

Abstract
Neuropilin-1 (NRP1) is a co-receptor for vascular endothelial growth factor (VEGF) that enhances the angiogenic signals cooperatively with VEGFR2. VEGF signaling is essential for physiological and pathological angiogenesis through its effects on vascular endothelial cells (ECs) and smooth muscle cells (SMCs), but the mechanisms coordinating this response are not well understood. Here we show that a substantial fraction of NRP1 is proteoglycan modified with either heparan sulfate or chondroitin sulfate on a single conserved Ser. The composition of the NRP1 glycosaminoglycan (GAG) chains differs between ECs and SMCs. Glycosylation increased VEGF binding in both cell types, but the differential GAG composition of NRP1 mediates opposite responsiveness to VEGF in ECs and SMCs. Finally, NRP1 expression and its GAG modification post-transcriptionally regulate VEGFR2 protein expression. These findings indicate that GAG modification of NRP1 plays a critical role in modulating VEGF signaling, and may provide new insights into physiological and pathological angiogenesis.
Keywords: glycosaminoglycan, neuropilin-1, VEGF, VEGFR2
Introduction
Neuropilin-1 (NRP1) was originally discovered as a co-receptor for semaphorin-3A (Sema3A), an axon repellent factor (Kolodkin et al, 1997). However, NRP1 also acts as a co-receptor for vascular endothelial growth factor (VEGF), a molecule with no sequence or structural homology to Sema3A (Soker et al, 1998). VEGF (also referred as VEGF-A) is an essential factor promoting both embryonic angiogenesis and postnatal neovascularization. Additionally, VEGF plays a significant role in causing pathological angiogenesis associated with tumor growth, age-related macular degeneration, diabetic retinopathy, and other conditions (Ferrara et al, 2003). Indeed, a blocking anti-VEGF antibody that disrupts VEGF signaling is a promising anticancer therapy currently in development (Hurwitz et al, 2004).
VEGF has three receptors, VEGF receptor 1 and 2 (VEGFR1, VEGFR2), and neuropilin (Veikkola and Alitalo, 1999; Ferrara et al, 2003). VEGFR2 is the primary receptor mediating the angiogenic activity of VEGF (Shalaby et al, 1995; Ferrara et al, 2003), and NRP1 functions as a co-receptor to enhance VEGFR2 signaling (Soker et al, 1998). Indeed, genetic ablation of NRP1 leads to severely impaired vascular development (Kawasaki et al, 1999; Takashima et al, 2002; Gu et al, 2003), indicating that NRP1 is essential for VEGF-mediated angiogenesis.
In addition to promoting angiogenesis, VEGF is now thought to be required for the maintenance and stabilization of mature blood vessels (Zachary, 2001; Saint-Geniez and D'Amore, 2004). Signaling through VEGFR2, VEGF induces not only endothelial cell (EC) proliferation but also cell survival (Gerber et al, 1998), and the loss of VEGF signals in the choroidal endothelium is one factor promoting age-related macular degeneration (Blaauwgeers et al, 1999). Smooth muscle cells (SMCs), another important component of the vessel wall, also express both NRP1 (Kitsukawa et al, 1995; Kawasaki et al, 1999) and VEGFR2 (Grosskreutz et al, 1999; Ishida et al, 2001). However, SMCs in mature vessels typically do not respond to VEGF signals except in certain conditions such as atherosclerosis (Carmeliet, 2003; Jain, 2003; Khurana et al, 2004). Therefore, we wished to identify the mechanism(s) responsible for different cellular responses to VEGF in ECs and SMCs.
In this study, we demonstrate that a substantial fraction of NRP1 is proteoglycan modified with either heparan sulfate (HS) or chondroitin sulfate (CS) attached to a single conserved Ser. The type of NRP1 glycosaminoglycan (GAG) chain modification differs between ECs and SMCs. Finally, we show that the type of NRP1 GAG modification critically and differentially modulates VEGFR2 signals in SMCs and ECs.
Results
A substantial fraction of NRP1 is proteoglycan modified with HS or CS
The differential responsiveness of ECs and SMCs to VEGF could be explained by a number of factors, and we initially investigated the ability of VEGF to bind to these cells. When human coronary artery smooth muscle cells (CASMCs) were incubated with 125I-labeled VEGF, we detected two distinct binding proteins after cell-surface crosslinking (Figure 1A). The lower band was also detected in human umbilical vein endothelial cells (HUVECs) and was identified as NRP1. However, the upper band was not found in HUVECs, and this band did not correspond to VEGFR2. In contrast, the upper band was also seen in bronchial smooth muscle cells (BSMCs) (Figure 1A), a non-vascular SMC, and, because NRP1 alone cannot transduce VEGF signals, we initially thought that this binding protein represented a new VEGF receptor. However, after transfection of CASMCs, but not HUVECs, with FLAG-tagged NRP1, we observed an identical upper molecular weight band when blotted with an anti-FLAG antibody (Figure 1B). The high molecular weight band was not simply a covalently linked homodimer of NRP1 (Figure 1C), and we reasoned NRP1 could undergo post-translational modification. Although NRP1 itself undergoes N-glycosylation, we found that the high molecular weight NRP1 was not a form of N-glycosylation or O-glycosylation by enzyme treatment and lectin blot (data not shown), but it did contain GAG chains. Indeed, treatment of NRP1 immunoprecipitates with both heparitinase and chondroitinase, which digest HS and CS, respectively, led to the disappearance of the upper band, whereas the lower band was not affected. Next, we investigated the composition of GAG-modified endogenous NRP1 in both CASMCs and HUVECs. Heparitinase slightly decreased the modified NRP1 band in CASMCs, whereas chondroitinase digested the majority of the GAG present on NRP1, indicating that CS was the dominant GAG modification of NRP1 (Figure 1D). In contrast, NRP1 in HUVECs was also modified, but to a much lesser extent than that seen in CASMCs, and HUVEC NRP1 contained almost equivalent amounts of CS and HS (Figure 1D). By analyzing the band intensity, we determined the degree of each modification relative to untreated samples (non-modified/HS-modified/CS-modified NRP1—CASMCs: 100%/79%/174%, HUVECs: 100%/27.3%/24.6%, respectively) (Figure 1D, right panel).
Figure 1.

A substantial fraction of cellular NRP1 is proteoglycan, composed of either HS or CS. (A) 125I-labeled VEGF is crosslinked to different proteins in ECs and SMCs. Arrow indicates VEGF-binding protein specifically seen in SMCs. CASMC: coronary artery smooth muscle cell; BSMC: bronchial smooth muscle cell; HUVEC: human umbilical vein endothelial cell. (B) Western blots of exogenously expressed NRP1 in either CASMCs or HUVECs. Adenovirus encoding FLAG-tagged NRP1 was transfected 2 days before analysis at the indicated MOI. LacZ-encoding adenovirus was used as a control. (C) The high molecular weight band was not simply a covalently linked homodimer of NRP1. Only FLAG-tagged NRP1 or both FLAG-tagged and V5-tagged NRP1 were transfected in CASMCs, and the cell lysates were immunoprecipitated and detected by the indicated antibody. (D) Endogenous NRP1 was modified by GAG chain addition in both SMCs and ECs. The upper band in CASMC immunoprecipitates disappeared following treatment with both HSase and CSase. HUVEC-expressed NRP1 is also GAG modified. The band intensity was analyzed and the proportion of each glycanated form of NRP1 was determined. Data are from three separate experiments. HSase: heparitinase; CSase: chondroitinase.
