

Abstract
Most serpins are associated with protease inhibition, and their ability to form loop–sheet polymers is linked to conformational disease and the human serpinopathies. Here we describe the structural and functional dissection of how a unique serpin, the non-histone architectural protein, MENT (Myeloid and Erythroid Nuclear Termination stage-specific protein), participates in DNA and chromatin condensation. Our data suggest that MENT contains at least two distinct DNA-binding sites, consistent with its simultaneous binding to the two closely juxtaposed linker DNA segments on a nucleosome. Remarkably, our studies suggest that the reactive centre loop, a region of the MENT molecule essential for chromatin bridging in vivo and in vitro, is able to mediate formation of a loop–sheet oligomer. These data provide mechanistic insight into chromatin compaction by a non-histone architectural protein and suggest how the structural plasticity of serpins has adapted to mediate physiological, rather than pathogenic, loop–sheet linkages.
Keywords: chromatin compaction, MENT, nucleosome, serpin
Introduction
In eukaryotes, DNA is repeatedly coiled around histone octamers to form nucleosomes (Richmond and Davey, 2003). The nucleosome zig-zag arrays fold into 30 nm higher order fibres (Schalch et al, 2005). The arrays and fibres are referred to as primary and secondary levels of chromatin folding, respectively. In the nucleus, the chromatin fibres are further folded by histone and non-histone architectural proteins to form more compact structures that are associated with tertiary, quaternary and higher levels of compaction (Woodcock and Dimitrov, 2001; Luger and Hansen, 2005). Although very little is known about chromatin organization at these higher levels of compaction, the structural transitions at this level play a crucial role in controlling transcriptional activity and genetic regulation.
Previous biochemical and ultrastructural analysis have shown that several chromatin architectural proteins such as linker histone H5 (Weintraub, 1984); SIR3 (Georgel et al, 2001), MeCP2 (Georgel et al, 2003) and the Myeloid and Erythroid Nuclear Termination stage-specific protein (MENT) (Grigoryev et al, 1999) are able to support a compact chromatin higher order structure via protein bridges connecting separate chromatin fibres. Biochemical and biophysical studies suggested that MENT mediates chromatin compaction by promoting tight intra-fibre folding of nucleosome arrays and, independently, inter-fibre nucleosome bridging (Springhetti et al, 2003).
Remarkably, the chromatin architectural protein MENT is a member of the serpin superfamily (Silverman et al, 2001). Alongside its ability to condense chromatin, MENT is an effective inhibitor of cathepsin L (Irving et al, 2002), a cysteine protease residing in lysosomes and the nucleus (Goulet et al, 2004). Inhibitory serpins are metastable molecules that utilize a dramatic conformational rearrangement termed the stressed to relaxed (S to R) transition to inhibit target enzymes. Upon interaction with a protease, the reactive centre loop (RCL), an exposed region in the native molecule, is cleaved and inserts into the centre of a large central β-sheet (the A-sheet) to form an additional β-strand. Throughout the S to R transition, the protease remains attached to the RCL via a covalent linkage and, in the final serpin–enzyme complex, the protease is trapped in a distorted inactive conformation (Huntington et al, 2000). As a result of this complex mechanism of inhibition, serpins are vulnerable to inactivating mutations that promote inappropriate conformational change and the formation of inter-molecular loop–sheet linked polymers (Lomas et al, 1992).
In vivo, the RCL sequence of MENT has been demonstrated to be essential for proper MENT-mediated chromatin compaction and controlling cell proliferation (Irving et al, 2002). Electron microscopy studies using purified nucleosome arrays and a non-inhibitory mutant of MENT revealed that the sequence and conformation of the RCL is crucial for chromatin bridging (Springhetti et al, 2003). Although RCL-mediated serpin polymerization is usually associated with disease (Lomas et al, 1992), the ability of serpins to form loop–sheet linkages suggested a possible mechanism of bridging through RCL-mediated contacts.
In this study, we have determined the X-ray crystal structure of the serpin MENT and describe how the molecule interacts with DNA and its location within nucleosome arrays. We also show that polymerization of MENT is likely to play a role in the physiological process of chromatin condensation.
Results
The X-ray crystal structure of native MENT reveals partial insertion of the RCL
We have determined the 2.7 Å crystal structure of native wild-type protein (MENTWT; Figure 1A and Supplementary Table 1). The molecule adopts a α/β fold and comprises three β-sheets (the five-stranded sheet (A), six-stranded sheet (B) and four-stranded sheet (C)) surrounded by nine α-helices (hA–hI; Figure 1A). β-Sheet hydrogen bonding between the fifth (s5A) and the sixth (s6A) strand of the A-sheet is disrupted in the centre, splitting s6A in two (Figure 1A). No electron density was observed for the majority of residues in the M-loop including the AT-hook motif, consistent with the flexible nature of this motif (Aravind and Landsman, 1998). The RCL is exposed and links the top of strand s4A with strand s1C of the four-stranded C β-sheet. Notably, the structure reveals that two residues of the RCL (P15–P14 (P1/P1′nomenclature described by Schechter and Berger, 1968)) are partially inserted into the A β-sheet (Figure 1A). The partial insertion is similar to that observed in the human serpins antithrombin and heparin cofactor II, both of which undergo cofactor-dependent conformational change and activation against protease targets (Jin et al, 1997; Baglin et al, 2002).
Figure 1.
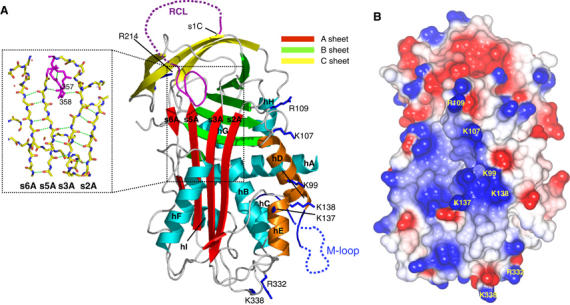
(A) Cartoon of native MENTWT showing the A β-sheet (red), B β-sheet (green), C β-sheet (yellow), RCL (purple) (the disordered region is dashed) and helices hA–hI (cyan). Residues K99, K107, R109, R332 and K338 are in dark blue bonds (labelled). The partial insertion of the RCL and the break in s6A (dotted rectangle) of the A-sheet are shown in the inset (hydrogen bonds as green broken lines). (B) CCP4MG (Potterton et al, 2002, 2004) electrostatic potential surface of native MENTWT. The positions of K99, K107, R109, K137, K138, R332 and K338 are shown.
Given the partial insertion of the RCL in the native structure, we investigated whether DNA was able to induce a conformational change in MENT or affect the inhibitory activity of the molecule. Our data reveal that DNA binding did not induce a change in tryptophan fluorescence in MENT consistent with RCL expulsion (Supplementary Figure 1) and had no effect on the inhibitory kinetics against cathepsin L (Supplementary Figure 1). Together, these data suggest that DNA is not able to induce RCL expulsion or substantially alter the conformation of the RCL of MENT.
