophoretic mobility of CTFs, as observed by WB, were excised from the silver stained gel and subjected to in-gel trypsin digestion. Tryptic digests were then analyzed by fractionation through a 75 μm i.d. 15 cm PepMap nanoseparation reversed phase column (LC Packings) by using an acetonitrile gradient (0–60%, 35 min), coupled to a nanospray ionization source. MS analysis of the separated peptides was performed on a Bruker Esquire HCT ion trap mass spectrometer. MS_MS fragmentation (100–2800m/z) was performed on the two most intense ions, as determined from a survey scan (1.5 s, 300–1500m/z), which were then actively excluded for 1.2 min. Peptide sequences were identified using MASCOT (MatrixScience), querying against the National Center for Biotechnology Information nonredundant protein database with human sequence filtering.
Northern blot
Total RNA was isolated with the RNeasy kit from Qiagen. For Northern analysis, 10 μg RNA were separated in 1% formaldehyde-agarose gels. Gels were transfered to Gene-Screen Plus nylon membranes and RNA fixed by UV-crosslinking. Blots were prehybridized at 65°C for 3 h in Rapid-Hyb buffer (Amersham Biosciences, Little Chafont, UK). cDNA probes for the extracellular or the cytoplasmic domain of human HER2 were labeled by random priming using [32P]-dCTP (esp. act. 3000 Ci/mmol) and Klenow DNA polymerase. Probes were purified, diluted to 8 × 105 c.p.m./ml in Rapid-Hyb buffer and incubated with the membranes for 4 h at 65°C. Background was reduced by sequential washing in 2 × SSC (3 M sodium chloride, 0.3 M sodium citrate, pH 7.5), 0.1% SDS at room temperature for 30 min and in 0.1 × SSC, 0.1% SDS at 65°C for another 30 min. Membranes were exposed and developed using a Molecular Imager FX imaging system (Bio-Rad Labs).
Metabolic labeling and immunoprecipitation
Exponentially growing CHO cells expressing different HER2 isoforms were metabolically labeled with 1 mCi/ml of 35S-Translabel, lysed and immunoprecipitated with anti-HA monoclonal antibodies as previously described (Arribas and Massagué, 1995). Immunoprecipitates were analyzed by SDS–PAGE and fluorography.
In vitro transcription and translation
Transcription and translation of wild-type HER2, different deletion or point mutants was performed using the TNT T7 coupled reticulocyte lysate system (Promega, Madison, WI) following the instructions of the manufacturer.
Immunofluorescence
Cells growing exponentially were fixed with 4% paraformaldehide for 30 min, permeabilized with saponin 0.1% during 30 min, and then immunostained with primary antibodies (1:100 dilution in PBS with 1% BSA and saponin 0.1%) for 1 h at room temperature. After washes with PBS, the FITC conjugated secondary antibody was applied for 1 h at room temperature. Cells were mounted in Mowiol and analyzed with confocal microscopy.
Biochemical fractionation
Exponentially growing cells expressing different HER2 species or breast tissue samples were detached with PBS with 10 mM EDTA and washed twice with PBS. Cells were resuspended in hypotonic buffer (20 mM HEPES pH=7.4, 10 mM KCl, 10% glycerol, 1 mM EDTA, 1 mM MgCl2, 50 μg/ml aprotinin, 2 mM phenylmethylsulfonyl fluoride, and 2 mM of sodium orthovanadate) and homogenized using a 30-gauge needle. The samples were then centrifuged at 1500 g during 10 min to remove nuclei and cell debris. Membranes were pelleted from the postnuclear supernatants by centrifugation for 1 h at 100 000 g and resuspended in lysis buffer.
Immunohistochemistry and FISH in tumor samples
Excised breast tumors were immediately fixed in 10% buffered neutral formalin for 18 h, and then were dehydrated and paraffin embedded under vacuum.
Immunohistochemistry was performed using 4-μm tissue sections placed on plus-charged glass slides. After deparaffinization in xylene and graded alcohols, antigen retrieval was performed by 10 mM pH 6 citrate buffer (DAKO, Carpinteria, CA) heating in a pressure cooker for 5 min. Endogenous peroxidase was blocked by immersing the sections in 0.03% hydrogen peroxide for 5 min. Slides were incubated with mouse monoclonal anti-HER2 intracellular domain antibody (clone CB11, BioGenex, SanRamon, CA) diluted 1:20 for 60 min at room temperature, followed by incubation with a mouse anti-immunoglobulin horseradish peroxidase-conjugated (DakoCytomation, Carpinteria, CA) to detect antigen–antibody reaction. Sections were then visualized with 3,3′-diaminobenzidine for 5 min and were counterstained with hematoxylin.