GAG chains are covalently added to Ser residues of the core protein contained within a Ser-Gly consensus sequence (Esko and Zhang, 1996). We mutated the nine consensus sequences present in NRP1, and the GAG chains are attached only to Ser612 (Figure 2A). As a single GAG chain cannot contain both HS and CS simultaneously, endogenous NRP1 exists as either an HS proteoglycan or CS proteoglycan but not as a hybrid proteoglycan. Moreover, as shown in Figure 1D, non-modified NRP1 (130 kDa, the core protein) was always detected in both CASMCs and HUVECs. Ser612 is located in the bridge region between the b1b2 and MAM domains of NRP1 (Figure 2D), and multiple sequence alignments suggested that Ser612 was remarkably conserved among vertebrates (Figure 2B), and the peptide sequence around Ser612 was also well conserved, especially the acidic amino acids that are important for HS attachment (Esko and Zhang, 1996). NRP2, a mammalian homolog of NRP1, does not have this conserved Ser residue, and adenovirus-mediated expression of NRP2 in CASMCs demonstrated that NRP2 is not GAG modified (Figure 2C).
Figure 2.

(A) NRP1 is GAG modified on a single Ser612 residue. CASMCs were transfected with adenoviral vectors encoding WT or S612A mutant NRP1. NRP1 S612A is not GAG modified. (B) Multiple alignments of NRP1 from different species. Ser612 is highly conserved among vertebrates. (C) NRP2, an NRP family member, is not GAG modified. (D) Design of siRNA and adenovirus constructs. Ser612 exists in the bridge region between the b1b2 and MAM domains. (E) Replacement of Ser612 by Ala612 of NRP1 did not change binding to VEGF. Cos7 cells were transfected with either NRP1 WT′ or S612A expression vector and preincubated with heparitinase (1.5 mU/ml), heparinase (1.5 mU/ml), and chondroitinase (20 mU/ml) in the culture medium at 37°C for 2 h to make NRP1 non-GAG form. After incubation with 125I-labeled VEGF for 30 min at room temperature, cell lysates were immunoprecipitated by anti-NRP1 antibody, and the bound radioactivity was quantitated using a gamma counter. Data are from three independent experiments. For panel E, error bars represent s.e.
GAG modification of NRP1 enhances VEGF binding
Addition of both chondroitinase and heparitinase to culture medium completely digests all GAGs attached to other core proteins on the cell surface, and this would dramatically complicate the interpretation of any experiments using this technique. Therefore, to further investigate the function of the GAG of only NRP1, we used RNAi to knock down endogenous NRP1 while expressing mutant NRP1. We designed an siRNA, named N-G, targeting the GAG attachment site of NRP1 (Figure 2D). We further generated two NRP1-encoding adenovirus constructs: NRP1 S612A, in which Ser612 was replaced by Ala612 and there was a three-base mismatch with N-G siRNA; and NRP1 WT′, which contains the glycan accepting residue but had a four-base mismatch with N-G siRNA (Figure 2D). In cells transfected with N-G siRNA, transfection of both adenovirus constructs led to the expression of the appropriate NRP1 molecules. We confirmed that addition of FLAG tag to NRP1 does not affect VEGF binding (data not shown) and that the mutation itself (NRP1 S612A) did not change VEGF binding to the core protein of NRP1 (Figure 2E).
We next examined the ability of VEGF to bind to experimentally replaced NRP1 in both SMCs and ECs. Transfection of both N-G siRNA and equal multiplicity of infection (MOI) adenoviral constructs successfully replaced endogenous NRP1 with either the GAG-acceptor (NRP1 WT′) or mutated (NRP1 S612A) NRP1 (Figure 3A). Throughout these experiments, MOIs were used to generate NRP1 WT′ or S612A expression levels comparable to endogenous NRP1. To determine whether GAG modifications affect the ability of NRP1 to bind VEGF, we measured the binding of 125I-labeled VEGF to FLAG-tagged NRP1 in these cells. After incubation with 125I-labeled VEGF, cell lysates were immunoprecipitated with an anti-FLAG antibody, and bound radioactivity was counted. NRP1 WT′ bound VEGF with 3.87- and 2.27-fold higher than NRP1 S612A in SMCs and ECs, respectively (Figure 3B). We found that heparitinase and chondroitinase treatment with these immunoprecipitates could not entirely eliminate the enhancement of VEGF binding (Figure 3B), different from the results of Figure 2E in which we showed that VEGF equally binds NRP1 WT′ and S612A pretreated with heparitinase and chondroitinase before the exposure to VEGF. These results suggest that GAG modifications of NRP1 in SMCs and ECs enhance VEGF binding mainly to NRP1 core protein and not to only GAG chain of NRP1.
Figure 3.
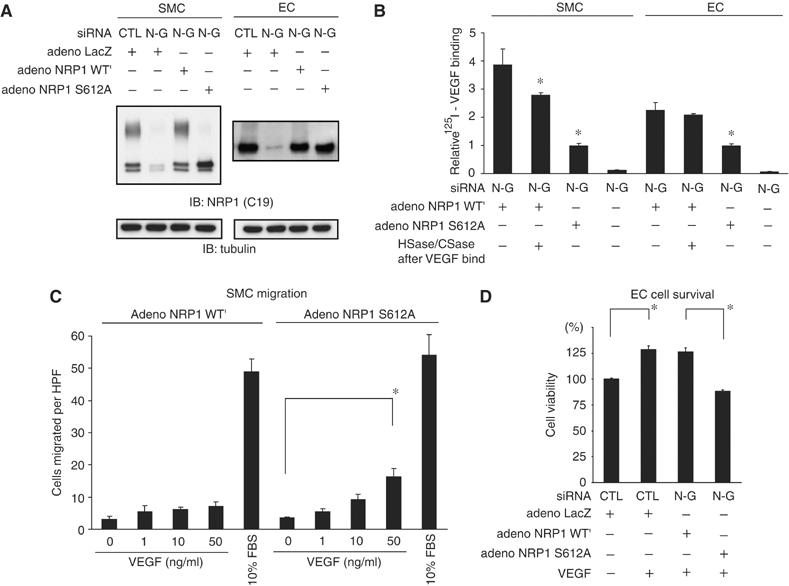
GAG modifications differentially affect NRP1 function in SMCs and ECs. (A) Experimental replacement of NRP1 in SMCs and ECs. After transfection with both N-G siRNA and adenoviral constructs, endogenous NRP1 was successfully replaced with either the glycanated form (NRP1 WT′) or non-glycanated form (NRP1 S612A) of NRP1. Tubulin was used as a loading control. (B) Addition of GAG to NRP1 enhances binding to VEGF in both types of cells. Two days after NRP1 replacement, cell lysates were immunoprecipitated with anti-FLAG antibody after incubation with 125I-labeled VEGF (25 ng/ml) for 40 min at room temperature, and bound radioactivity was quantitated using a gamma counter. Heparitinase and chondroitinase treatment with these immunoprecipitates could not entirely eliminate the enhancement of VEGF binding. Data are from three independent experiments. (C) VEGF (50 ng/ml) induced greater cell migration in SMCs expressing non-modified NRP1 S612A than those expressing NRP1 WT′. Migrated cells were quantified by counting cells in three random high-power fields (HPF, × 200). Similar results were obtained from additional two independent experiments. (D) VEGF (50 ng/ml) increased cell viability in ECs expressing NRP1 WT′ to a greater extent than in ECs expressing NRP1 S612A. Data are from three independent experiments. For panels B–D, error bars represent s.e. *P<0.05, versus adeno-NRP1 WT′ in panel B.