Identification of DNA-binding sites on MENT
MENT is a basic molecule (pI=9.2) capable of binding and condensing DNA in a non-sequence-specific manner (Springhetti et al, 2003). The electrostatic potential surface of native MENT reveals a large (∼20 Å diameter) positively charged patch centred on and around the D- (K99, K107 and R109) and E-helices (K137 and K138; Figure 1A and B), suggesting a potential DNA-binding surface. Twelve additional basic amino acids that contribute to smaller positively charged patches on the surface of MENT were also identified (Supplementary Table 2). Collectively, these residues were mutated to test their contribution to DNA binding by MENT. The integrity of all the MENT mutants was verified using (1) far-UV circular dichroism spectroscopy, (2) thermal stability and (3) inhibitory activity (the association rate constant, kass, and the stoichiometry of inhibition, SI) (Supplementary Table 2 and data not shown). Serpin inhibitory activity is sensitive to structural perturbation and all mutant proteins retained inhibitory activity. The ability of the mutants to interact with naked DNA was determined by electrophoretic mobility shift assays (EMSAs) and chromatin association assays and compared to control proteins. In an EMSA, MENTWT binds DNA with an apparent KD value of 0.67 μM (where KD is defined as 50% of DNA bound—see Materials and methods), whereas the deleted M-loop mutant (MENTΔMloop) has negligible DNA-binding affinity (KD value in excess of 10 μM; Figure 2A and B). The mutant MENTR78Q contains a substitution of the conserved arginine in the AT-hook that impairs its ability to bind DNA (Figure 2A and B) and associate with chromatin (Figure 2C). These data confirm the essential role of the AT-hook in chromatin interactions.
Figure 2.
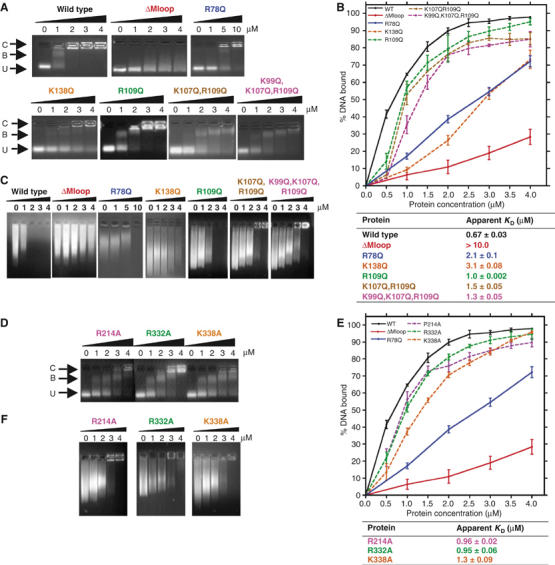
Analysis of purified MENT proteins incubated with DNA and chromatin. Gel mobility shift analysis of D- and E-helix mutants (A) and alanine mutants (D) in comparison to MENTWT (WT) and two negative control proteins, MENTΔMloop (ΔMloop) and a single AT-hook mutant, MENTR78Q (R78Q) (control proteins only shown in panels A and C). Proteins were incubated with DNA and analysed by agarose gel electrophoresis. The final concentration (μM) of purified proteins, as indicated, is shown at the top of each gel panel. Arrows indicate the position of unbound DNA (U), bound DNA/complex (B) and large nucleoprotein species (C). Quantitative analysis of EMSAs of D- and E-helix (B) and alanine mutants (E) is shown graphically. Graphs represent the % of bound DNA at increasing MENT concentrations (as indicated at the bottom of graph). Graphed data of the control proteins MENTWT, MENTΔMloop and MENTR78Q are shown as solid lines and the mutant proteins as dashed lines. Affinities (KD) were calculated from gel electrophoresis experiments as described in Materials and methods and are defined as the concentration (μM) of protein required to bind 50% of DNA. (C, F) Chromatin association assays with increasing concentrations of D- and E-helix mutant (C) and alanine mutant proteins (F). The final concentration (μM) of purified protein, as indicated, added to soluble erythrocyte chromatin (OD260=1.6) is shown at the top of each gel panel.
The E-helix mutant MENTK138Q was impaired in its ability to bind DNA (Figure 2A) with a KD value >3 μM (Figure 2B). In addition, MENTK138Q was unable to condense chromatin in vitro (Figure 2C). K138 is located at the top of the E-helix, and points across the face of the neighbouring D-helix (Figure 1A). In contrast, MENTK137Q was unaffected and was not investigated further (Supplementary Figure 2).
Two of the single D-helix mutants, MENTK99Q and MENTK107Q, have the same affinity for DNA as the wild-type protein (Supplementary Figure 2). In contrast, MENTR109Q showed a reduced ability to bind DNA and compact chromatin (Figure 2A–C). Additional effects were noted when multiple mutations were present (MENTK107Q,R109Q and MENTK99Q,K107Q,R109Q; Figure 2A–C). Whereas both MENTK107Q,R109Q and MENTK99Q,K107Q,R109Q show reduced DNA-binding affinity (KD values of 1.5 and 1.3 μM, respectively; Figure 2B), MENTK99Q,K107Q,R109Q in particular was also unable to form the large nucleoprotein species (Figure 2A, band C) identified using MENTWT.
The mutants MENTR214A, MENTR332A and MENTK338A showed an almost two-fold decrease in DNA/chromatin-binding affinity: MENTK338A was the most compromised with a KD value of 1.3 μM (Figure 2D–F). Interestingly, R332 and K338 are solvent exposed and are located at the bottom of the molecule, away from the RCL, on the loop between the I-helix and strand s5A (Figure 1A). Both are in close proximity (∼10 Å) to the visible C-terminal segment of the M-loop, suggesting that these residues may aid DNA coordination by the AT-hook motif. In contrast, R214 is located at the top of the molecule on strand s4C (Supplementary Figure 3). The residue is solvent exposed; in native MENTWT, this residue forms a salt bridge with D384 at the N-terminus of strand s1C and D263 on the loop between strand s3B and the G-helix (Supplementary Figure 3). Three positively charged residues (K217, K265 and K382) are also located on s3C/s4C. However, mutagenesis revealed that K217 and K382, which are located ∼6 Å from R214, are not important for DNA binding. Thus, while R214 may directly interact with DNA, we cannot exclude the possibility that mutation of this residue has altered the structure of the serpin gate. Collectively, these data suggest that there are at least two major DNA-binding sites on the surface of MENT, one on and around the AT-hook and a second centred on a positively charged patch encompassing the D- and E-helix.
MENT has a high affinity for mononucleosomes containing two DNA linkers
Our data suggest that, like linker histones (Goytisolo et al, 1996), MENT contains at least two DNA-binding sites and that two juxtaposed DNA linkers would be predicted to constitute a strong binding site for MENT. In order to investigate MENT linker binding, three mononucleosome constructs were generated using a strong 146-bp-long nucleosome positioning sequence from clone 601 (Lowary and Widom, 1998) and containing either two DNA linkers (two-linker nucleosome), one linker containing the same DNA length as in the two-linker nucleosome or a nucleosome core with no linkers (Figure 3).
Figure 3.
