

Abstract
The dynamin-like GTPase OPA1, a causal gene product of human dominant optic atrophy, functions in mitochondrial fusion and inner membrane remodeling. It has several splice variants and even a single variant is found as several processed forms, although their functional significance is unknown. In yeast, mitochondrial rhomboid protease regulates mitochondrial function and morphology through proteolytic cleavage of Mgm1, the yeast homolog of OPA1. We demonstrate that OPA1 variants are synthesized with a bipartite-type mitochondrial targeting sequence. During import, the matrix-targeting signal is removed and processed forms (L-isoforms) are anchored to the inner membrane in type I topology. L-isoforms undergo further processing in the matrix to produce S-isoforms. Knockdown of OPA1 induced mitochondrial fragmentation, whose network morphology was recovered by expression of L-isoform but not S-isoform, indicating that only L-isoform is fusion-competent. Dissipation of membrane potential, expression of m-AAA protease paraplegin, or induction of apoptosis stimulated this processing along with the mitochondrial fragmentation. Thus, mammalian mitochondrial function and morphology is regulated through processing of OPA1 in a ΔΨ-dependent manner.
Keywords: AAA-protease, mitochondrial fission, mitochondrial fusion, OPA1, rhomboid protease
Introduction
Mitochondria are extremely dynamic, changing their size and shape through frequent fusion and fission in response to the cell environment, cellular differentiation, or pathologic conditions (Yaffe, 1999; Jensen et al, 2000; Griparic and van der Bliek, 2001; Shaw and Nunnari, 2002; Mozdy and Shaw, 2003; Westermann, 2003; Chen and Chan, 2004). Recent studies with cultured mammalian cells revealed that mitochondrial activity or cellular signaling are reflected in the mitochondrial morphology (Yaffe, 1999; Karbowski and Youle, 2003). Dissipation of the mitochondrial inner membrane potential (ΔΨ) induces mitochondrial fragmentation by inhibiting mitofusin (Mfn)-dependent mitochondrial fusion or activating dynamin-related protein (Drp1/Dlp1)-dependent mitochondrial fission (Legros et al, 2002; Ishihara et al, 2003). The re-establishment of ΔΨ recovers filamentous mitochondrial structures by fusion; the recovery requires de novo protein synthesis (Ishihara et al, 2003). Proapoptotic stimuli also activate mitochondrial fission and, conversely, inhibit mitochondrial fusion, resulting in mitochondrial fragmentation (Frank et al, 2001; Karbowski and Youle, 2003). In this context, mitochondrial fragmentation is intimately correlated to the progression of apoptosis (Frank et al, 2001; Lee et al, 2004; Sugioka et al, 2004).
OPA1 is a causal gene product of autosomal dominant optic atrophy, which features a progressive loss of retinal ganglion cells that leads to legal blindness (Alexander et al, 2000; Delettre et al, 2000). OPA1 is a dynamin-like protein with a sequence homology to yeast Mgm1, which is an essential factor for maintaining mitochondrial DNA and inner membrane structures in yeast (Shepard and Yaffe, 1999; Wong et al, 2003). Mgm1 interacts with Fzo1 through Ugo1 and is involved in mitochondrial fusion (Wong et al, 2003; Sesaki and Jensen, 2004). In mammalian cells, exogenous expression of OPA1 affects mitochondrial morphology, and depletion of OPA1 by RNA interference (RNAi) induces mitochondrial fragmentation with aberrant inner membrane structures (Misaka et al, 2002; Olichon et al, 2003; Cipolat et al, 2004; Griparic et al, 2004). Furthermore, the OPA1-depleted cells become sensitive to exogenous apoptotic induction, suggesting that OPA1 is involved in the apoptotic process (Olichon et al, 2003; Lee et al, 2004).
Recent studies in yeast indicate that the function of Mgm1 is regulated by proteolytic cleavage (Herlan et al, 2003; McQuibban et al, 2003; Sesaki et al, 2003). Mgm1 exists in two forms of different lengths: the large isoform (l-Mgm1) representing the mitochondrial processing peptidase (MPP)-processed mature form, and the small isoform (s-Mgm1) produced after cleavage of the mature form by the mitochondrial rhomboid protease Pcp1/Rbd1/Mdm37/Ugo2. The expression of both isoforms, but not either one alone, weakly complements the yeast mgm1 phenotype. Processing of the large isoform and the presence of both isoforms are essential for the maintenance of both normal mitochondrial morphology and mitochondrial DNA. Presenilin associated rhomboid-like (PARL) is a mammalian mitochondrial rhomboid protease (Pellegrini et al, 2001; McQuibban et al, 2003), but it is not known if it is involved in OPA1 processing.
In human cells, there are eight OPA1 splice variants (Delettre et al, 2001), each of which seems to be subsequently processed to form several isoforms with distinct molecular sizes. In the present study, we used two OPA1 splice variants, rat variants 1 and 7, as the substrates, expressed in mammalian cells, and analyzed their processing and effects upon mitochondrial morphology. OPA1 was synthesized as a precursor with a bipartite mitochondrial targeting signal: the matrix-targeting signal (MTS) followed by a transmembrane domain (TM). The N-terminal MTS is removed by MPP in the matrix during import to form the mature OPA1 isoform (L-isoform of variants 1 and 7), which was subjected to further processing to form the short isoforms: S1 for variant 1, and S1 and S2 for variant 7. Here, we demonstrated that OPA1 processing occurred in the matrix was important for the regulation of mitochondrial morphology. Stimulation of the processing strongly correlated with stimulation of mitochondrial fragmentation, and conversely, inhibition of the processing was reflected in the extension of the mitochondrial tubular networks. Interestingly, the processing was activated by dissipation of the ΔΨ across the inner membrane and by proapoptotic stimuli. In this relation, we demonstrated that the L-isoform had mitochondrial fusion stimulating activity, which was lost by proteolytic processing to be the S-isoform. Our present results suggest that m-AAA-protease is involved in some way in this processing reaction.