NRP1 GAG modifications lead to differential VEGF responsiveness in SMCs and ECs
We next investigated whether GAG modifications of NRP1 in both SMCs and ECs affected cellular responsiveness to VEGF. VEGF increases the motility of vascular SMCs (Grosskreutz et al, 1999; Ishida et al, 2001), and induces proliferation, migration, and cell survival in ECs (Ferrara et al, 2003). These actions are primarily mediated through the VEGFR2 signaling pathway likely in conjunction with NRP1.
Notably, VEGF induced the migration of SMCs expressing NRP1 S612A (non-modified) stronger than those expressing NRP1 WT′ (GAG modified) (Figure 3C). In contrast, VEGF increased the viability of ECs expressing NRP1 WT′ to a greater extent than those expressing NRP1 S612A (Figure 3D). The observed increased viability seen in ECs expressing NRP1 WT′ in response to VEGF is consistent with the increased VEGF binding shown in Figure 3B. However, the decreased motility seen in SMCs expressing NRP1 WT′ was unexpected. To further explore this discrepancy, we examined the influence of different GAG chains on the expression of VEGR2 and the formation of the VEGF–VEGFR2–NRP1 ternary complex, both important determinants of VEGF signaling (Soker et al, 2002).
We first analyzed VEGFR2 protein expression in cells expressing either NRP1 WT′ or S612A. In SMCs expressing S612A mutant, VEGFR2 expression was two-fold higher than in cells expressing NRP1 WT′ (Figure 4A (left) and B), but expression of either NRP1 WT′ or S612A did not affect VEGFR2 expression in ECs (Figure 4A (right) and B). The increased VEGFR2 protein expression in SMCs was not accompanied by changes in mRNA levels (Figure 4C), indicating that post-transcriptional mechanisms regulate VEGFR2 expression.
Figure 4.
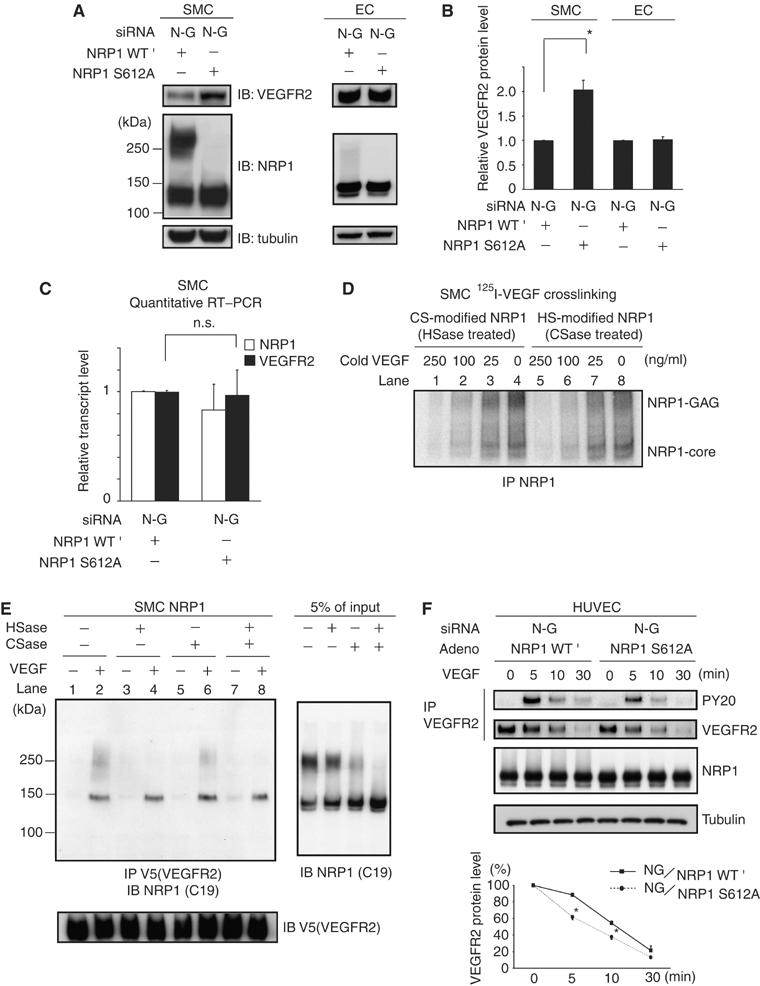
Different roles of the GAG of NRP1 on VEGFR2 in SMCs. (A) Experimental replacement with NRP1 S612A increased VEGFR2 expression in SMCs, but replacement did not affect VEGFR2 expression in ECs. Two days after transfection with siRNA and adeno-NRP1, cells were analyzed by Western blotting. Data are representative of at least three independent experiments. (B) Quantitative results of Western blot. (C) Experimental replacement with NRP1 S612A increased VEGFR2 protein expression without any transcriptional change in SMCs. Each sample was analyzed in duplicate and the experiments were performed in triplicate for the full set of genes. (D) CS-modified NRP1 had the same affinity for VEGF as HS-modified NRP1 and non-modified NRP1. Note that 125I-labeled VEGF bound CS-modified NRP1 with a similar ratio before crosslink (upper band in lanes 4 and 8, CS-modified NRP1:HS-modified NRP1=2:1). Increasing amounts of cold VEGF equally inhibited 125I-labeled VEGF binding to all forms of NRP1. (E) Co-immunoprecipitation of NRP1 with VEGFR2. CS-modified NRP1 (left panel, about 250 kDa in lane 4) minimally associated with VEGFR2 compared to non-modified (130 kDa, in lanes 2, 4, 6, 8) and HS-modified NRP1 (about 250 kDa in lane 6), although there was a two-fold excess of CS-modified NRP1 compared to HS-modified NRP1 at input (right panel). The membrane was stripped and re-probed with anti-V5 as a loading control. Data are representative of at least three independent experiments. (F) The rate of VEGFR2 degradation was decreased in NRP1 WT′ ECs compared to NRP1 S612A ECs. Phosphorylated VEGFR2 was also much higher in NRP1 WT′ ECs than in NRP1 S612A ECs at any time point after VEGF. Data are representative of at least three independent experiments. For panels B, C, F, error bars represent s.e. *P<0.05, versus NG/ NRP1 S612A at the same period as in panel F. HSase: heparitinase; CSase: chondroitinase.
Both the extent of GAG modification and the predominant GAG chain added (i.e. HS or CS) differ between ECs and SMCs. Therefore, we examined whether the type of GAG modification affected ternary complex formation. We used enzymatic digestions and 125I-labeled VEGF binding to assess the contribution of CS- and HS-modified NRP1 to VEGF binding, and found that all forms of NRP1 bound VEGF equally well (Figure 4D). We next examined the ability of GAG-modified NRP1 to associate with VEGFR2 in the presence of VEGF by co-immunoprecipitation. After pretreatment with heparitinase and/or chondroitinase, V5-tagged VEGFR2 was precipitated from SMC lysates in the presence of VEGF. As shown in Figure 4E, CS-modified NRP1 minimally associated with VEGFR2 compared to non-modified or HS-modified NRP1.
Thus, in SMCs, GAG-modified NRP1 post-transcriptionally downregulates VEGFR2 expression, and CS-modified NRP1 may act as a decoy receptor, rather than a co-receptor. It is likely that a combination of these factors explains the differences in VEGF activity seen in SMCs expressing NRP1 WT′ or S612A (Figure 3C).