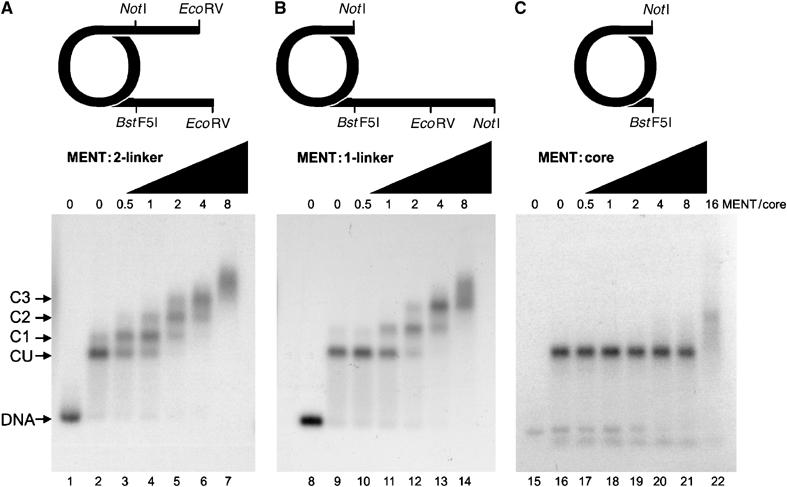
Native agarose gel electrophoresis of MENT interactions with reconstituted mononucleosomes. Mononucleosomes with two linkers (A, lanes 2–7), one linker (containing the same quantity of free DNA as panel A) (B, lanes 9–14) and no linkers (C, lanes 16–22) were reconstituted from histones and clone 601 DNA (Lowary and Widom, 1998) labelled with [32P]ATP. Lanes 1, 8, and 15 contain free DNA used for the two-linker (lane 1), one-linker (lane 8) and no-linker (lane 15) nucleosomes, respectively. MENT/nucleosome core ratios are indicated at the top of the agarose gel panels. Arrows indicate the position of complexes corresponding to unbound nucleosomes (CU) and discrete MENT–nucleosome complexes (C1–C3).
When MENT was added to the two-linker mononucleosomes, discrete protein–nucleosome complexes were resolved with increasing MENT concentration (marked as C1, C2, C3 in Figure 3A). At a MENT/nucleosome ratio approaching one, MENT caused a complete retardation of free mononucleosomes to form a discrete complex (C1). A further increase in the concentration of MENT yielded a second complex with lower nucleosome mobility (C2), and higher ratios resulted in complexes C3 and nucleosome smearing indicative of higher order nuclear–protein complexes (Figure 3A, lanes 6 and 7). Interestingly, MENT demonstrated a significantly reduced ability to bind the one-linker mononucleosomes in particular at protein/core ratio of 1:1 (Figure 3B, lanes 11). A two-linker mononucleosome therefore represents a stronger binding partner for MENT than the one-linker mononucleosome. Furthermore, the nucleosome core had a sharply reduced MENT affinity compared to the linker-containing nucleosomes as no MENT-retarded nucleosomes were detected (Figure 3C). These data reveal that MENT has a high affinity for the DNA stem structure formed by linkers at the entry/exit point of nucleosomes and are consistent with the molecule containing two DNA-binding sites.
MENT protects linker DNA from digestion by DNase I
We employed clone 601 DNA to map the location of MENT on a DNA array with precisely defined nucleosome positioning (Lowary and Widom, 1998). We also aimed to map MENT on the linkers protected by nucleosome cores from both sides so that linker organization would be similar to those in native nucleosome arrays. For this purpose, nucleosome trimers were reconstituted from clone 601 DNA and core histones. The 146-bp-long nucleosome positioning sequence from clone 601 was placed between nucleotides 46 and 192, 259 and 405 and 470 and 616 in the 636-bp-long template. To test that the clone 601 correctly positions nucleosomes under our experimental conditions, we treated the reconstituted nucleosome trimers with increasing concentrations of DNase I (Figure 4A, lanes 1–8). The digestion patterns show a clear and equal protection of the three nucleosome cores at the expected positions.
Figure 4.
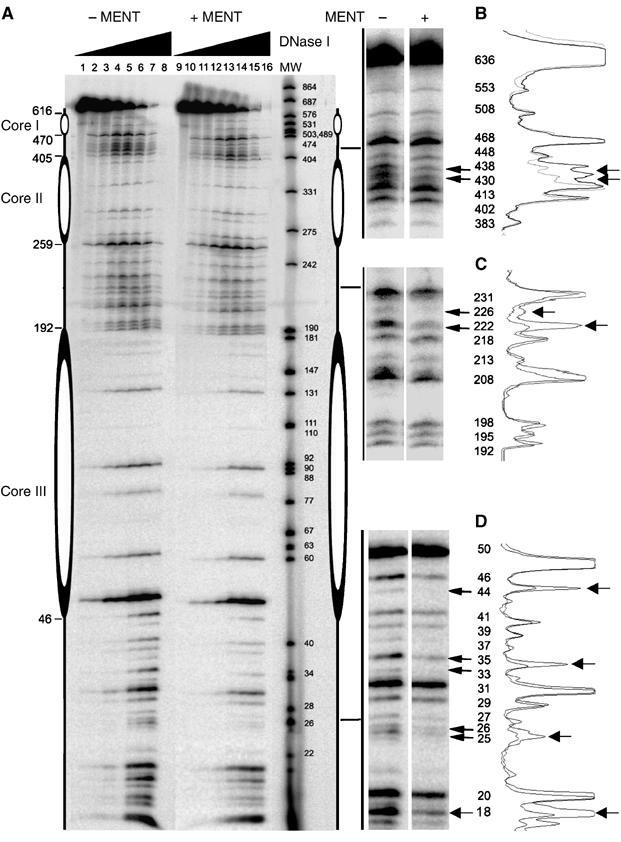
DNase I footprinting of MENT reconstituted with trinuclesomes. (A) Reconstituted nucleosome trimers labelled with 32P-ATP without MENT (lanes 1–8) or reconstituted with two molecules of MENT per nucleosome (lanes 9–16) were incubated with DNase I for 0 (1,9), 1 (2, 10), 2 (3, 11), 5 (4, 12), 10 (5, 13), 20 (6, 14), 30 (7, 15) and 40 (8, 16) min and analysed on a 6% polyacrylamide/urea gel. The molecular size markers (lane MW) represent 32P-labelled DNA of pUC19 (GenBank accession no. L09137) and pEGFP-C3 (GenBank accession no. U57607) simultaneously digested with MspI restriction enzyme. (B–D) Magnified regions of the gel where MENT interferes with DNase I digestion. The molecular sizes of the DNA bands (bp) are indicated. On the left of panels B–D quantitation of DNase I footprints is shown. The extent of protection of each region in trinucleosome from DNase I without (heavy line) and with MENT (light line) was quantitated using ImageJ.
We reconstituted the nucleosome trimers with MENT (2 molecules/nucleosome) and digested MENT-reconstituted nucleosome with DNase I under identical conditions (Figure 4A, lanes 9–16). All MENT-induced changes in the DNase I cleavage were observed within the linkers. At the first linker, not protected by a nucleosome on one side, MENT protection was spread between more sites (25/26/33/35/44 in Figure 4D), indicating that MENT's position is less precise than on the linkers situated between two nucleosomes. A similar widespread pattern of MENT protection was observed on the mononucleosome linkers (data not shown). In each of the two ‘closed' linkers (flanked by nucleosome cores on both sides), MENT protected two sites (430/438 in Figure 4B and 222/226 in Figure 4C) located in the middle of the nucleosome linkers. This location is consistent with (a) nucleosome linker juxtaposition in MENT-containing chromatin (Grigoryev et al, 1999), (b) MENT-induced tramlines of juxtaposed naked DNA duplexes (Springhetti et al, 2003) and (c) the higher affinity of MENT for linker DNA in the two-linker nucleosome (Figure 3). Taken together, these data strongly support the action of MENT as an architectural factor organizing nucleosome linker stems in condensed chromatin.