Results
Multiple processing of rat OPA1 variants at the N-terminal portion
Human OPA1 exists as eight variants due to alternative splicing at the N-terminal portion (Delettre et al, 2001), in which variants 1 and 7 are dominantly expressed, although their differences in function and processing mechanisms remain unknown. In the present study, we used two rat OPA1 isoforms, rat variant 1 (960 amino-acid residues) corresponding to human OPA1 variant 1, and rat variant 7 (997 amino-acid residues) corresponding to human OPA1 variant 7, which has a 37-residue insertion encoded by exon 5b (209–245). Hydrophobicity profiles indicated that variant 1 has a hydrophobic segment (residues 93–113) with moderate hydrophobicity that might function as membrane-anchoring domain (TM1) followed by two segments with lower hydrophobicity (Figure 1A and B).
Figure 1.
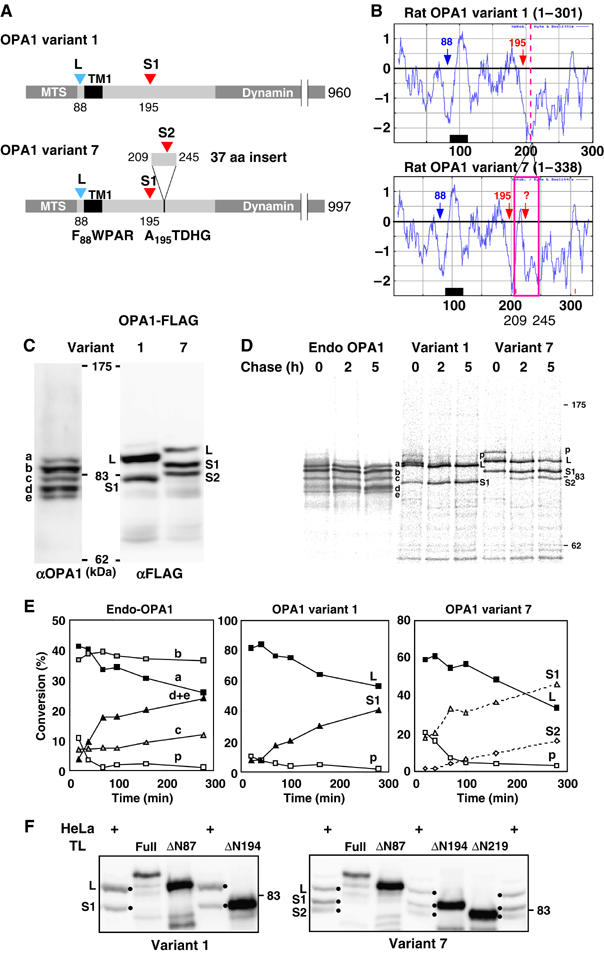
Processing of OPA1 splice variants. (A) Schematic representation of rat OPA1 variants 1 and 7, and their processing sites. Processing sites are indicated by arrowheads, and numbers represent the amino-acid residues of the rat OPA1 precursor. The N-terminal amino-acid residues of L-OPA1 and S1-OPA1 are shown. (B) Hydrophobicity profiles of the N-terminal domain of two OPA1 variants were analyzed by Kite and Doolittle program (window 15). (C) Endogenously expressed OPA1 and exogenously expressed C-terminal FLAG-tagged OPA1 variants in HeLa cells were analyzed by immunoblotting using anti-OPA1 or anti-FLAG antibodies. (D, E) Processing of OPA1 proteins as analyzed by pulse-chase experiments. HeLa cells expressing endogenous OPA1 or exogenous OPA1 variants 1 or 7 were labeled with 35S-Met/Cys mix for 30 min, then chased for the indicated time periods. The cell lysates were subjected to immunoprecipitation using anti-OPA1 (endo-OPA1) or anti-FLAG (variant 1 and variant 7) antibodies. The indicated band intensities were quantified. As bands d and e were electrophorased closely, they were quantified together. p: precursor. (F) The N-terminal deletion mutants of OPA1-FLAG (ΔN87, ΔN194, and ΔN219) synthesized in vitro by PURESYSTMEM and the total lysates of the cells expressing OPA1-FLAG variants were subjected to SDS–PAGE and analyzed by immunoblotting using anti-FLAG antibodies.
Immunoblot analysis of HeLa cells with OPA1 antibodies for endogenous OPA1 detected at least five bands (Figure 1C). To analyze whether these multiple forms resulted from proteolytic cleavage of OPA1 variants, rat OPA1 variants 1 and 7 were FLAG-tagged at the C-terminus, expressed in HeLa cells, and analyzed by Western blotting using anti-FLAG antibody. Two bands (L: ∼100 kDa and S1: ∼80 kDa) or three bands (L: ∼110 kDa, S1: ∼90 kDa and S2: ∼85 kDa) were detected for variants 1 and 7, respectively (Figure 1C). These results indicated that proteolytic cleavage of each OPA1 splice variant produced multiple forms, and the processing occurred at their N-terminal segments.
To analyze biosynthetic process of these bands, pulse-chase experiments were performed for both endogenously- and exogenously expressed OPA1 variants. L-isoforms of variants 1 and 7 converted over time to S-isoforms (Figure 1D and E). Size changes of newly synthesized endogenous OPA1 proteins were, however, not clear; band a of endogenous OPA1 decreased appreciably with chase time, and conversely band d accumulated, whereas bands b and c did not change.
Processing sites of OPA1 variants
The amino-acid sequence of OPA1 residues 86–89 (RNFW) matched well with the consensus sequence for the MPP processing site (Taylor et al, 2001). To determine the processing site, several N-terminal deletion constructs were synthesized in vitro, and their mobility in SDS–PAGE was compared with that of the proteins expressed in HeLa cells (Figure 1F). The mobility of in vitro-synthesized OPA1ΔN87 of each variant coincided with that of variant 1-L (100-kDa band) and variant 7-L (110-kDa band). Protein sequencing of variant 7-L immunoisolated from HeLa cells revealed that its N-terminal sequence starts from Phe-88. We further confirmed that the N-terminal 90 amino-acid residues were sufficient to function as the MPP-cleavable MTS. The OPA1(1–90)-DHFR and su9(1–69)-DHFR proteins imported into mitochondria in vitro and those incubated with purified MPP gave mature forms having similar gel-mobility as DHFR (Supplementary Figure S1). These results clearly indicated that the presequence cleavage of both OPA1 variants occurred at the N87/F88 site to produce the L-isoforms.