Based on the results of receptor complex formation in the presence of VEGF in Figure 4E, we hypothesized that NRP1 might affect VEGFR2 internalization/degradation after ligand binding, because degradation of the receptor tyrosine kinase is an important regulator of signaling intensity (Duval et al, 2003; Rubin et al, 2005). Before exposure to VEGF, VEGFR2 expression was not different between ECs expressing NRP1 WT′ and S612A (Figure 4A). However, the rate of VEGFR2 degradation was decreased in NRP1 WT′ ECs compared to NRP1 S612A ECs. Phosphorylated VEGFR2 was also much higher in ECs expressing NRP1 WT′ than those expressing NRP1 S612A at any time points after VEGF (Figure 4F). These results suggested that the GAG modification of NRP1 enhances VEGF signaling in ECs by delaying the degradation of VEGFR2 in the presence of VEGF, and not just by the enhancement of VEGF binding.
NRP1 post-transcriptionally modulates VEGFR2 expression
NRP1 knockout mice exhibit severely impaired vascular development and die around E13.5 (Kitsukawa et al, 1995; Kawasaki et al, 1999). VEGF has several splicing isoforms (its major forms in mice are VEGF120, 164, 188) and NRP1 does not bind VEGF120. In contrast, VEGFR2 can bind all of VEGF isoforms. Although NRP1 is a common receptor for both VEGF and Sema3A, impaired VEGF signaling is responsible for the observed vascular defects in these mice (Gu et al, 2003). However, the vascular defect in NRP1−/− mice is more severe than that seen in VEGF120/120 mice, in which only VEGF120 is expressed (Carmeliet et al, 1999; Stalmans et al, 2003). Thus, NRP1 appears to play a more prominent role in VEGF signaling than simply functioning as a co-receptor for some VEGF isoforms. Based on the results that CS-dominant GAG of NRP1 negatively affects VEGFR2 expression levels in SMCs, we hypothesized that NRP1 basically stabilizes VEGFR2 leading to increased expression. Thus, VEGFR2 expression should be lower in NRP1−/− mice, leading to a more pronounced vascular phenotype.
To test this hypothesis in cells, we knocked down NRP1 in ECs using siRNA. Before the addition of VEGF, VEGFR2 expression was substantially decreased in cells transfected with NRP1 siRNAs (Figure 5A). Two siRNAs targeting NRP1 were used to exclude the possibility of an off-target effect of RNAi. VEGFR2 mRNA levels were unaffected by NRP1 knockdown, however (Figure 5B). Additionally, VEGFR1, another VEGF receptor, was not affected by either NRP1 siRNA, suggesting that NRP1 specifically regulates VEGFR2 expression (Figure 5A). As VEGFR2 protein level was not associated with transcription level, we conducted pulse–chase experiments in HUVECs treated with NRP1 siRNA to determine the rate of VEGFR2 degradation in the absence of VEGF. Notably, we found that the rate of VEGFR2 degradation was not changed by NRP1 knockdown (Figure 5C), which was different from the results in the presence of VEGF (Figure 4F).
Figure 5.
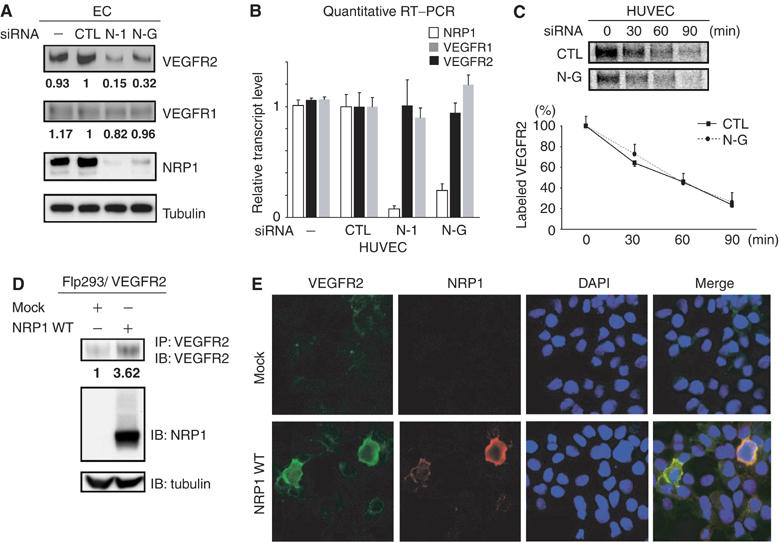
NRP1 post-transcriptionally regulates the expression of VEGFR2. (A) Both NRP1 siRNAs (N-G, N-1) decreased VEGFR2 expression. In contrast, VEGFR1 was not influenced by NRP1 knockdown. Tubulin was used as a loading control. Data are representative of at least three independent experiments. (B) Transcription levels of both VEGFR1 and VEGFR2 were not influenced by NRP1 knockdown. Each sample was analyzed in duplicate and experiments were performed in triplicate for the full set of genes. (C) Pulse–chase experiments in HUVECs. The rate of degradation of VEGFR2 was not changed by NRP1 knockdown. Data are from four independent experiments. (D) NRP1 significantly upregulated VEGFR2 protein levels in Flp293/VEGFR2 cells. Transfected NRP1 in Flp293/VEGFR2 cells was GAG modified similar to ECs. Data are representative of two independent experiments. (E) NRP1 regulates cell-surface VEGFR2 expression. Transient expression of FLAG-tagged NRP1 WT upregulated the cell membrane-associated VEGFR2 expression compared to adjacent non-transfected cells. Flp293/VEGFR2 cells were transfected with either NRP1 WT or mock and stained without permeabilization using anti-VEGFR2 (green) and anti-FLAG-Cy3 (red). Blue: DAPI nuclear staining. For panels A and C, numeric represents the mean of band intensity of three experiments. For panel B, error bars represent s.e.
We next examined whether the ability of NRP1 to promote VEGFR2 expression was specific for ECs, and we generated Flp293/VEGFR2 cells stably expressing VEGFR2. These cells express much less NRP1 than either ECs or SMCs. When these cells were transfected with NRP1, NRP1 was GAG modified similar to ECs (Figure 5D), and VEGFR2 expression was substantially upregulated (Figure 5D). VEGFR2 transcription was not altered by NRP1 expression (data not shown). Finally, when transfected cells were examined by confocal microscopy, NRP1-expressing cells had substantially higher cell-surface VEGFR2 levels compared to non-transfected adjacent cells (Figure 5E).
Discussion
NRP1 GAG modifications differentially regulate VEGF responsiveness in SMCs and ECs
In this study, we showed that a substantial fraction of NRP1 is proteoglycan modified with either HS or CS on a single conserved Ser residue. Additionally, both the degree and length of GAG modification and the predominant side chain added differ between ECs and SMCs. In both ECs and SMCs, GAG modifications enhanced VEGF binding, but GAG addition to NRP1 in ECs enhanced VEGF–VEGFR2 signaling. In contrast, GAG-modified NRP1 negatively affected VEGF activity in SMCs. Interestingly, in SMCs, GAG modification of NRP1 post-transcriptionally downregulates VEGFR2 expression, and CS-modified NRP1, the major form of NRP1 in SMCs (about 50% of total NRP1), may act as a decoy receptor, rather than a co-receptor.