Structural consequences of serpin conformational change for MENT function
Inhibitory serpins undergo RCL cleavage and conformational rearrangement throughout the molecule as part of their inhibitory function. The rearrangement of the RCL in MENT is of interest, as in vitro and in vivo studies show that the sequence and conformation of the RCL is critical for mediating chromatin bridging. Furthermore, several serpins utilize conformational change to alter their affinities for target ligands: for example, native and cleaved antithrombin bind heparin pentasaccharide with high and low affinity, respectively, as a result of conformational change in heparin-binding residues (Olson and Bjork, 1992). To investigate such rearrangements in MENT, we solved the structure of cleaved wild-type protein at 1.7 Å resolution (Figure 5). The structure reveals that the P17–P1 region of the RCL is buried in the centre of the A β-sheet, forming an additional β-strand s4A (Figure 5A). To accommodate the insertion of the RCL into the A β-sheet, all structurally characterized inhibitory serpins undergo major conformational rearrangements in which the D-, E- and F-helices shift in concert with the first three strands of the A-sheet (s1A–s3A) (Stein and Chothia, 1991). However, a comparison of cleaved and native MENTWT structures reveals that the molecule does not undergo the characteristic structural repositioning of the D-, E- and F-helices in response to RCL insertion (Figure 5B). Instead, strands s1A–s3A and s5A–s6A, which are distorted in the native structure of MENT (Figure 1A), spring apart to accept the RCL as the fourth strand of the A β-sheet (Figure 5B). The absence of conformational rearrangement is reflected in the relatively good superposition of the native and cleaved forms of MENTWT (1.29 Å over 352 Cα atoms; Figure 5B). Accordingly, the electrostatic potential surfaces of native and cleaved MENT are very similar (Figure 5C).
Figure 5.
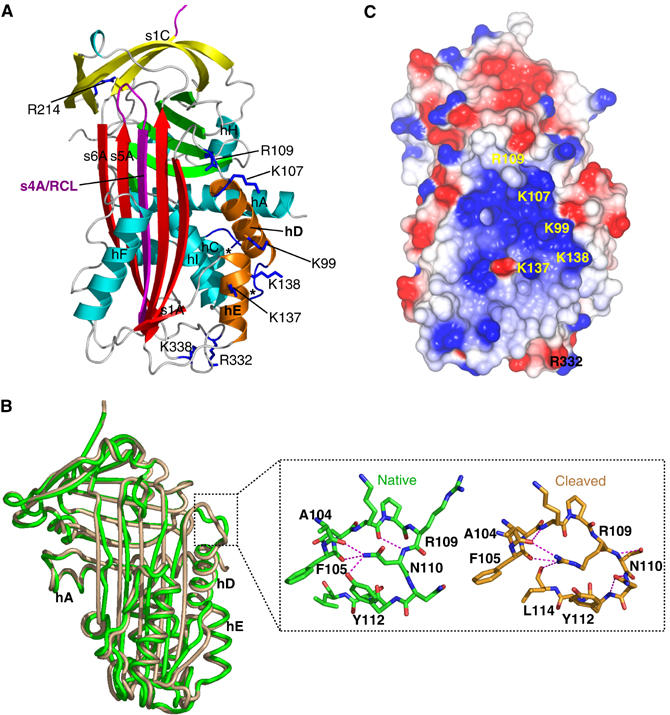
(A) The structure of cleaved MENTWT labelled as in Figure 1A. The termini of the M-loop (between hC and hD) are indicated by *. (B) Superposition of native (green) and cleaved (brown) MENTWT. The change in conformation at the top of the D-helix is indicated by a dotted square and shown in the inset. Hydrogen bonds are shown by dashed lines and R109, N110, Y112, F105 and A104 are labelled. (C) CCP4MG (Potterton et al, 2002, 2004) electrostatic potential surface of cleaved MENTWT, coloured as in Figure 1B.
As a consequence of this unusual lack of structural rearrangement, all but one of the DNA-binding residues identified in this study adopt identical conformations in the wild-type and cleaved structures of MENT (Figure 5B). The exception, R109, is located in a loop joining the C-terminus of the D-helix to s2A that undergoes plastic deformation (Figure 5B). In the native conformation, R109 is solvent exposed, and the neighbouring N110 adopts a buried conformation (Figure 5B). In the cleaved state, these two residues switch position, with N110 becoming surface exposed, and R109 more buried, forming side-chain-mediated hydrogen bonds with the backbone of A104 (hD), F105 (hD) and L114 (s2A). Thus, while R109 may exert subtle effects on MENT DNA binding, insertion of the RCL into the centre of the A β-sheet is not expected to dramatically change the DNA-binding function of MENT.
The structure of native MENTΔMloop reveals intermolecular loop–sheet interactions
Protein–protein interactions and oligomerization of linker histones and non-histone chromatin architectural proteins may play a role in mediating chromatin bridging (Luger and Hansen, 2005). However, the structural basis of such an interaction remains obscure. We have previously shown that within MENT, the sequence of the RCL is crucial for chromatin bridging. Specifically, a MENT construct (MENTOV) in which the sequence of the RCL has been changed to that of the non-inhibitory serpin, ovalbumin, binds to DNA with an affinity indistinguishable from MENTWT, but is unable to promote chromatin bridging in vitro in a defined system (Springhetti et al, 2003). In contrast, MENTWT formed large self-associated particles of extensively bridged chromatin and in complex with DNA or chromatin, it could be crosslinked in dimers and higher order oligomers suggesting polymerization within the complex (Springhetti et al, 2003). Given the ability of serpins to form loop–sheet oligomers mediated by the RCL, we suggest that loop–sheet polymerization may be central to MENT function. Furthermore, we have shown that the M-loop blocked the spontaneous formation of MENT oligomers and that the MENTΔMloop mutant spontaneously polymerizes (Springhetti et al, 2003). Thus, in order to investigate MENT oligomerization, we determined the structures of native and cleaved MENTΔMloop.
Native and cleaved MENTΔMloop adopt similar conformations to their wild-type counterparts: in particular, the RCL of native MENTΔMloop is partially inserted into the top of the A β-sheet. The structure of native MENTΔMloop revealed four molecules in the asymmetric unit, with two modes of interaction (Figure 6A). Firstly, a ‘back-to-back' MENT interaction is present, whereby the A-, C-, G-, H- and I-helices of two molecules are in contact (Figure 6A). Secondly, a loop–sheet interaction is seen where the RCL of one molecule forms β-strand interactions with strand s6A of the A-sheet of its neighbour, to form a seventh strand (s7A′; Figure 6A and B; Supplementary Table 3 and Supplementary Figure 4). Such ‘strand s7A' interactions have been previously observed in the structure of native PAI-1 and represent a reversible mechanism of loop–sheet polymerization (Sharp et al, 1999). In the cleaved form of MENTΔMloop no such RCL mediated protein–protein interaction is present, as the RCL is buried as s4A in the A β-sheet.
Figure 6.