We then determined the downstream processing sites of OPA1 variants that produce S-isoforms. Mobility of ΔN194 of both variants coincided well with that of variant 1-S1 and variant 7-S1 (Figure 1F). Sequence analysis revealed that the N-terminal sequence of variant 7-S1 started from Ala-195, demonstrating that both OPA1 variants were processed at position R194/A195 after MPP processing. Variant 7-S1 was further processed at the position around residue 220 because variant 7-S2 and in vitro-synthesized OPA1 Δ219 exhibited similar gel-mobility (Figure 1F). Although the exact S2 processing site was not determined by protein sequencing, the N-terminal segment of variant 7-S2 contained several Gln residues, and we speculated that the S2-processing site localized around 217LQQQIQE223. Variant 1 seemed to have lost the S2-processing site because exon 5b was skipped. When expressed in HeLa cells, variant 1 dominantly produced the L-isoform, whereas variant 7 efficiently produced the S1 and S2 isoforms (Figure 1C), suggesting that insertion of the 37-residue sequence affected processing efficiency.
Localization and membrane topology of OPA1 in mitochondria
Subcellular localization of OPA1 isoforms as analyzed for OPA1-FLAG-expressing HeLa cells revealed that they were weakly anchored to the inner membrane (Figure 5D), exposing the bulk portion to the inter membrane space (IMS) (Supplementary Figure S2A), confirming the previous results (Satoh et al, 2003; Griparic et al, 2004).
Figure 5.
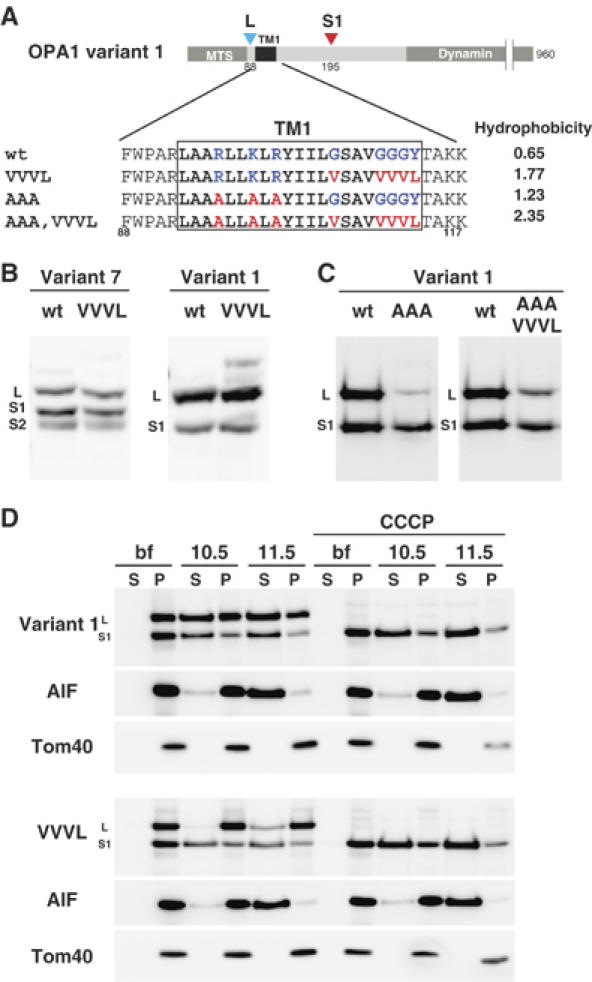
Increment of hydrophobicity of TM1 does not inhibit OPA1 processing. (A) Schematic representation of OPA1 constructs mutated in TM1. Average hydrophobicity of mutated TM1 was also shown. (B, C) HeLa cells expressing the indicated constructs were analyzed by immunoblottiong using anti-FLAG antibodies. (D) The indicated constructs were transfected to HeLa cells, and cultured with or without CCCP for 1 h. Isolated mitochondria were converted to mitoplasts in hypotonic buffer, then treated with sodium carbonate at pH 10.5, or pH 11.5. The membrane pellets (P) and soluble fractions (S) were subjected to immunoblotting using antibodies against FLAG or the indicated proteins.
We then analyzed the membrane topology of the N-terminal portion of OPA1, using the tobacco etch virus (TEV) protease as the probe (Faber et al, 2001) (Supplementary Figure 2B). The OPA1 variant 7 precursor, in which the S1 site region (170–200) was replaced by the TEV-cleavage sequence ((Δ170–200)-tev), was processed only when mitochondrial matrix-targeted version of TEV (mit-TEV) was coexpressed (Supplementary Figure S2C). Alternatively, (Δ170–200)-tev in the isolated mitochondria was cleaved by externally added TEV protease only when both outer and inner membranes were solubilized by detergent (Supplementary Figure S2D). Thus, OPA1 variant 7 is a type I inner membrane protein exposing the N-terminal segment to the matrix and the bulk of the C-terminal domain to the IMS (Supplementary Figure S2E).
Although OPA1 proteins assumed a transmembrane topology, a significant fraction was extracted from the inner membrane by alkaline treatment (Figure 5D). This is probably because the hydrophobicity of the putative TM was too low to firmly anchor them to the inner membrane. The mitochondrial apoptosis-inducing factor (AIF), a type I inner membrane protein with the bulk of the C-terminal segment exposed to the IMS, had similar properties, although it was completely extracted from the membrane under pH 11.5 (See Figure 5D; Otera et al, 2005).
OPA1 processing is affected by membrane potential and proapoptotic stimuli
As reported previously (Ishihara et al, 2003), treatment of cultured mammalian cells with protonophore carbonylcyanide m-chlorophenylhydrazone (CCCP) led to mitochondrial fragmentation, and removal of CCCP recovered the filamentous mitochondrial networks (Figure 2A). This recovery depended on de novo protein synthesis, because cycloheximide (CHX) inhibited the morphologic recovery. These results indicated that mitochondrial morphology is controlled by ΔΨ. We found, in this context, that dissipation of ΔΨ stimulated OPA1 processing. When HeLa cells were cultured in medium containing CCCP for 15 min, the longer forms of the endogenous OPA1 rapidly disappeared, and the shorter forms accumulated (Figure 2B). Similar results were obtained with the exogenously expressed variant 1-FLAG and variant 7-FLAG (Figure 2C), suggesting that OPA1 processing was activated by the dissipation of ΔΨ. The OPA1 L-isoforms were regenerated following 4-h culture after the removal of CCCP, depending on de novo protein synthesis (Figure 2B). These results coincided well with the CCCP-induced mitochondrial morphologic changes, and suggested that OPA1 processing and mitochondrial fragmentation were correlated.