The mechanism by which the addition of GAGs to NRP1 enhanced VEGF signals in ECs remains unclear. We speculate that the addition of HS chains to NRP1 promotes multimerization. Exogenous heparin and HS bind NRP1 via its b1b2 domain and increase VEGF binding to NRP1 and VEGFR2 (Gitay-Goren et al, 1992; Mamluk et al, 2002). Additionally, heparin can induce NRP1 multimerization in the presence or absence of ligands (Fuh et al, 2000; Mamluk et al, 2002). Binding between the NRP1 b1b2 domain and HS requires only eight highly sulfated monosaccharide units (Mamluk et al, 2002). Generally, in a single HS chain, sulfated sugar residues occur in multiple clusters (containing 6–10 sugars) separated by regions of low sulfation (Gallagher, 2001). We estimated the length of a single HS chain of NRP1 as about 50 kDa in ECs including at least 200 monosaccharide units, and this modification is sufficient to bind multiple NRP1 molecules. Therefore, we speculate that a single NRP1 HS chain could bind multiple NRP1 molecules and promote NRP1 clustering. Such an NRP1 cluster could recruit substantial amounts of VEGFR2, and, in the presence of VEGF, increase the binding frequency without affecting the dissociation constant. When the receptor complex with VEGF and VEGFR2 is formed, VEGFR2 might stabilize and escape internalization/degradation, and as a result it enhances VEGF signal (as in Figure 4F). In contrast to HS, the role of CS in VEGF signaling has not been well investigated. We found that only chondroitin sulfate-E (CS-E, a subclass of CS chains) enhances VEGF binding to NRP1 in ECs like heparin (Supplementary data A). However, CS-E is a very rare modification, and we found no CS-E on the GAG chains of NRP1 in both SMCs and ECs in our preliminary analysis (data not shown). Furthermore, chondroitinase treatment with immunoprecipitated NRP1 after VEGF did not change the VEGF binding to NRP1 in both ECs and SMCs, whereas heparitinase treatment decreased (Supplementary data B). These results suggested that endogenous CS, which usually does not contain CS-E, has no beneficial or unprofitable effect on VEGF binding to NRP1 core and it is unlikely that endogenous CS on the cell surface including CS chains of NRP1 could induce NRP1 multimerization.
We have not ruled out the possibility that GAG modification of NRP1 induces conformational changes in the binding surface between VEGF and NRP1, or between VEGFR2 and NRP1, and the addition of CS to NRP1 might hamper such allosteric effects in SMCs. Further structural studies examining the interaction of GAG-modified NRP1 with VEGFR2 will clarify this issue.
NRP1 is also a receptor for Sema3A (Kolodkin et al, 1997). It was recently found that GAG regulates the function of Sema5A, a member of a different class of the semaphorin family. Sema5A is able to exert both attractive and repulsive effects, depending on association with HS and CS, respectively (Kantor et al, 2004). This intriguing report raises the possibility that GAG chains of NRP1 might also regulate Sema3A binding and function. Further study is needed to clarify the role of GAG modifications on NRP1/Sema3A signaling. Furthermore, it was recently reported that FGF2 binds NRP1 (West et al, 2005). It might be interesting to search the role of GAG modifications of NRP1 on FGF2 signaling.
What is the physiological relevance of GAG-mediated differences in VEGF responsiveness in vascular cells?
The addition of GAG chains to NRP1 led to opposite effects on VEGFR2 signaling in ECs and SMCs. SMCs migrate in response to VEGF, and this migration is mediated mainly through VEGFR2. However, cell density or passage number affects SMC response to VEGF (Ishida et al, 2001). We identified a specific lot of SMCs expressing comparable levels of VEGFR2 as ECs. In these cells, the pattern of NRP1 GAG modification was similar to that of ECs seen in this study (Y Shintani and S Takashima, unpublished observations). Additionally, GAG chain modifications changed with time in culture or by incubating in ischemic conditions (preliminary data). Thus, different ill-defined culture conditions can cause changes in GAG addition to NRP1, and this can affect VEGF signaling in both SMCs and ECs.
Based on the present results, it appears that differential GAG chain addition to NRP1 can mediate opposite responses to VEGF in ECs and SMCs in mature blood vessels. In contrast, during active angiogenesis, SMCs need to migrate and enclose ECs to form complete vessels. Under these circumstances, both ECs and SMCs might express NRP1 with short GAG chains and respond similarly to VEGF for efficient vessel formation. Indeed, VEGF has also been recently implicated in the normal development of SMC-surrounded coronary arteries and pericyte coverage in the retinal vasculature (Benjamin et al, 1998; Carmeliet et al, 1999). Further investigation will be needed to clarify the in vivo composition of GAG chains of NRP1.
NRP1 post-transcriptionally regulates the expression of VEGFR2
We demonstrated that knockdown of NRP1 by RNAi post-transcriptionally reduced VEGFR2 expression in ECs, suggesting that NRP1 positively regulates the expression of VEGFR2. In contrast, GAG addition to NRP1 in SMCs eliminated this effect. As the transcription of VEGFR2 was not affected by NRP1 expression or GAG modification, we conclude that VEGFR2 expression is post-transcriptionally regulated by NRP1.
Cell-surface proteins undergo a complex process including co-translational folding, post-translational modifications, and transport through various cellular compartments including the ER and Golgi apparatus. Recent reports suggest that stable cell-surface receptor expression in the absence of ligand sometimes requires a specific adaptor protein or co-receptor (McLatchie et al, 1998; Loconto et al, 2003; Saito et al, 2004). Most identified receptors are complex proteins with seven transmembrane domains, but a type I membrane receptor such as VEGFR2 could also require such an adaptor or co-receptor, for example, NRP1. NRP1 and VEGFR2 might interact in the absence of VEGF and the addition of CS to NRP1 interferes with the trafficking and/or stability of VEGFR2.
Our findings suggested that NRP1 is a key modulator of VEGF signaling. Further studies and technical advances are needed to precisely characterize the importance of particular GAG modifications on VEGF signaling in vitro and in vivo. However, the role of NRP1 and its post-translational modifications may provide new insights into the growth of vascular networks in physiological and pathological conditions.
Materials and methods
Materials
We utilized the following commercially available antibodies: anti-NRP1 antibody (C-19, Santa Cruz Inc.), anti-human VEGFR2 for Western blot (A-3, Santa Cruz Inc.), for immunofluorescent study (ab9530, Abcam), for immunoprecipitation (C-1158, Santa Cruz Inc.), anti-alpha-tubulin (clone B-5-1-2, Sigma), anti-VEGFR1 (C-17, Santa Cruz Inc.), anti-FLAG M2 (Sigma), anti-V5 (Invitrogen), PY20-HRP-conjugated antibody (BD Biosciences). Heparitinase, heparinase, and chondroitinase were purchased from Seikagaku Corp.
Preparation, radioiodination of VEGF, and chemical crosslinking
Recombinant human VEGF165 was prepared in Hi5 cells using the baculovirus system (Invitrogen) and purified with two-step chromatography to over 95% purity as determined by silver stain. Na125I was purchased from Amersham Biosciences, and 125I-labeled VEGF was prepared using IODO-BEADS (Pierce). 125I-labeled VEGF crosslinking study was performed as described previously (Soker et al, 1998). Briefly, cells were grown to 90–95% confluence in a 60 mm collagen 1-coated dish and labeled with 125I-labeled VEGF (25 ng/ml) using DSS (Pierce) according to the manufacturer's instructions. Cells were lysed with 1% Nonidet P-40 containing buffer (1% Nonidet P-40, 0.15 M NaCl, 20 mM Tris pH 7.2, including protease inhibitor cocktail (Nacalai)) and then subjected to SDS–PAGE using 5–10% gradient gel (Bio-Rad). Bound 125I-labeled VEGF was detected by autoradiography using the BAS system (Fuji). For competitive binding analysis (Figure 4D), CASMC was transfected with FLAG-tagged NRP1 at MOI 10 2 days before the experiment. After crosslinking, cells lysates were immunoprecipitated with anti-FLAG M2 antibody, and immunoprecipitates were then subjected to heparitinase (1.25 mU/ml) or chondroitinase (250 mU/ml) treatment. Cold VEGF was added 5 min before 125I-labeled VEGF for assessing binding affinity.