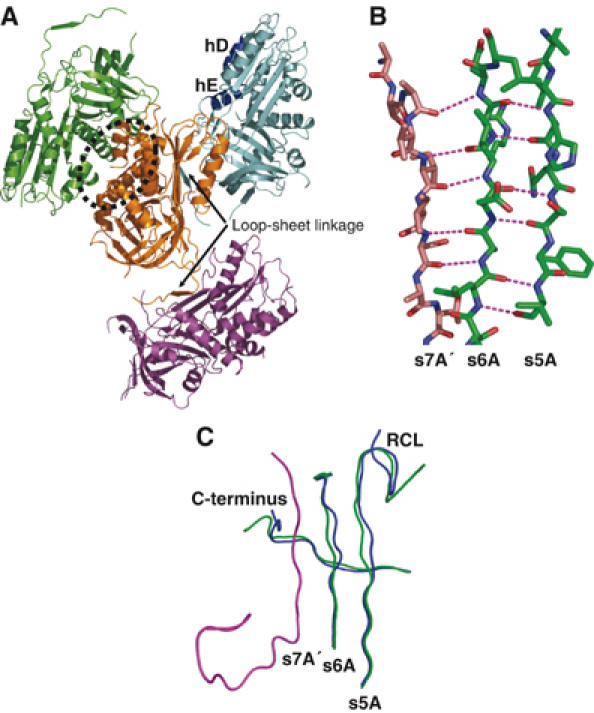
(A) Structure of the native MENTΔMloop tetramer in the asymmetric unit. Each monomer is coloured differently; the orange and green monomers form a ‘back to back' dimer (indicated by a dotted oval); the orange molecule forms a loop–sheet linkage (arrow) to the cyan molecule and a loop–sheet linkage to the magenta molecule. hD and hE are labelled. (B) Loop–sheet hydrogen bonds formed by the RCL (brown) of one molecule with the s6A of an adjoining molecule (green). Hydrogen bonds are shown as magenta broken lines. (C) Comparison of native MENTWT (blue) and MENTΔMloop (green) reveals conformational change in the C-terminus (labelled) and s5A/s6A of the A β-sheet of MENTΔMloop in response to the interaction with the RCL of a neighbouring molecule (magenta). Note also the different trajectory of the RCL (labelled).
The RCL-mediated interaction creates a β-sheet polymer running down the three-fold screw axis throughout the crystal lattice. Comparison of the structures of native forms of MENTΔMloop and MENTWT reveals significant conformational changes in the C-terminus as well as strands s6A and s5A in the A-sheet in order to allow the RCL to interact with a neighbouring molecule (Figure 6C). Furthermore, the trajectory of the RCL changes from a central orientation to one in which the P14–P9 (358–363) residues lie across the top of strand s3A (Figure 6C).
MENT oligomers are functionally active against cathepsin L
The observation that MENTΔMloop was able to form an s7A polymer raised the question of the role of such an interaction in MENT function and the impact of this interaction on the inhibitory activity of the molecule. Sharp et al (1999) have previously observed that s7A-mediated polymers of PAI-1 were reversible and did not affect inhibitory activity. We induced MENT polymerization by incubating both MENTWT and MENTΔMloop at 44°C for 16 h (Supplementary Figure 5), producing oligomers similar to those we have previously reported (Springhetti et al, 2003). We also noted that MENTΔMloop polymerized more readily than MENTWT (Springhetti et al, 2003). Consistent with an s7A loop–sheet interaction, the formation of MENT oligomers at 44°C had no effect on the inhibitory activity of the molecule against cathepsin L (Supplementary Figure 5). In contrast, and in common with numerous other serpins characterized to date (e.g. Devlin et al, 2001), incubation of MENT at a higher temperature (55°C) resulted in the formation of typical inactive serpin polymers and aggregated material (data not shown). Thus, we show that MENT is able to form active MENT oligomers in vitro consistent with an s7A-mediated interaction; however, our data also reveal that, like other serpins, MENT is also able to form inactive s4A-mediated loop–sheet polymers in response to heating at temperatures close to the thermal melting point of the molecule (Irving et al, 2002).
An RCL/s7A peptide is able to inhibit MENT activity in vitro
We investigated the possibility that an RCL/s7A peptide might be capable of interfering with MENT's condensing ability. We reasoned that a 12-mer peptide, encoding the RCL of MENT, should be able to form similar s7A β-strand interactions with strand s6A and would block the formation of the s7A-mediated oligomerization. To assess the effect of the peptide on MENT activity, protein was mixed with an RCL/s7A peptide and then reconstituted either with 1092 bp DNA (Figure 7A) or with soluble chromatin (Figure 7B).
Figure 7.
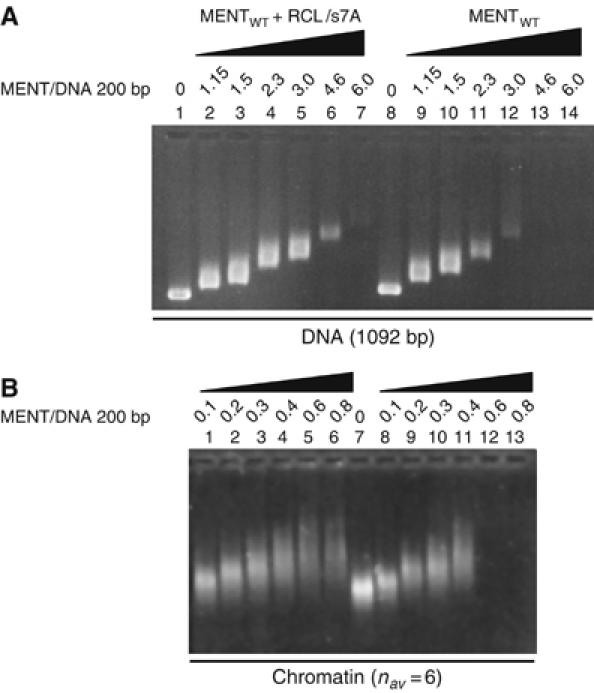
An RCL/s7A peptide interferes with MENT-induced self-association of naked DNA and chromatin. (A) Agarose gel electrophoresis of DNA (1092 bp long) reconstituted with control MENT (lanes 8–14) and MENT reconstituted with an RCL/s7A peptide (1–7). (B) Agarose gel electrophoresis of soluble erythrocyte chromatin reconstituted with control MENT (lanes 8–13) and MENT reconstituted with RCL/s7A peptide (lanes 1–6). The MENT/DNA ratios (molecule/200 bp) were as indicated at the top of the lane markers.
When reconstituted with DNA, MENT+RCL/s7A was unable to condense DNA with the same efficiency as MENT alone. As the protein/DNA ratio increased, MENT was able to alter the mobility of the DNA (Figure 7A, lanes 8–12), and by a ratio approaching 3.0, was able to condense the DNA to the point where solubility was lost (Figure 7A, lanes 13 and 14). In contrast, when the peptide was present, the protein/DNA ratio required to completely condense the DNA was greater than 5.0 (Figure 7A, lanes 6 and 7). A similar result was noted when the peptide was added to chromatin association assays where MENT was capable of complete chromatin condensation at a ratio of 0.4 (Figure 7B, lane 11) whereas soluble chromatin was still present at a ratio of 0.8 (Figure 7B, lane 6) when the RCL/s7A peptide was present. The combined results of these assays indicate that a 12-mer RCL/s7A peptide is indeed able to significantly inhibit the condensing ability of MENT.