Figure 2.
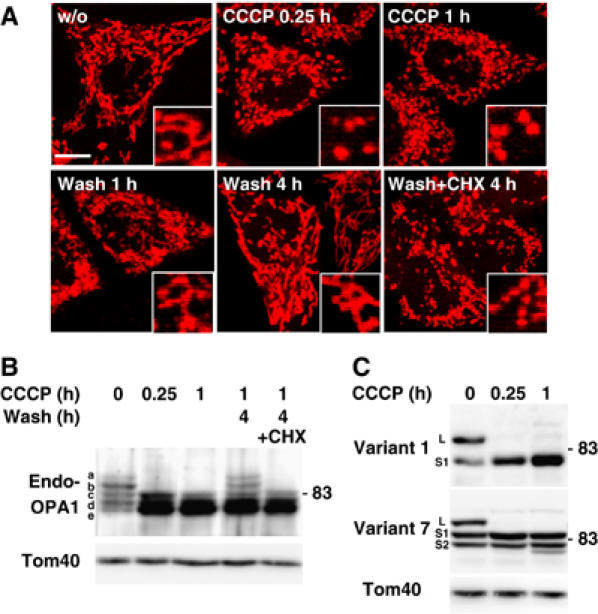
Processing of OPA1 after dissipation of membrane potential. (A) HeLa cells stained by MitoTracker were cultured in the presence of 20 μM CCCP for 0.25 or 1 h. After 1 h culture, CCCP was washed out and the cells were further cultured for 1 or 4 h with or without 1 mM cycloheximide (CHX). Images were obtained by confocal microscopy. Inset: magnified images. Scale bar: 10 μm. (B) HeLa cells were treated as in (A). Lysates obtained from these cells were subjected to immunoblotting using anti-OPA1 antibodies. (C) HeLa cells expressing OPA1-FLAG variants were cultured under the indicated conditions, and the cell lysates were subjected to immunoblotting using anti-FLAG antibodies.
Mitochondria have a central role in apoptosis, and mitochondrial fragmentation activates the progression of apoptosis and increases sensitivity to proapoptotic stimuli (Frank et al, 2001; Lee et al, 2004). In this context, when HeLa cells were treated with actinomycin D, mitochondrial fragmentation was induced (Supplementary Figure S3A). During this process, the long forms of endogenous OPA1 disappeared (Supplementary Figure S3B). The same results were obtained by actinomycin D treatment in the presence of the broad caspase inhibitor zVAD-fmk (data not shown). The actinomycin D-induced OPA1 processing was blocked, however, by expressing antiapoptic factor Bcl-XL (Supplementary Figure S3B). Processing of the exogenously expressed OPA1 variants was also stimulated by proapoptotic stimuli (Supplementary Figure S3C). These results indicated that OPA1 processing is activated in response to proapoptotic stimuli and might be involved in mitochondrial fragmentation in the progression of apoptosis.
OPA1 processing and mitochondrial morphology
Thus, mitochondrial morphology in HeLa cells seemed to be influenced by the expression levels of the L- and S-isoforms. To address the functional difference of the L- and S-isoforms, we tried to express a single isoform, either the L- or S-isoform, within the IMS, to analyze their effects on mitochondrial morphology (Figure 3A). Deletion from variant 7 of residues 190–200 (variant 7-ΔS1) prevented formation of the S1-isoform, and formed the L- and S2-isoforms, indicating that processing at the S2-site occurred independently of the S1-site (Figure 3B). On the other hand, deletion of the S1-site (residues 190–200) from variant 1 (variant 1-ΔS1) produced a single L-isoform (Figure 3B). A single S-isoform could be expressed in the IMS using cDNA in which residues 1–229 of variant 7 was replaced with the IMS-targeting signal of AIF (AIF-230 in Figure 3A and B)(Otera et al, 2005).
Figure 3.
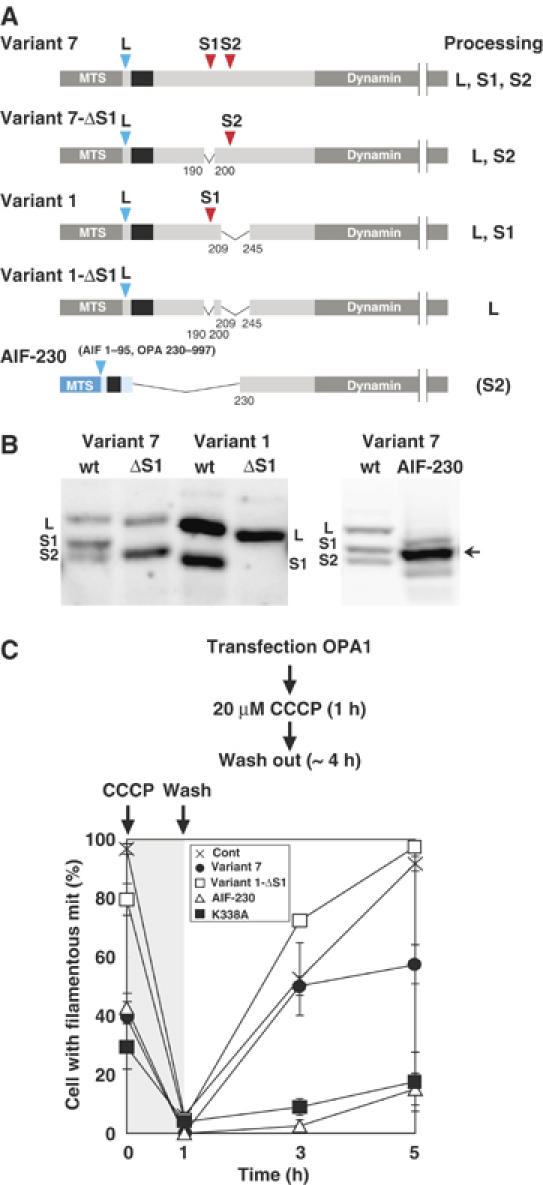
Exogenous expression of processing-defect mutants of OPA1 and their effects on mitochondrial fusion reaction. (A) Schematic representation of OPA1 constructs and their process sites. See Figure 1A for details. (B) HeLa cells expressing the indicated constructs were analyzed by immunoblotting using anti-FLAG antibodies. (C) The indicated OPA1 constructs were exogenously expressed in HeLa cells and the cells were treated with CCCP for 1 h. After removal of CCCP, the cells were incubated for the indicated times. At least 100 cells were counted for the cells with filamentous network structures of mitochondria for three distinct fields. Note that ‘cell with filamentous mit' in this figure (ordinate) corresponds to ‘filamentous/network' plus ‘intermediate' in Supplementary Figure S4B.