Immunofluorescence was performed on two sequential 4-μm tissue sections placed on plus-charged glass slides. After deparaffinization in xylene and graded alcohols, antigen retrieval was performed by 10 mM pH 6 citrate buffer (DAKO, Carpinteria, CA) heating in water bath for 40 min. Nonspecific protein bindings were blocked by immersing the sections in a TBS and BSA 5% solution for 10 min.
First section was incubated with a mouse monoclonal anti-HER2 intracellular domain antibody (clone CB11, BioGenex, SanRamon, CA) diluted 1:20 during 60 min at room temperature, followed by incubation with an Alexa fluor 568 goat anti-mouse IgG (Molecular Probes, Eugene, OR) diluted 1:700 for 30 min to detect antigen–antibody reaction.
Second section was assayed with a goat anti-HER2 extracellular domain antibody (R&D Systems Inc., Minneapolis, MN) diluted 1:25 for 120 min at room temperature, followed by incubation with an Alexa fluor 568 rabbit anti-goat IgG (Molecular Probes, Eugene, OR) diluted 1:350 for 30 min to detect antigen–antibody reaction. All of the immuhistochemical and fluorescence assays were performed in a Dako Autostainer.
FISH was performed according to the PathVysion (Vysis Inc., Downers Grove, IL) protocol, described in the package insert as approved by the United States Food and Drug Administration. In brief, the PathVysion protocol involves rehydration of paraffin-embedded 5-μm thick section. Section was air-dried, pretreated, and digested with proteinase-K before being hybridized with fluorescent-labeled probes for HER2 gene and α-satellite DNA for chromosome 17. The nuclei were routinely counterstained with an intercalating fluorescent counterstain 4′,6-diamidino-2-phenylindole (DAPI). For each tumor, 60 tumor cell nuclei were identified and scored for both HER2 and chromosome 17 centromere numbers. HER2 gene amplification was defined as a HER2-to-chromosome 17 ratio >2.0 as required by the manufacturer.
Peroxidase-based immunohistochemistry was evaluated using a Nikon Eclipse E400 optical microscopy, immunofluorescence slides were observed in a confocal microscopy, and FISH in a fluorescence microscopy with a Rhodamine and fluorescein isothiocyanate (FITC) double filter locating the invasive tumour areas at low magnification and reading the signals a high magnification with × 100 oil-immersion objectives.
Kinase assay
MDA-MB-468 cells stably expressing HER2 full-length or CTFs were lysed in lysis buffer (20 mM Tris–HCl pH 7.4, 137 mM NaCl, 2 mM EDTA, 10% glycerol, 1% NP40, 10 mg/ml aprotinin and 10 mg/ml leupeptin). Cell lysates were immunoprecipitated with the anti-HER2 antibody CB11 O.N. at 4°C, then rabbit anti-mouse antibody was added during 2 h and finally protein A was added during 1 h. Immunoprecipitates were washed three times with washing buffer (50 mM HEPES pH 7.5, 150 mM NaCl, 0.1% Triton X-100, 2 mM EDTA, 10 μg/μl aprotinin, 0.03 mM Sodium Orthovanadate), resuspended in assay buffer (20 mM HEPES, 5 mM MnCl2, 0.1% Triton X-100, 0.03 mM sodium orthovanadate, 20 μg/μl aprotinin, 1 μl [γ-32P]ATP) and incubated during 30 min at room temperature. Immunoprecipitates were eluted by addition of 2 × SDS–PAGE loading buffer and proteins were separated by SDS–PAGE and visualized by autoradiography using a Molecular Imager FX imaging system (Bio-Rad Labs).
Tumor xenografts in nude mice
All of the experimental procedures were approved, and mice were maintained and treated in accordance with institutional guidelines of Vall d'Hebron University Hospital Care and Use Committee. Six- to eight-week-old female BALB/c athymic (nu+/nu+) mice were purchased from Charles Rivers Laboratories (Paris, France). Mice were housed in air-filtered laminar flow cabinets and handled with aseptic procedures with a 12-h light cycle and food and water ad libitum and allowed to acclimatize to local conditions for 1 week. A 17β-estradiol pellet (Innovative Research of America, Sarasota, FL) was introduced subcutaneously in mice 1 day before the injection of T47D parental cells expressing HER2 full-length or CTFs subcutaneously into the right flank.