Discussion
Serpins are the largest and most diverse family of protease inhibitors. Within multicellular eukaryotes, these proteins have evolved to control important extra- and intracellular processes such as coagulation, inflammation and apoptosis. In rare cases, the serpin fold has been adapted for non-inhibitory function (Silverman et al, 2001). Here we investigate an extreme example of this adaptation and reveal the role of MENT in the fundamental process of chromatin condensation.
The hallmark of serpins is their ability to undergo large conformational rearrangements. Primarily, these structural transitions drive inhibition of target proteases (Huntington et al, 2000). However, uniquely among protease inhibitors, conformational change also allows for exquisite modulation of inhibitory activity. The native structure of MENT reveals that the RCL is partially inserted into the top of the A β-sheet. To date, this feature has been identified only in serpins that undergo RCL expulsion in response to the binding of specific cofactors (Jin et al, 1997). Although our studies revealed that MENT is able to coordinate DNA via residues on the D- and E-helices, we were unable to detect any change in tryptophan fluorescence within the MENT molecule in response to DNA binding. Furthermore, DNA binding did not affect the inhibitory activity of MENT against the protease, cathepsin L. Together, these data suggest that DNA does not promote RCL expulsion in MENT.
Our biophysical studies coupled with the crystal structure of MENT reveal that there are two clusters of DNA-binding residues >30 Å apart. Mutagenesis data confirmed that MENT coordinates DNA via residues on the D- and E-helices, as well on and around the AT-hook in the M-loop. It is shown that MENT demonstrates a strong affinity for reconstituted nucleosomes that contain two linkers and that when reconstituted with nucleosome trimers, MENT is predominantly located in the middle of the nucleosome linkers. Together, these data support a role for MENT in organizing nucleosome linker stems where MENT contains two DNA-binding sites and may be able to simultaneously coordinate two linkers. We cannot, however, completely exclude an alternative model where linkers are coordinated by two or more MENT molecules in concert.
Surprisingly, the structure of cleaved MENT revealed that the molecule does not undergo substantial rearrangement of the ligand-binding region in response to the S to R transition. Only one residue, R109, undergoes conformational change; however, movement of this residue alone would not be predicted to seriously compromise DNA binding. Thus, although we cannot exclude conformational control of MENT by a cofactor other than DNA, it is suggested that MENT adopts the partially inserted conformation in order to minimize the structural transitions that the molecule has to undergo during inhibitory function. Such a feature of MENT would allow the molecule to assemble onto chromatin fibres and also function to control a protease without major perturbation of the DNA-binding surface.
Considerable biophysical and biochemical data indicate the importance of protein oligomerization for chromatin condensation by non-homologous histone and non-histone architectural proteins. In particular, linker histone H5 and the non-histone MeCP2 proteins have both been shown to oligomerize in the presence of DNA (Carter and van Holde, 1998; Georgel et al, 2003), the heterochromatin protein 1 can self-associate in solution in the absence of DNA (Wang et al, 2000) and it has been postulated that nucleosome arrays in yeast heterochromatin are constrained by SIR polymers spreading along the chromatin fibres (Strahl-Bolsinger et al, 1997; Rudner et al, 2005). In common with these proteins, MENT forms large nucleoprotein suprastructures and it is possible to crosslink MENT oligomers in the presence of DNA or chromatin (Springhetti et al, 2003). This activity is important for the bridging of distinct chromatin arrays. Electron microscopy as well as extensive biochemical data reveals that the sequence and conformation of the RCL is important for this bridging function, as mutations in this region compromise chromatin bridging in vitro (Springhetti et al, 2003) and in vivo (Irving et al, 2002).
The ability of MENT to form higher order oligomers suggested that MENT may have adapted loop–sheet linkages for function (Figure 8). The serpin fold is susceptible to forming long chain, strand s4A-mediated loop–sheet polymers; however, to date, serpin polymerization has been exclusively linked with pathogenesis and disease (Carrell and Lomas, 1997).
Figure 8.
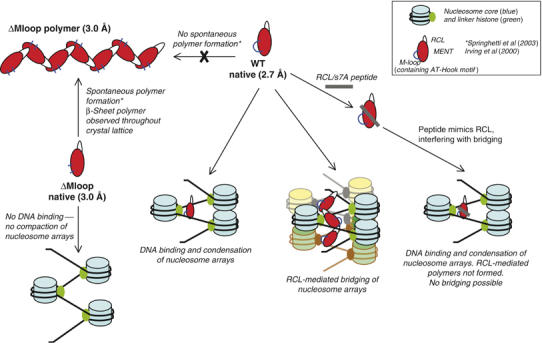
Model for MENT function. Studies on mutants of MENT (red ovals) reveal that deletion of the M-loop (and hence the AT-hook motif) (blue lines) results in the loss of DNA binding and therefore MENTΔMloop (ΔMloop) is unable to condense nucleosome arrays. Linker histone H5 is depicted with the nucleosome as MENT and linker histone meet together in chicken blood cells and act synergistically (Springhetti et al, 2003). Deletion of the M-loop in MENT leads to spontaneous polymerization of the molecule (Springhetti et al, 2003) demonstrating that the M-loop in the absence of DNA prevents MENT polymerization. In contrast, the MENTWT (WT) is unable to spontaneously form polymers; however, we have previously shown that oligomeric MENTWT structures form in the presence of DNA (Springhetti et al, 2003). It is suggested that MENTWT, utilizing dynamic s7A–s6A interactions to form oligomers, can bridge distinct nucleosome arrays. Consistent with this model, an RCL/s7A peptide (grey line) is able to disrupt the edge-strand interactions and partially block MENT function.
We investigated the mechanism of MENT polymerization by determining the structure of a MENT mutant that spontaneously oligomerizes. Our data reveal that rather than forming inactive archetypal s4A serpin linkages, MENTΔMloop is able to form RCL β-strand (s7A) interactions with the edge β-strand of the A β-sheet of a neighbouring molecule. These data rationalize the observation that the sequence and conformation of the MENT RCL is important for mediating chromatin bridging in vitro and in vivo (Irving et al, 2002; Springhetti et al, 2003), as mutations that would be expected to disrupt the β-strand conformation of the RCL would no longer be expected to properly interact with the A-sheet (Figures 6 and 8). In support of these structural data, we show that an RCL/s7A peptide is able to inhibit the ability of MENTWT to condense chromatin and coordinate DNA in vitro, presumably by mimicking RCL and effectively blocking polymer formation (Figures 7 and 8).
The structure of the native MENTΔMloop oligomer and comparison with the native MENTWT structure reveals modest conformational changes in the C-terminus and strands s6A and s5A. The RCL is partially inserted in the structures of both native MENTWT and native MENTΔMloop; however, the broad trajectory of the RCL in the MENTΔMloop structure is altered to allow the RCL to interact with the neighbouring molecule. The reason why MENTΔMloop and not MENTWT is able to spontaneously polymerize is not immediately apparent; however, it is suggested that the M-loop may prevent oligomerization via steric interactions. It is presumed that upon or after DNA binding, the position of the M-loop changes, allowing the s7A-mediated oligomerization.