Expression vector and adenovirus constructs
Human NRP1 cDNA was obtained as described previously (Soker et al, 1998). In this experiment, all construction was performed using the Gateway system (Invitrogen) according to the manufacturer's instructions. With PCR primer designed to include or delete stop codon of NRP1, the amplified fragment was inserted into pENTR/D-TOPO (Invitrogen), named pENTR/NRP1 or pENTR/NRP1-cV5, respectively. To generate N-terminal FLAG-tagged NRP1 (NRP1 FLAG), FLAG epitope (DYKDDDDK) was inserted just after the signal sequence of NRP1 (between Lys26 and Cys27) by PCR-based mutagenesis using pENTR/NRP1 as a template. Both NRP1 WT′ and S612A were also generated by PCR-based mutagenesis (primer design is shown in Figure 2C). All the NRP1 constructs were recombined to mammalian expression vector, pEF-DEST51 (Invitrogen) and pAd/CMV/V5-DEST (Invitrogen). Adenovirus constructs were generated using ViraPower Adenoviral Expression System (Invitrogen) essentially as described by the manufacturer. Recombined vectors along with the supplied pAd/CMV/V5-DEST/lacZ were transfected into host HEK293A cells (Invitrogen). NRP2 and VEGFR2 cDNAs were cloned into pENTR/D-TOPO from HUVEC cDNA. Both adenoviruses and expression vectors of non-tagged and V5-tagged NRP2 and VEGFR2 were generated as those of NRP1.
Cell culture
HUVECs, human CASMCs, human BSMCs, and human aortic smooth muscle cells (AoSMCs) were obtained from Clonetics. They were cultured in endothelial and smooth muscle cell medium (Clonetics) and used up to passage 5.
Treatment with heparitinase and chondroitinase, and analysis of the proportion of each glycanated NRP1
Cells were lysed with lysis buffer (0.15 M NaCl, 1 mM EDTA, 20 mM Tris pH 7.2, including protease inhibitor cocktail (Nacalai)). The aliquots of lysates were incubated with anti-NRP1 antibody (C-19), followed by addition of Protein G Sepharose (Amersham Bioscience) for 1–2 h at 4°C. Bound NRP1 was then subjected to heparitinase and/or chondroitinase treatment in enzyme-contained buffer (1% Nonidet P-40, 0.15 M NaCl, 5 × 10−5 M Ca2+, 20 mM Tris pH 7.2, including heparitinase (1.25 mU/ml) and/or chondroitinase (25 mU/ml)) at 37°C for 1 h. Enzyme-treated immunoprecipitates were subjected to SDS–PAGE and Western blotting. By analyzing the band intensity using ImageJ software (version 1.34s), the proportion of each glycanated form of NRP1 was determined.
RNAi and adenovirus transfection, experimental replacement of NRP1 with/without GAG
HUVECs at 50–70% confluency were transfected with the indicated siRNA duplexes using Optifect (Invitrogen) according to the manufacturer's instructions. AoSMCs (for Western blot, RT–PCR, VEGF binding and migration assay) or CASMCs (for 125I-labeled VEGF crosslinking and co-immunoprecipitation) at 80–90% confluency were transfected with Lipofectamine 2000 (Invitrogen). SiRNA was transfected at 50 nM in a 60 or 100 mm dish 4–6 h after plating. After another 4 h, adeno- LacZ, NRP1 WT′, or S612A was infected at an MOI of 2 (for ECs) or 4–6 (for SMCs). NRP1 siRNAs were synthesized by Dharmacon Inc., and siRNAs sequences were as follows: N-G: sense 5′-cugccacaguggaacaggu-dTdT, N-1: sense 5′-gagagguccugaauguucc-dTdT. N-1 was the same sequence that had been previously reported (Bachelder et al, 2003). SiRNA for non-silencing control used in this study was targeted for GL2: sense 5′-cguacgcggaauacuucga-dTdT (Elbashir et al, 2001). We chose this oligonucleotide as control because VEGFR2 and NRP1 expression level was not affected as compared with non-transfected cells, different from other several non-silencing control siRNAs that are commercially available.
125I-labeled VEGF binding to NRP1
Two days after transfection with siRNA (N-G) and adeno-NRP1 WT′ or S612A, both SMCs and ECs were incubated with 125I-labeled VEGF (25 ng/ml) for 40 min at room temperature. The cells were washed twice with PBS and lysed with lysis buffer (0.15 M NaCl, 1 mM EDTA, 20 mM Tris pH 7.2, including protease inhibitor cocktail (Nacalai)). Cell lysates were immunoprecipitated with anti-FLAG M2 agarose (Sigma) for 1 h at 4°C. After washing with buffer twice, the immunoprecipitates were subjected to gamma counter (Beckman).
Cell migration assay
Effects of VEGF on SMCs migration were studied using 24-well Transwell® microplate (Corning Inc.). Pore (8.0 μm) polystyrene filters were treated with 10 μg/ml fibronectin (Sigma). Two days after the transfection with RNAi (N-G) and respective adenovirus (NRP1 WT′ or S612A), AoSMCs were trypsinized and then loaded into the inner chamber at 1 × 104 cells/well. After incubation with or without VEGF (1, 10, 50 ng/ml) or 10% FBS at 37°C for 6 h in a CO2 incubator, the upper side of the filters containing non-migrated cells was wiped and rinsed. The filters were fixed and stained with Diff-Quik®. Migrating cells were quantified by counting cells in each well at three random high-power fields ( × 200). All groups were studied in triplicate.
Cell viability
Cell viability was assessed with a CellTiter 96 Aqueous One Solution Cell Proliferation Assay System (Promega). HUVECs were plated in a 96-well culture plate at a density of 5 × 104 cells/well in 0.5% FBS in EBM2 medium (Clontech). Six hours after plating, siRNA was transfected using Optifect at 100 nM; consequently, adenovirus addition was performed at MOI 2. Serum starvation with or without VEGF (50 ng/ml) was performed 24 h after transfection and MTS reagent was added to each well 24 h later, and optical absorbance at 490 nm was measured with a microplate reader.
Quantitative RT–PCR
Total RNA was extracted using RNA-Bee-RNA Isolation Reagent (Tel-Test Inc.). Then, 1 μg of total RNA was reverse-transcribed using Omniscript RT (Qiagen) according to the manufacturer's protocol. Quantitative RT–PCR was performed with TaqMan technology using the ABI Prism 7000 detection system (Applied Biosystems) according to the manufacturer's instructions. RT–PCR conditions were 2 min at 50°C, 10 min at 95°C, and 40 cycles of 15 s at 95°C and 1 min at 60°C. Data were normalized to 18S ribosome or GAPDH level. Each sample was analyzed in duplicate and the experiments were replicated twice for the full set of genes. For 18S ribosome, GAPDH, VEGFR1, and VEGFR2, primers and probes were obtained using TaqMan Assays-on-Demand gene expression products (Applied Biosystems). For NRP1, primer sequences were as follows: sense 5′-CAAGGTGTTCATGAGGAAGTTCAA, antisense 5′-CCGCAGCTCAGGTGTATCATAGT, probe FAM-5′-TGACAGCAAACGCAAGGCGAAGTCTT-TAMRA.