In these experiments, tumors were allowed to reach a predetermined size before the start of treatment with Herceptin or PBS as a control given by intraperitoneal injection twice a week and Lapatinib or the vehicle (0.5% hydroxypropylmethylcellulose, 0.1% Tween-80) as a control given orally twice a day. Tumor growth was assessed twice weekly by caliper measurement. Tumor volume (mm3) was calculated by the formula π/6 × larger diameter × (smaller diameter)2. Results are presented as mean±s.d.
Supplementary Material
Supplementary Figure 1
{kind=link}
Supplementary Figure 2
{kind=link}
Supplementary Figure 3
{kind=link}
Supplementary Figures and Table Legends
Acknowledgments
This work was supported by grants from La Marató de TV3, the Spanish Ministry of Education and Science (SAF2005-03939) and the Spanish Association Against Cancer (AECC) to JA.
References
- Arribas J, Borroto A (2002) Protein ectodomain shedding. Chem Rev 102: 4627–4638 [DOI] [PubMed] [Google Scholar]
- Arribas J, Coodly L, Vollmer P, Kishimoto TK, Rosejohn S, Massagué J (1996) Diverse cell surface protein ectodomains are shed by a system sensitive to metalloprotease inhibitors. J Biol Chem 271: 11376–11382 [DOI] [PubMed] [Google Scholar]
- Arribas J, Massagué J (1995) Transforming growth factor alpha and beta amyloid precursor protein share a secretory mechanism. J Cell Biol 128: 433–441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagatell R, Khan O, Paine-Murrieta G, Taylor CW, Akinaga S, Whitesell L (2001) Destabilization of steroid receptors by heat shock protein 90-binding drugs: a ligand-independent approach to hormonal therapy of breast cancer. Clin Cancer Res 7: 2076–2084 [PubMed] [Google Scholar]
- Baselga J, Norton L (2002) Focus on breast cancer. Cancer Cell 1: 319–322 [DOI] [PubMed] [Google Scholar]
- Baselga J, Norton L, Albanell J, Kim YM, Mendelsohn J (1998) Recombinant humanized anti-HER2 antibody (Herceptin) enhances the antitumor activity of paclitaxel and doxorubicin against HER2/neu overexpressing human breast cancer xenografts. Cancer Res 58: 2825–2831 [PubMed] [Google Scholar]
- Burgess AW, Cho H-S, Eigenbrot C, Ferguson KM, Garrett TPJ, Leahy DJ, Lemmon MA, Sliwkowski MX, Ward CW, Yokoyama S (2003) An open-and-shut case? Recent insights into the activation of EGF/ErbB receptors. Mol Cell 12: 541–552 [DOI] [PubMed] [Google Scholar]
- Carpenter G (2003) Nuclear localization and possible functions of receptor tyrosine kinases. Curr Opin Cell Biol 15: 143–148 [DOI] [PubMed] [Google Scholar]
- Codony-Servat J, Albanell J, Arribas J, Baselga J (1999) Activation of HER ectodomain shedding by pervanadate and inhibition by the tissue inhibitor of matrix metalloproteases TIMP-1 in breast cancer cells. Cancer Res 59: 1196–1201 [PubMed] [Google Scholar]
- Franco SJ, Huttenlocher A (2005) Regulating cell migration: calpains make the cut. J Cell Sci 118: 3829–3838 [DOI] [PubMed] [Google Scholar]
- Giri DK, Ali-Seyed M, Li LY, Lee DF, Ling P, Bartholomeusz G, Wang SC, Hung MC (2005) Endosomal transport of ErbB-2: mechanism for nuclear entry of the cell surface receptor. Mol Cell Biol 25: 11005–11018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson HM, Subramaniam PS, Olsnes S, Jans DA (2004) Trafficking and signaling pathways of nuclear localizing protein ligands and their receptors. Bioessays 26: 993–1004 [DOI] [PubMed] [Google Scholar]
- Komar AA, Hatzoglou M (2005) Internal ribosome entry sites in cellular mRNAs: mystery of their existence. J Biol Chem 280: 23425–23428 [DOI] [PubMed] [Google Scholar]