The s7A MENT–RCL interactions observed in the structure of native MENTΔMloop do not block the central (s4A) region of the A-sheet and thus would not be predicted to affect inhibitory function (Zhou et al, 2001). Consistent with this type of polymerization, we have shown that oligomerization of MENTΔMloop and MENTWT in vitro at 44°C does not affect inhibitory function against cathepsin L (Supplementary Figure 5). The use of a reversible s7A-type oligomerization raises the possibility that MENT polymerization may be dynamic within the nucleus and possibly influenced by other controlling factors.
The recent discovery of nuclear localization by cathepsins (Goulet et al, 2004) suggests that the interplay between MENT and proteases may control chromatin structure. We have also shown that nuclear cathepsin L stimulates the relocation of MENT away from heterochromatin, its subsequent association with euchromatin and also repression of cell proliferation (Irving et al, 2002). Remarkably, a cathepsin L null mutant dramatically reduces histone H3 methylation showing a surprising link between the protease and chromatin epigenetics (Bulynko et al, 2006). Although the precise role of cathepsin L requires clarification, we speculate that the protease could trigger MENT exit from heterochromatin by destabilizing histone H3 methylation and allowing spreading of MENT molecules into uncondensed euchromatin. As cathepsin L is present in the nuclei of dividing but not quiescent cells, this might be especially important to prevent cells from escaping differentiation and resuming proliferation.
The MENT structure reveals that the RCL of MENT performs two important, but distinct functions. Like all inhibitory serpins, it is responsible for interacting with target proteases. Uniquely, however, the structural data reveal that the native, exposed RCL of MENT can be utilized for mediating physiological linkages within condensed chromatin, rather than the pathogenic β-strand interactions normally associated with serpin oligomerization. It would appear that both functions apparently evolved to fulfil one goal—to remodel and condense heterochromatin in its transition from proliferation to terminal differentiation.
Materials and methods
Protein expression and purification
Plasmid constructs expressing MENTWT, MENTOV and MENTΔMloop were described previously and were used to over-express and purify MENT mutants as described previously (Irving et al, 2002). The purity of the proteins was verified using SDS–PAGE.
Crystallography
Crystals were grown in all cases using the hanging drop method and diffraction data collected using both in-house and synchrotron X-ray sources. The structures of all forms were solved by molecular replacement. Data collection and refinement statistics are detailed in Supplementary Table 1. Further details of structural determination are found in Supplementary Methods.
Mutagenesis and analysis of MENT mutants
Details of the mutagenesis and analysis of the mutants are given in Supplementary data.
Determination of kinetic parameters
The stoichiometry of inhibition (SI) and second-order association rate constants (kass) were determined as described previously and in Supplementary Figure 1 (Irving et al, 2002).
Electrophoretic mobility shift assays
For EMSAs, 20 μl reactions containing 10 mM Hepes (pH 7.0), 0.5 mM EDTA, 40 mM NaCl, 0.5% (w/v) Brij-35, <0.3 μM of annealed oligonucleotides (65 bp) (5′-GATCCTTGAAAATACAGGTTTTCGGTACCAG ATCTACCATGATGATGATGATGATGAGAACCCCGCA-3′) and between 1 and 10 μM MENT protein were used. Reactions were incubated at room temperature for 15 min. Samples were loaded onto 2% agarose (Amresco) gels after the addition of 5 μl of 50% glycerol. Electrophoresis was performed for 30 min at 100 V. Gels were visualized using ethidium bromide and imaged on an AlphaImager (Alpha Innotech). DNA that remained unbound in each reaction was quantified (ImageJ) from digital images. Affinities were calculated by plotting the log of the protein concentration versus the log of (b/1−b), where b is the fraction of bound DNA (Allain et al, 1999). When the value of log(b/1−b) is 0, 50% of DNA is bound. Apparent equilibrium dissociation constants (KD) were estimated from an average of at least three independent experiments. MENTWT bound DNA rapidly (Figure 2A) with an apparent KD of 0.67 μM (Figure 2B). The smearing seen at 1 μM protein is indicative of the presence of multiple nucleoprotein species (Carruthers et al, 1998; Georgel et al, 2001, 2003). Incubation with >1 μM MENTWT produced nucleoprotein complexes that were too large to migrate through an agarose gel (0.5–2.0% agarose, data not shown).
Chromatin association assays
Chromatin association assays were performed in a similar fashion to those described previously (see details in Supplementary Methods).
MENT linker positioning and DNase I protection experiments
Details of the nucleosome reconstitution, deoxynucleoprotein electrophoresis and DNase I protection experiments are detailed in Supplementary Methods.
Preparation of MENT complex with an RCL/s7a peptide
MENT preincubated with the 12-mer peptide M12 (TEAAAATAVIIS) was used in EMSA and chromatin association studies (as detailed in Supplementary data).
Figures
Figures 1A, 5A, C and 6A–C and Supplementary Figure 2 were prepared with Pymol (http://www.pymol.org). Figures 1B and 5C were prepared with CCP4MG (Potterton et al, 2002, 2004). Supplementary Figure 4 was prepared using O (Jones et al, 1991).
Structural coordinates
The coordinates of all four structures have been deposited in the Protein Data Bank (PDB identifiers: native MENTWT, 2H4R; cleaved MENTWT, 2H4P; native MENTΔMloop, 2H4S; cleaved MENTΔMloop, 2H4Q).
Supplementary Material
Supplementary Figures
Supplementary Tables and Methods
Acknowledgments
This work was supported by the NHMRC, the ARC and by NIH grant GM-59118. We thank Jonathan Widom for the generous gift of the nucleosome positioning ‘601' DNA. We thank Phil Bird and Michelle Dunstone for critical reading of the manuscript and helpful discussion. We thank the staff at BIOCARS (The Advanced Photon Source, Chicago) for technical assistance.
Competing Interests Statement The authors declare that they have no competing financial interests.