Exogenously expressed variant 1-ΔS1, which produced only the L-isoform, had an increased number of filamentous network mitochondria compared with the other constructs which produced both L- and S-isoforms (Supplementary Figure S4A and B). In contrast, when AIF-230 was expressed in HeLa cells, it localized in the IMS (data not shown) as a processed form with mobility similar to that of the S1-isoform (Figure 3B), and induced extensive mitochondrial fragmentation (Supplementary Figure S4A and B). Less than 20% of the expressing cells had normal filamentous mitochondrial structures. Taken together, these results indicated that the short isoforms induced mitochondrial fragmentation, whereas the long isoforms induced filamentous mitochondrial network extension.
As OPA1 is strictly required for mitochondrial fusion (Chen et al, 2005), we then examined mitochondrial fusion activity for the mitochondria harboring various OPA1 constructs. We took advantage of CCCP-treated mitochondria and measured recovery of mitochondrial network structures by fusion reaction after removal of the protonophore (Ishihara et al, 2003). The mitochondria harboring all these constructs responded to CCCP to become fragmented, indicating that these constructs did not affect mitochondrial fission reaction (Figure 3C). Mitochondria in mock-transfected control cells recovered normal network structures in 4 h after CCCP removal. However, the mitochondria harboring GTPase-domain mutant of OPA1 (OPA1K338A) failed to recover the network structures, indicating that OPA1K338A inhibited mitochondrial fusion. Similarly, the fusion reaction was inhibited for the mitochondria harboring AIF-230. In marked contrast, the mitochondria harboring OPA1 variant 1-ΔS1 efficiently recovered elongated network structures. Together, these results indicated that L-isoform had mitochondrial fusion stimulating activity, whereas the S-isoform rather inhibited fusion activity.
In the above experiments, the effects of exogenously expressed OPA1 isoforms were examined under the endogenous OPA1 background. We therefore examined the effects in OPA1-repressed cells. Knockdown of human OPA1 in HeLa cells by small interference RNA (siRNA) induced mitochondrial fragmentation through inhibition of mitochondrial fusion, confirming the previous report (Figure 4A and B) (Griparic et al, 2004; Chen et al, 2005). Expression of rat OPA1 variant 1, which does not have target sequence of this siRNA, recovered filamentous mitochodrial structures to a similar extent as in mock-RNAi cells (Figure 4A and B), suggesting that rat OPA1 variant 1 could substitute for human OPA1 function. Expression of rat OPA1 variant 7 partially, but significantly, recovered the filamentous mitochondrial structure. Variant 1-ΔS1 clearly induced filamentous network structures. In marked contrast, AIF-230 did not have any effects on the mitochondrial morphology in endogenous OPA1 knockdown cells (Figure 4A and B). Together, these results clearly indicated that the L-isoforms are the fusion-active species of OPA1.
Figure 4.
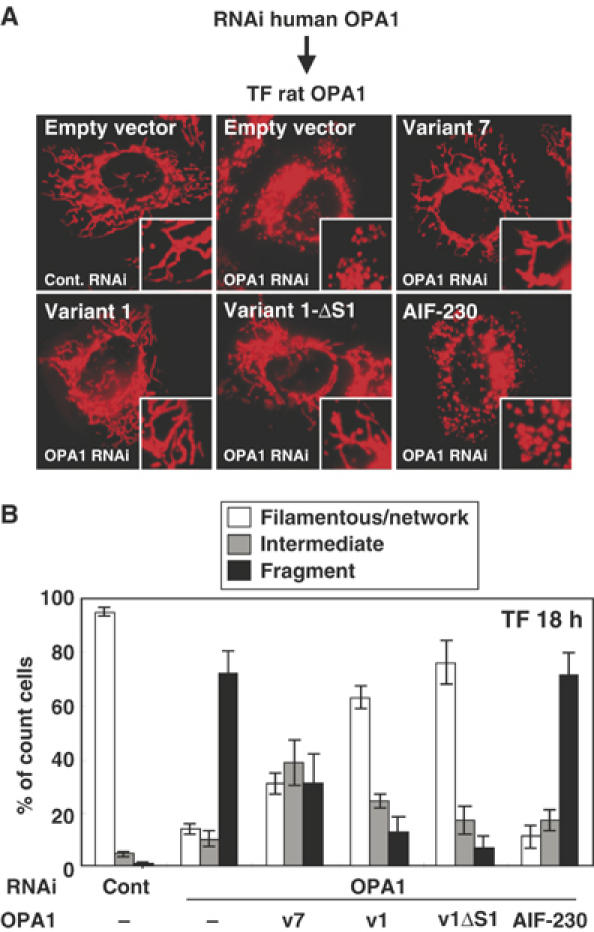
Complementation of mitochondrial morphology by L-OPA1 in OPA1-RNAi cells. (A) HeLa cells were transfected with the siRNA for GFP (cont.) or for human OPA1 three times with 48 h intervals. Then the indicated constructs were transfected and cultured for 18 h. The cells were stained with MitoTracker. (B) HeLa cells with filamentous-network, intermediate, or completely fragmented mitochondrial structures in (A) were counted. At least 100 cells were counted from three different optical fields.