- Kopan R, Ilagan MX (2004) Gamma-secretase: proteasome of the membrane? Nat Rev Mol Cell Biol 5: 499–504 [DOI] [PubMed] [Google Scholar]
- Lauring AS, Overbaugh J (2000) Evidence that an IRES within the Notch2 coding region can direct expression of a nuclear form of the protein. Mol Cell 6: 939–945 [DOI] [PubMed] [Google Scholar]
- Lin S-Y, Makino K, Xia W, Matin A, Wen Y, Kwong KY, Bourguignon L, Hung M-C (2001) Nuclear localization of EGF receptor and its potential new role as a transcription factor. Nat Cell Biol 3: 802–808 [DOI] [PubMed] [Google Scholar]
- Maier D, Nagel AC, Preiss A (2002) Two isoforms of the Notch antagonist Hairless are produced by differential translation initiation. Proc Natl Acad Sci 99: 15480–15485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marambaud P, Shioi J, Serban G, Georgakopoulos A, Sarner S, Nagy V, Baki L, Wen P, Efthimiopoulos S, Shao Z, Wisniewski T, Robakis NK (2002) A presenilin-1/gamma-secretase cleavage releases the E-cadherin intracellular domain and regulates disassembly of adherens junctions. EMBO J 21: 1948–1956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Salas E, Fernandez-Miragall O, Reigadas S, Pacheco A, Serrano P (2004) Internal ribosome entry site elements in eukaryotic genomes. Curr Genomics 5: 259–277 [Google Scholar]
- Molina MA, Codony-Servat J, Albanell J, Rojo F, Arribas J, Baselga J (2001) Trastuzumab (Herceptin®), a humanized anti-HER2 receptor monoclonal antibody, inhibits basal and activated HER2 ectodomain cleavage in breast cancer cells. Cancer Res 61: 4744–4749 [PubMed] [Google Scholar]
- Molina MA, Saez R, Ramsey EE, Garcia-Barchino MJ, Rojo F, Evans AJ, Albanell J, Keenan EJ, Lluch A, Garcia-Conde J, Baselga J, Clinton GM (2002) NH(2)-terminal truncated HER-2 protein but not full-length receptor is associated with nodal metastasis in human breast cancer. Clin Cancer Res 8: 347–353 [PubMed] [Google Scholar]
- Ni CY, Murphy MP, Golde TE, Carpenter G (2001) Gamma-secretase cleavage and nuclear localization of ErbB-4 receptor tyrosine kinase. Science 294: 2179–2181 [DOI] [PubMed] [Google Scholar]
- Offterdinger M, Schofer C, Weipoltshammer K, Grunt TW (2002) C-erbB-3: a nuclear protein in mammary epithelial cells. J Cell Biol 157: 929–939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oksvold M, Huitfeldt H, Stang E, Madshus I (2002) Localizing the EGF receptor. Nat Cell Biol 4: E22; author reply E22–E23 [DOI] [PubMed] [Google Scholar]
- Wang SC, Lien HC, Xia W, Chen IF, Lo HW, Wang Z, Ali-Seyed M, Lee DF, Bartholomeusz G, Ou-Yang F, Giri DK, Hung MC (2004) Binding at and transactivation of the COX-2 promoter by nuclear tyrosine kinase receptor ErbB-2. Cancer Cell 6: 251–261 [DOI] [PubMed] [Google Scholar]
- Wells A, Marti U (2002) Signalling shortcuts: cell-surface receptors in the nucleus? Nat Rev Mol Cell Biol 3: 697–702 [DOI] [PubMed] [Google Scholar]
- Williams CC, Allison JG, Vidal GA, Burow ME, Beckman BS, Marrero L, Jones FE (2004) The ERBB4/HER4 receptor tyrosine kinase regulates gene expression by functioning as a STAT5A nuclear chaperone. J Cell Biol 167: 469–478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie YM, Hung M C (1994) Nuclear localization of P185neu tyrosine kinase and its association with transcriptional transactivation. Biochem Biophys Res Commun 203: 1589–1598 [DOI] [PubMed] [Google Scholar]
- Yarden Y, Sliwkowski MX (2001) Untangling the ErbB signalling network. Nat Rev Mol Cell Biol 2: 127–137 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1
Supplementary Figure 2
Supplementary Figure 3
Supplementary Figures and Table Legends