Co-immunoprecipitation assay
HEK293T cells in a 60 mm dish were transfected with 5 μg of pEF-DEST51/VEGFR2 V5 using Lipofectamine 2000. Two days after transfection, we treated the cells with both 10 mU heparitinase and 200 mU chondroitinase in serum-free medium to eliminate extracellular GAG for 2 h at 37°C. Then, cells were lysed in lysis buffer (1% Nonidet P-40, 0.15 M NaCl, 20 mM Tris pH 7.2, including protease inhibitor cocktail (Nacalai)). We also prepared four sets of endogenous CASMCs (100 mm plate, each), which were treated with heparitinase alone, chondroitinase alone, both heparitinase and chondroitinase, or none in serum-free medium for 2 h at 37°C. We then mixed with the aliquot of enzyme-treated VEGFR2 V5 cell lysates and the lysate of each enzyme-treated CASMCs and incubated with anti-V5 agarose (Sigma) in the presence or absence of VEGF (50 ng/ml) for 3 h at 4°C. After extensive washing, immunoprecipitated samples were subjected to SDS–PAGE and Western blotting.
Pulse–chase experiments
HUVECs were transfected with control or N-G siRNA. Two days after transfection, cells were labeled for 20 min at 37°C with 20 μCi [35S]methionine per milliliter in methionine-free Dulbecco's modified Eagle's medium (DMEM; Invitrogen). The cells were then washed and chased in DMEM containing 10% FBS for the indicated time periods. At each time point of the chase, cell lysates were immunoprecipitated with anti-VEGFR2 antibody (C-1158) for 2 h at 4°C. The immunoprecipitates were subjected to SDS–PAGE using 5% polyacrylamide gel. Labeled VEGFR2 was visualized by autoradiography and quantified using the BAS system (Fuji).
Generation of stable cell line
We generated 293 cells which stably expressed VEGFR2 using the Flp-In system (Invitrogen) according to manufacturer's instructions. After VEGFR2 cDNA in the pENTR/D-TOPO was recombined to pEF5/FRT/V5-DEST (Invitrogen), we transfected this construct to Flp293 cells (Invitrogen) with Lipofectamine 2000 (Invitrogen) and established the 293/VEGFR2 cells by selection with 250 μg/ml hygromycin (Invitrogen).
Immunofluorescent staining
Flp293/VEGFR2 cells were transfected with pDEST51-NRP1 FLAG using Optifect (Invitrogen) on a poly-D-lysine-coated chamber-slide (Nunc). Two days later, cells were fixed with 2% paraformaldehyde in PBS for 15 min at room temperature, washed twice in 0.1 M glycine/PBS, and blocked with 10% FBS/PBS for 30 min at room temperature. The cells were probed with anti-VEGFR2 antibody (1:400 dilution in 10% FBS/PBS) for 1 h, then washed and incubated with AlexaFluor 488-conjugated goat antibody against mouse IgG (1:1000 dilution in 10% FBS/PBS; Molecular Probes) for 1 h. The cells were washed thoroughly and then probed with anti-FLAG M2 antibodies conjugated with Cy3 (1:1000 dilution in 10% FBS/PBS; Sigma) for 1 h. We mounted the preparations using PermaFluor Mountant Medium (Thermo) and took images with Radiance 2100 (Bio-Rad).
Data analysis
Statistical significance was assessed with ANOVA using the Fisher's post hoc test. A value of P<0.05 was considered to be statistically significant.
Supplementary Material
Supplementary Data
Acknowledgments
We thank Drs M Takahashi, N Taniguchi, S Yamada, and H Kitagawa for thoughtful discussion, and A Ogal, Y Nagamachi, H Okuda, and M Nakamura for technical assistance. We thank Drs T Toyofuku and R Iwamoto for reading the manuscript. This study is supported by Grant-in-aid for Scientific Research (nos.16390225, 17390229) from the Ministry of Education, Science and Culture, Japan, a Grant from Japan Cardiovascular Research Foundation, and the Human Frontier Science Program.
References
- Bachelder RE, Lipscomb EA, Lin X, Wendt MA, Chadborn NH, Eickholt BJ, Mercurio AM (2003) Competing autocrine pathways involving alternative neuropilin-1 ligands regulate chemotaxis of carcinoma cells. Cancer Res 63: 5230–5233 [PubMed] [Google Scholar]
- Benjamin LE, Hemo I, Keshet E (1998) A plasticity window for blood vessel remodelling is defined by pericyte coverage of the preformed endothelial network and is regulated by PDGF-B and VEGF. Development 125: 1591–1598 [DOI] [PubMed] [Google Scholar]
- Blaauwgeers HG, Holtkamp GM, Rutten H, Witmer AN, Koolwijk P, Partanen TA, Alitalo K, Kroon ME, Kijlstra A, van Hinsbergh VW, Schlingemann RO (1999) Polarized vascular endothelial growth factor secretion by human retinal pigment epithelium and localization of vascular endothelial growth factor receptors on the inner choriocapillaris. Evidence for a trophic paracrine relation. Am J Pathol 155: 421–428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmeliet P (2003) Angiogenesis in health and disease. Nat Med 9: 653–660 [DOI] [PubMed] [Google Scholar]
- Carmeliet P, Ng YS, Nuyens D, Theilmeier G, Brusselmans K, Cornelissen I, Ehler E, Kakkar VV, Stalmans I, Mattot V, Perriard JC, Dewerchin M, Flameng W, Nagy A, Lupu F, Moons L, Collen D, D'Amore PA, Shima DT (1999) Impaired myocardial angiogenesis and ischemic cardiomyopathy in mice lacking the vascular endothelial growth factor isoforms VEGF164 and VEGF188. Nat Med 5: 495–502 [DOI] [PubMed] [Google Scholar]
- Duval M, Bedard-Goulet S, Delisle C, Gratton JP (2003) Vascular endothelial growth factor-dependent down-regulation of Flk-1/KDR involves Cbl-mediated ubiquitination. Consequences on nitric oxide production from endothelial cells. J Biol Chem 278: 20091–20097 [DOI] [PubMed] [Google Scholar]
- Elbashir SM, Harborth J, Lendeckel W, Yalcin A, Weber K, Tuschl T (2001) Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature 411: 494–498 [DOI] [PubMed] [Google Scholar]
- Esko JD, Zhang L (1996) Influence of core protein sequence on glycosaminoglycan assembly. Curr Opin Struct Biol 6: 663–670 [DOI] [PubMed] [Google Scholar]
- Ferrara N, Gerber HP, LeCouter J (2003) The biology of VEGF and its receptors. Nat Med 9: 669–676 [DOI] [PubMed] [Google Scholar]
- Fuh G, Garcia KC, de Vos AM (2000) The interaction of neuropilin-1 with vascular endothelial growth factor and its receptor flt-1. J Biol Chem 275: 26690–26695 [DOI] [PubMed] [Google Scholar]
- Gallagher JT (2001) Heparan sulfate: growth control with a restricted sequence menu. J Clin Invest 108: 357–361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerber HP, Dixit V, Ferrara N (1998) Vascular endothelial growth factor induces expression of the antiapoptotic proteins Bcl-2 and A1 in vascular endothelial cells. J Biol Chem 273: 13313–13316 [DOI] [PubMed] [Google Scholar]