m-AAA protease is involved in OPA1 processing in yeast
Yeast Mgm1, the homolog of OPA1, is present as the l-isoform and the processed s-isoform, whose balance is important for mitochondrial function and the conversion is mediated by mitochondrial rhomboid-like protease Pcp1/Rbd1/Mdm37/Ugo2 (Herlan et al, 2003; McQuibban et al, 2003; Sesaki et al, 2003; Herlan et al, 2004). When OPA1 variants 1 and 7 were expressed in yeast cells, they were both processed to smaller isoforms with the size corresponding to S1-OPA1 and S2-OPA1 detected in HeLa cells (Supplementary Figure S5A), suggesting that OPA1 proteins were cleaved similarly as in mammalian cells. We therefore probed the protease responsible for OPA1 processing using mitochondrial protease-deficient yeast mutants (Supplementary Figure S5B and C).
Of two known yeast mitochondrial rhomboid-like proteases (Pcp1/Rbd1 and Rbd2), Rbd2 was not involved in the processing of OPA1, as the processing proceeded normally in Δrbd2 cells. The processing was compromised significantly in Δpcp1 cells (Supplementary Figure S5B). Consistent with previous reports, the endogenous Mgm1 processing was completely blocked in Δpcp1 cells (Herlan et al, 2003; McQuibban et al, 2003; Sesaki et al, 2003). Unexpectedly, the OPA1 processing was efficiently blocked in the m-AAA protease-deficient mutants (Supplementary Figure S5B). m-AAA protease is an ATP-dependent metalloprotease anchored to the inner membrane exposing the active site to the matrix (Langer et al, 2001). It is a hetero-multimeric complex composed of Yta10/Afg3 and Yta12/Rca1. In Δyta10 and Δyta12 strains, processing of the exogenously expressed variants 1 and 7 was severely blocked, whereas processing of endogenous Mgm1 was not affected. Furthermore, the processing defect in Δyta10 was partially, but clearly, suppressed by YTA10 on multicopy plasmids (Supplementary Figure S5C). Processing of endogenous Mgm1 was not affected by this manipulation. Thus, m-AAA protease is essential for OPA1 processing in yeast cells. On the other hand, OPA1 processing proceeded normally in cells deficient in Yme1, the i-AAA protease that exposes the catalytic site to the IMS (Supplementary Figure S5B). These results were consistent with the finding described above that the OPA1 processing site is exposed to the matrix in mammalian cells (see Supplementary Figure S2). These results suggested that m-AAA protease and rhomboid-like protease Pcp1/Rbd1 are involved in OPA1 processing in yeast cells.
OPA1 processing occurs irrespective of TM1 hydrophobicity
Mgm1 has two hydrophobic segments at the N-terminal portion and is anchored to the inner membrane through the first hydrophobic segment (TM1). Processing by Pcp1/Rbd1 occurs only when the cleavage site in the second hydrophobic segment localizing in the IMS reaches the inner membrane, which is driven by matrix ATP and preprotein import motor (Herlan et al, 2004). In this case, TM1 functions as a stop-transfer sequence and the hydrophobicity of TM1 determines the processing efficiency; TM1 with increased hydrophobicity strongly inhibits the processing.
If OPA1 processing is regulated by the similar mechanism, introduction of hydrophobic amino-acid residues in its TM1 should affect the processing (Figure 5A). However, the processing was not inhibited by increment of TM1 hydrophobicity (Figure 5A–C). Of note, increment of TM1 hydrophobicity increased membrane-anchoring efficiency of OPA1 as assessed by alkaline treatment (Variant 1-VVVL; Figure 5D). Furthermore, it was completely processed to the S1-isoform by CCCP treatment of the cells (Figure 5D). These results strengthened our notion that the L-isoforms of OPA1 are anchored to the inner membrane with type I topology. Furthermore, these results suggested that OPA1 processing was drived by a mechanism distinct from that of yeast Mgm1.
Metal-dependent processing of OPA1 in mammalian mitochondria
If m-AAA protease also functions in OPA1 processing in mammalian cells, bivalent metal chelators should inhibit the processing. We therefore examined the effect of 1,10-phenanthroline (o-phe) on the CCCP-induced OPA1 processing. Processing of FLAG-tagged OPA1 variant 1 (Figure 6A) and variant 7 (data not shown) was efficiently inhibited by pretreatment of the cells with o-phe. We further analyzed the processing reaction using isolated mitochondria harboring variants 1-FLAG or 7-FLAG. When the isolated mitochondria were incubated at 30°C, OPA1 processing occurred in a time-dependent manner (Figure 6B and C). In contrast to the in vivo results, the processing reaction proceeded in the absence of CCCP; mitochondria might be in some uncoupled states in the in vitro assay system. This processing reaction was also significantly blocked by o-phe (Figure 6B and C). In contrast, serine protease inhibitor TPCK, which inhibits Drosophila rhomboid protease, had no effect on the OPA1 processing (Figure 6C). These results indicated that OPA1 processing is catalyzed by metalloprotease.
Figure 6.