References
- Allain FH, Yen YM, Masse JE, Schultze P, Dieckmann T, Johnson RC, Feigon J (1999) Solution structure of the HMG protein NHP6A and its interaction with DNA reveals the structural determinants for non-sequence-specific binding. EMBO J 18: 2563–2579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aravind L, Landsman D (1998) AT-hook motifs identified in a wide variety of DNA-binding proteins. Nucleic Acids Res 26: 4413–4421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baglin TP, Carrell RW, Church FC, Esmon CT, Huntington JA (2002) Crystal structures of native and thrombin-complexed heparin cofactor II reveal a multistep allosteric mechanism. Proc Natl Acad Sci USA 99: 11079–11084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bulynko YA, Hsing LC, Mason RW, Tremethick D, Grigoryev SA (2006) Cathepsin L stabilizes histone modification landscape on Y chromosome and pericentromeric heterochromatin. Mol Cell Biol 26: 4172–4184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrell RW, Lomas DA (1997) Conformational disease. Lancet 350: 134–138 [DOI] [PubMed] [Google Scholar]
- Carruthers LM, Bednar J, Woodcock CL, Hansen JC (1998) Linker histones stabilize the intrinsic salt-dependent folding of nucleosomal arrays: mechanistic ramifications for higher-order chromatin folding. Biochemistry 37: 14776–14787 [DOI] [PubMed] [Google Scholar]
- Carter GJ, van Holde K (1998) Self-association of linker histone H5 and of its globular domain: evidence for specific self-contacts. Biochemistry 37: 12477–12488 [DOI] [PubMed] [Google Scholar]
- Devlin GL, Parfrey H, Tew DJ, Lomas DA, Bottomley SP (2001) Prevention of polymerization of M and Z1-antitrypsin (1-AT) with trimethylamine N-oxide. Implications for the treatment of 1-AT deficiency. Am J Respir Cell Mol Biol 24: 727–732 [DOI] [PubMed] [Google Scholar]
- Georgel PT, Horowitz-Scherer RA, Adkins N, Woodcock CL, Wade PA, Hansen JC (2003) Chromatin compaction by human MeCP2: assembly of novel secondary chromatin structures in the absence of DNA methylation. J Biol Chem 278: 32181–32188 [DOI] [PubMed] [Google Scholar]
- Georgel PT, Palacios DeBeer MA, Pietz G, Fox CA, Hansen JC (2001) Sir3-dependent assembly of supramolecular chromatin structures in vitro. Proc Natl Acad Sci USA 98: 8584–8589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goulet B, Baruch A, Moon NS, Poirier M, Sansregret LL, Erickson A, Bogyo M, Nepveu A (2004) A cathepsin L isoform that is devoid of a signal peptide localizes to the nucleus in S phase and processes the CDP/Cux transcription factor. Mol Cell 14: 207–219 [DOI] [PubMed] [Google Scholar]
- Goytisolo FA, Gerchman SE, Yu X, Rees C, Graziano V, Ramakrishnan V, Thomas JO (1996) Identification of two DNA-binding sites on the globular domain of histone H5. EMBO J 15: 3421–3429 [PMC free article] [PubMed] [Google Scholar]
- Grigoryev SA, Bednar J, Woodcock CL (1999) MENT, a heterochromatin protein that mediates higher order chromatin folding, is a new serpin family member. J Biol Chem 274: 5626–5636 [DOI] [PubMed] [Google Scholar]
- Huntington JA, Read RJ, Carrell RW (2000) Structure of a serpin–protease complex shows inhibition by deformation. Nature 407: 923–926 [DOI] [PubMed] [Google Scholar]
- Irving JA, Shushanov SS, Pike RN, Popova EY, Bromme D, Coetzer TH, Bottomley SP, Boulynko IA, Grigoryev SA, Whisstock JC (2002) Inhibitory activity of a heterochromatin-associated serpin (MENT) against papain-like cysteine proteinases affects chromatin structure and blocks cell proliferation. J Biol Chem 277: 13192–13201 [DOI] [PubMed] [Google Scholar]
- Jin L, Abrahams JP, Skinner R, Petitou M, Pike RN, Carrell RW (1997) The anticoagulant activation of antithrombin by heparin. Proc Natl Acad Sci USA 94: 14683–14688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones TA, Zou JY, Cowan SW, Kjeldgaard M (1991) Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr A 47: 110–119 [DOI] [PubMed] [Google Scholar]
- Lomas DA, Evans DL, Finch JT, Carrell RW (1992) The mechanism of Z alpha 1-antitrypsin accumulation in the liver. Nature 357: 605–607 [DOI] [PubMed] [Google Scholar]
- Lowary PT, Widom J (1998) New DNA sequence rules for high affinity binding to histone octamer and sequence-directed nucleosome positioning. J Mol Biol 276: 19–42 [DOI] [PubMed] [Google Scholar]
- Luger K, Hansen JC (2005) Nucleosome and chromatin fiber dynamics. Curr Opin Struct Biol 15: 188–196 [DOI] [PubMed] [Google Scholar]
- Olson ST, Bjork I (1992) Regulation of thrombin by antithrombin and heparin cofactor II. In Thrombin: Structure and Function, Berliner LJ (ed) pp 159–217. New York: Plenum Press [Google Scholar]
- Potterton E, McNicholas S, Krissinel E, Cowtan K, Noble M (2002) The CCP4 molecular-graphics project. Acta Cryst D 58: 1955–1957 [DOI] [PubMed] [Google Scholar]
- Potterton L, McNicholas S, Krissinel E, Gruber J, Cowtan K, Emsley P, Murshudov GN, Cohen S, Perrakis A, Noble M (2004) Developments in the CCP4 molecular-graphics project. Acta Cryst D 60: 2288–2294 [DOI] [PubMed] [Google Scholar]
- Richmond TJ, Davey CA (2003) The structure of DNA in the nucleosome core. Nature 423: 145–150 [DOI] [PubMed] [Google Scholar]
- Rudner AD, Hall BE, Ellenberger T, Moazed D (2005) A nonhistone protein–protein interaction required for assembly of the SIR complex and silent chromatin. Mol Cell Biol 25: 4514–4528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schalch T, Duda S, Sargent DF, Richmond TJ (2005) X-ray structure of a tetranucleosome and its implications for the chromatin fibre. Nature 436: 138–141 [DOI] [PubMed] [Google Scholar]
- Schechter I, Berger A (1968) On the active site of proteases. 3. Mapping the active site of papain; specific peptide inhibitors of papain. Biochem Biophys Res Commun 32: 898–902 [DOI] [PubMed] [Google Scholar]
- Sharp AM, Stein PE, Pannu NS, Carrell RW, Berkenpas MB, Ginsburg D, Lawrence DA, Read RJ (1999) The active conformation of plasminogen activator inhibitor 1, a target for drugs to control fibrinolysis and cell adhesion. Struct Fold Des 7: 111–118 [DOI] [PubMed] [Google Scholar]
- Silverman GA, Bird PI, Carrell RW, Church FC, Coughlin PB, Gettins PG, Irving JA, Lomas DA, Luke CJ, Moyer RW, Pemberton PA, Remold-O'Donnell E, Salvesen GS, Travis J, Whisstock JC (2001) The serpins are an expanding superfamily of structurally similar but functionally diverse proteins. Evolution, mechanism of inhibition, novel functions, and a revised nomenclature. J Biol Chem 276: 33293–33296 [DOI] [PubMed] [Google Scholar]
- Springhetti EM, Istomina NE, Whisstock JC, Nikitina T, Woodcock CL, Grigoryev SA (2003) Role of the M-loop and reactive center loop domains in the folding and bridging of nucleosome arrays by MENT. J Biol Chem 278: 43384–43393 [DOI] [PubMed] [Google Scholar]
- Stein P, Chothia C (1991) Serpin tertiary structure transformation. J Mol Biol 221: 615–621 [DOI] [PubMed] [Google Scholar]
- Strahl-Bolsinger S, Hecht A, Luo K, Grunstein M (1997) SIR2 and SIR4 interactions differ in core and extended telomeric heterochromatin in yeast. Genes Dev 11: 83–93 [DOI] [PubMed] [Google Scholar]
- Wang G, Ma A, Chow CM, Horsley D, Brown NR, Cowell IG, Singh PB (2000) Conservation of heterochromatin protein 1 function. Mol Cell Biol 20: 6970–6983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weintraub H (1984) Histone-H1-dependent chromatin superstructures and the suppression of gene activity. Cell 38: 17–27 [DOI] [PubMed] [Google Scholar]
- Woodcock CL, Dimitrov S (2001) Higher-order structure of chromatin and chromosomes. Curr Opin Genet Dev 11: 130–135 [DOI] [PubMed] [Google Scholar]
- Zhou A, Faint R, Charlton P, Dafforn TR, Carrell RW, Lomas DA (2001) Polymerization of plasminogen activator inhibitor-1*. J Biol Chem 276: 9115–9122 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figures
Supplementary Tables and Methods