- Gitay-Goren H, Soker S, Vlodavsky I, Neufeld G (1992) The binding of vascular endothelial growth factor to its receptors is dependent on cell surface-associated heparin-like molecules. J Biol Chem 267: 6093–6098 [PubMed] [Google Scholar]
- Grosskreutz CL, Anand-Apte B, Duplaa C, Quinn TP, Terman BI, Zetter B, D'Amore PA (1999) Vascular endothelial growth factor-induced migration of vascular smooth muscle cells in vitro. Microvasc Res 58: 128–136 [DOI] [PubMed] [Google Scholar]
- Gu C, Rodriguez ER, Reimert DV, Shu T, Fritzsch B, Richards LJ, Kolodkin AL, Ginty DD (2003) Neuropilin-1 conveys semaphorin and VEGF signaling during neural and cardiovascular development. Dev Cell 5: 45–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurwitz H, Fehrenbacher L, Novotny W, Cartwright T, Hainsworth J, Heim W, Berlin J, Baron A, Griffing S, Holmgren E, Ferrara N, Fyfe G, Rogers B, Ross R, Kabbinavar F (2004) Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. N Engl J Med 350: 2335–2342 [DOI] [PubMed] [Google Scholar]
- Ishida A, Murray J, Saito Y, Kanthou C, Benzakour O, Shibuya M, Wijelath ES (2001) Expression of vascular endothelial growth factor receptors in smooth muscle cells. J Cell Physiol 188: 359–368 [DOI] [PubMed] [Google Scholar]
- Jain RK (2003) Molecular regulation of vessel maturation. Nat Med 9: 685–693 [DOI] [PubMed] [Google Scholar]
- Kantor DB, Chivatakarn O, Peer KL, Oster SF, Inatani M, Hansen MJ, Flanagan JG, Yamaguchi Y, Sretavan DW, Giger RJ, Kolodkin AL (2004) Semaphorin 5A is a bifunctional axon guidance cue regulated by heparan and chondroitin sulfate proteoglycans. Neuron 44: 961–975 [DOI] [PubMed] [Google Scholar]
- Kawasaki T, Kitsukawa T, Bekku Y, Matsuda Y, Sanbo M, Yagi T, Fujisawa H (1999) A requirement for neuropilin-1 in embryonic vessel formation. Development 126: 4895–4902 [DOI] [PubMed] [Google Scholar]
- Khurana R, Zhuang Z, Bhardwaj S, Murakami M, De Muinck E, Yla-Herttuala S, Ferrara N, Martin JF, Zachary I, Simons M (2004) Angiogenesis-dependent and independent phases of intimal hyperplasia. Circulation 110: 2436–2443 [DOI] [PubMed] [Google Scholar]
- Kitsukawa T, Shimono A, Kawakami A, Kondoh H, Fujisawa H (1995) Overexpression of a membrane protein, neuropilin, in chimeric mice causes anomalies in the cardiovascular system, nervous system and limbs. Development 121: 4309–4318 [DOI] [PubMed] [Google Scholar]
- Kolodkin AL, Levengood DV, Rowe EG, Tai YT, Giger RJ, Ginty DD (1997) Neuropilin is a semaphorin III receptor. Cell 90: 753–762 [DOI] [PubMed] [Google Scholar]
- Loconto J, Papes F, Chang E, Stowers L, Jones EP, Takada T, Kumanovics A, Fischer Lindahl K, Dulac C (2003) Functional expression of murine V2R pheromone receptors involves selective association with the M10 and M1 families of MHC class Ib molecules. Cell 112: 607–618 [DOI] [PubMed] [Google Scholar]
- Mamluk R, Gechtman Z, Kutcher ME, Gasiunas N, Gallagher J, Klagsbrun M (2002) Neuropilin-1 binds vascular endothelial growth factor 165, placenta growth factor-2, and heparin via its b1b2 domain. J Biol Chem 277: 24818–24825 [DOI] [PubMed] [Google Scholar]
- McLatchie LM, Fraser NJ, Main MJ, Wise A, Brown J, Thompson N, Solari R, Lee MG, Foord SM (1998) RAMPs regulate the transport and ligand specificity of the calcitonin-receptor-like receptor. Nature 393: 333–339 [DOI] [PubMed] [Google Scholar]
- Rubin C, Gur G, Yarden Y (2005) Negative regulation of receptor tyrosine kinases: unexpected links to c-Cbl and receptor ubiquitylation. Cell Res 15: 66–71 [DOI] [PubMed] [Google Scholar]
- Saint-Geniez M, D'Amore PA (2004) Development and pathology of the hyaloid, choroidal and retinal vasculature. Int J Dev Biol 48: 1045–1058 [DOI] [PubMed] [Google Scholar]
- Saito H, Kubota M, Roberts RW, Chi Q, Matsunami H (2004) RTP family members induce functional expression of mammalian odorant receptors. Cell 119: 679–691 [DOI] [PubMed] [Google Scholar]
- Shalaby F, Rossant J, Yamaguchi TP, Gertsenstein M, Wu XF, Breitman ML, Schuh AC (1995) Failure of blood-island formation and vasculogenesis in Flk-1-deficient mice. Nature 376: 62–66 [DOI] [PubMed] [Google Scholar]
- Soker S, Miao HQ, Nomi M, Takashima S, Klagsbrun M (2002) VEGF165 mediates formation of complexes containing VEGFR-2 and neuropilin-1 that enhance VEGF165-receptor binding. J Cell Biochem 85: 357–368 [DOI] [PubMed] [Google Scholar]
- Soker S, Takashima S, Miao HQ, Neufeld G, Klagsbrun M (1998) Neuropilin-1 is expressed by endothelial and tumor cells as an isoform-specific receptor for vascular endothelial growth factor. Cell 92: 735–745 [DOI] [PubMed] [Google Scholar]
- Stalmans I, Lambrechts D, De Smet F, Jansen S, Wang J, Maity S, Kneer P, von der Ohe M, Swillen A, Maes C, Gewillig M, Molin DG, Hellings P, Boetel T, Haardt M, Compernolle V, Dewerchin M, Plaisance S, Vlietinck R, Emanuel B, Gittenberger-de Groot AC, Scambler P, Morrow B, Driscol DA, Moons L, Esguerra CV, Carmeliet G, Behn-Krappa A, Devriendt K, Collen D, Conway SJ, Carmeliet P (2003) VEGF: a modifier of the del22q11 (DiGeorge) syndrome? Nat Med 9: 173–182 [DOI] [PubMed] [Google Scholar]
- Takashima S, Kitakaze M, Asakura M, Asanuma H, Sanada S, Tashiro F, Niwa H, Miyazaki Ji J, Hirota S, Kitamura Y, Kitsukawa T, Fujisawa H, Klagsbrun M, Hori M (2002) Targeting of both mouse neuropilin-1 and neuropilin-2 genes severely impairs developmental yolk sac and embryonic angiogenesis. Proc Natl Acad Sci USA 99: 3657–3662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veikkola T, Alitalo K (1999) VEGFs, receptors and angiogenesis. Semin Cancer Biol 9: 211–220 [DOI] [PubMed] [Google Scholar]
- West DC, Rees CG, Duchesne L, Patey SJ, Terry CJ, Turnbull JE, Delehedde M, Heegaard CW, Allain F, Vanpouille C, Ron D, Fernig DG (2005) Interactions of multiple heparin binding growth factors with neuropilin-1 and potentiation of the activity of fibroblast growth factor-2. J Biol Chem 280: 13457–13464 [DOI] [PubMed] [Google Scholar]
- Zachary I (2001) Signaling mechanisms mediating vascular protective actions of vascular endothelial growth factor. Am J Physiol Cell Physiol 280: C1375–C1386 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Data