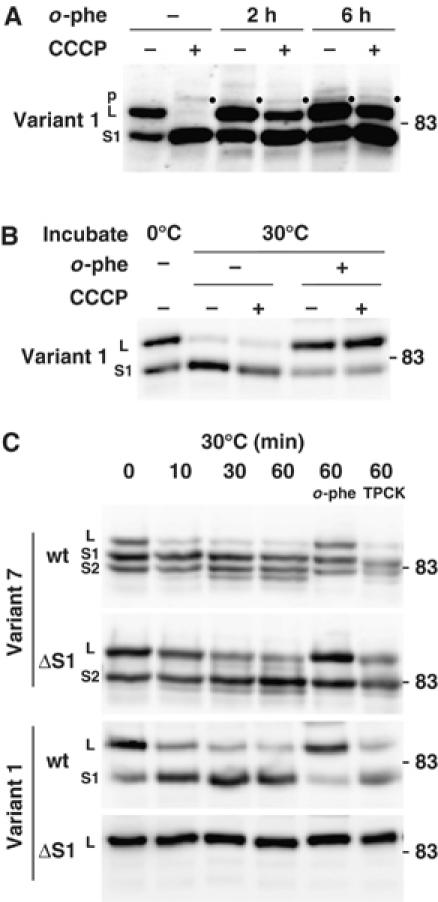
Inhibition of CCCP-induced processing of OPA1 variants by o-phenanthroline. (A) HeLa cells expressing FLAG-tagged OPA1 variant 1 were cultured in the presence of 0.5 mM 1,10-phenanthroline (o-phe) for 2 or 6 h, then 20 μM CCCP was added. After 1 h culture, cell lysates were prepared and subjected to immunoblotting using anti-FLAG antibodies. p: precursor. (B) Isolated mitochondria from HeLa cells expressing FLAG-tagged OPA1 variant 1 were incubated with or without 20 μM CCCP in the presence of 1 mM o-phe at 30°C for 60 min. The mitochondria were subjected to immunoblotting using anti-FLAG antibodies. (C) The isolated mitochondria from HeLa cells expressing the OPA1-FLAG constructs were incubated with or without 1 mM o-phe or 1 mM TPCK at 30°C for the indicated time periods. The mitochondria were subjected to immunoblotting using anti-FLAG antibodies.
m-AAA protease paraplegin is involved in OPA1 processing and mitochondrial fragmentation
We then examined whether m-AAA protease is involved in OPA1 processing in mammalian cells. Two m-AAA proteases, paraplegin and AFG3L2, are expressed in human and form a high molecular weight complex in the mitochondrial inner membrane (Atorino et al, 2003). Although mutation in paraplegin causes an oxidative phosphorylation defect and neuronal degeneration (Casari et al, 1998; Ferreirinha et al, 2004), its specific substrates are not fully understood. The function of AFG3L2 is not known. Recently Langer and collaborators demonstrated in yeast that m-AAA protease Yta12/Yta10 is involved in processing of mitochondrial ribosomal protein MrpL32 and ribosome assembly (Nolden et al, 2005). Similarly, mMrpL32 processing and mitochondrial protein synthesis are affected in paraplegin-deficient mice (Nolden et al, 2005).
Here we analyzed the effect of the exogenous expression of m-AAA proteases on the processing of OPA1. When HA-tagged paraplegin was coexpressed with OPA1 variant 1 or variant 7, the L-isoforms were clearly decreased, and instead the S1-isoforms accumulated (Figure 7A and B), suggesting that the expressed paraplegin activated conversion of the L-isoforms to S1-isoforms. The conversion was not stimulated by expression of paraplegin carrying a mutation within the metal-binding motif (E575Q) of the protease domain, suggesting that protease activity was essential (Figure 7A). Afg3L2 and mammalian mitochondrial rhomboid-like protease PARL did not affect the conversion (Figure 7A–C). The same results were obtained for variant 7 (Figure 7C and data not shown). To analyze kinetics of the OPA1 processing, HeLa cells that had been transfected with paraplegin, AFG3L2, or PARL cDNAs were subjected to pulse-chase reaction with [35S]methionine (Supplementary Figure S6). Conversion of the L- to S1- isoforms of variant 1 was clearly stimulated by exogenous expression of paraplegin, whereas no stimulation was observed with AFG3L2 or PARL (Supplementary Figure S6A). The same was true for the endogenous OPA1 proteins (Supplementary Figure 6B).
Figure 7.
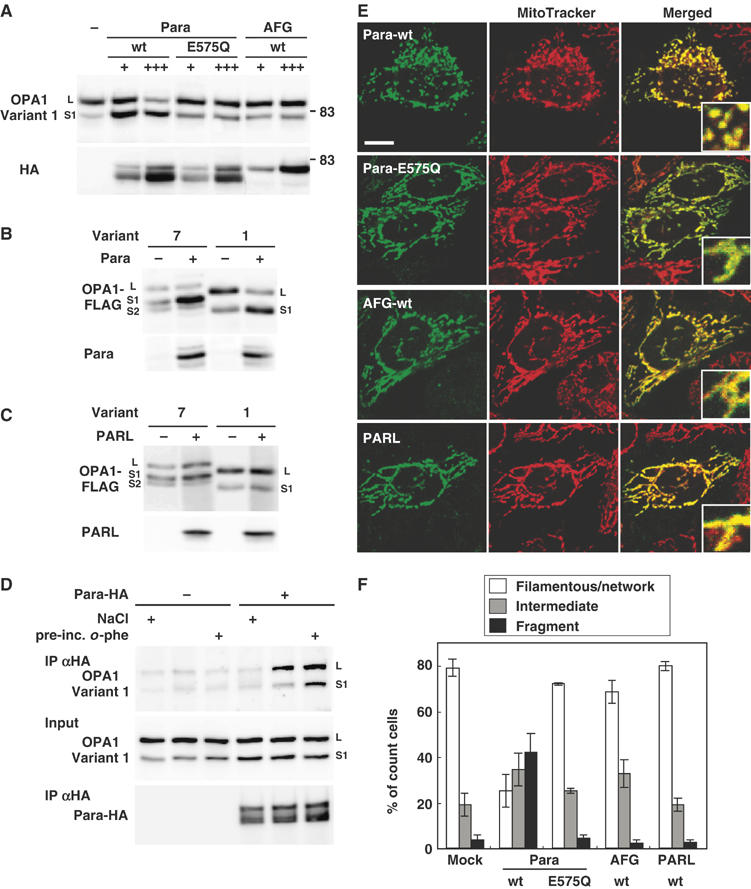
Effect of m-AAA proteases on OPA1 processing and mitochondrial morphology. (A) HA-tagged paraplegin (Para), AFG3L2 (AFG), or paraplegin mutant on metal-binding motif (E575Q) was cotransfected with FLAG-tagged OPA1-variant 1 into HeLa cells. The mitochondrial fractions were subjected to immunoblotting using anti-FLAG or anti-HA antibodies. For cotransfection, OPA1-expression plasmid (0.5 μg) was mixed with 0.5 μg (+) or 1.5 μg (+++) of the protease-expression plasmids. Total amounts of DNA were adjusted to 2.0 μg with empty vector. The upper bands detected in paraplegin-HA-expressing cells seemed to be the precursor. (B, C) Paraplegin (Para) or PARL were cotransfected with OPA1 variants into HeLa cells and analyzed as in (A). (D) OPA1-FLAG (variant 1) was transfected with or without paraplegin-HA. Isolated mitochondria were preincubated with 2 mM o-phe at 30°C (pre-inc.), then solubilized in 1% digitonin and immunoprecipitated with anti-HA IgGs. The precipitants were washed with 150 or 500 mM (NaCl ‘+') NaCl. They were analyzed by immunoblotting using anti-HA or anti-FLAG IgGs. (E) HeLa cells expressing the indicated constructs were labeled with MitoTracker and subjected to immunofluorescence microscopy. Scale bar: 10 μm. (F) HeLa cells with filamentous-network, intermediate, or completely fragmented mitochondrial structures in (E) were counted. More than 100 cells were counted for three different optical fields.
The physical interaction of paraplegin with OPA1 in inner membrane was then examined. The mitochondria isolated from HeLa cells cotransfected with OPA1-FLAG and paraplegin-HA were preincubated with o-phe, solubilized with digitonin, and subjected to immunoprecipitation using anti-HA. OPA1 was co-precipitated dependent on expressed paraplegin, and recovery of the complex was stimulated by preincubation of the mitochondria with o-phe (Figure 7D). The interaction was also observed in the presence of Triton X-100 (data not shown).
In this context, exogenous expression of paraplegin induced mitochondrial fragmentation depending on the functional protease domain (Figure 7E and F), suggesting that overexpression of paraplegin stimulated OPA1 processing, which resulted in mitochondrial fragmentation. Intriguingly, expression of the protease-site mutant of paraplegin (E575Q), AFG3L2, or PARL did not affect mitochondrial morphology (Figure 7E and F). Taken together, these results suggest that the m-AAA protease paraplegin stimulates OPA1 processing in mammalian cells.
Knockdown of paraplegin affects OPA1 processing and mitochondrial morphology
To further corroborate the above findings, we performed knockdown experiments for paraplegin using RNAi. The siRNA treatment efficiently reduced the expression of paraplegin (Figure 8A and B). However, the effect of paraplegin knockdown on processing of endogenously- or exogenously expressed OPA1 at steady-state level was only weak, if any (Figure 8A). We therefore analyzed the effect of RNAi on the kinetics of OPA1 processing by pulse-chase reaction. The processing rate was significantly compromised by paraplegin knockdown (Figure 8C). Similar results were obtained for endogenous OPA1 species (Figure 8D). We further examined the effect of PARL knockdown on OPA1 processing (Figure 8A–D). Steady-state population of endogenously- or exogenously expressed OPA1 proteins, as well as the processing rate of the L-isoforms was not inhibited; rather, the processing rate was stimulated for unknown reasons (Figure 8C and D).
Figure 8.
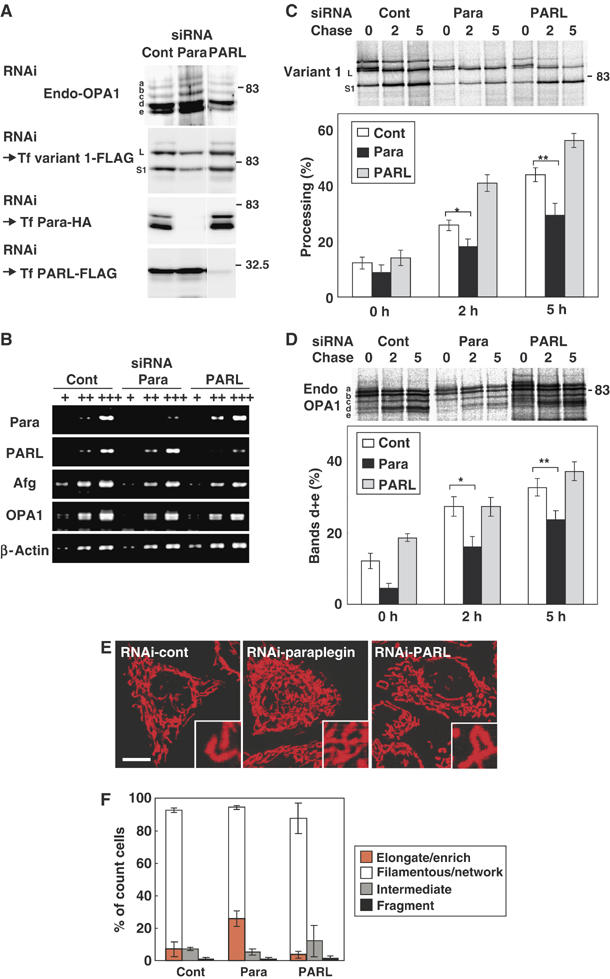
Knockdown of paraplegin and PARL by RNAi in HeLa cells. (A) HeLa cells were transfected with the siRNA duplex for GFP (control) or the indicated proteases three times. An aliquot of the cells was subjected to immunoblot analysis using anti-OPA1 antibodies (for endogenous OPA1). The other aliquots were transfected with OPA1-variant 1-FLAG, paraplegin-HA, or PARL-FLAG, then analyzed by immunoblotting using anti-FLAG (for OPA1 variant 1 and PARL) or anti-HA antibodies (for paraplegin). (B) The amount of mRNAs for paraplegin, PARL, Afg3L2, and OPA1 were examined by RT–PCR using 100- (+) or 10-fold (++) diluted templates. In all, 200–400 bp fragments were amplified. (C, D) The RNAi cells in (A) were labeled with 35S-Met/Cys mix for 30 min, then chased for the indicated time periods (h). The cell lysates were subjected to immunoprecipitation using anti-FLAG (for OPA1 variant 1; C) or anti-OPA1 (for endogenous OPA1; D). The immunoprecipitates were analyzed by SDS–PAGE and subsequent digital autoradiography as in Figure 1D and E. *P and **P<0.05 in (C). *P and **P<0.05 in (D). (E) HeLa cells subjected to RNAi as above were stained with MitoTracker and analyzed by fluorescence microscopy. Scale bar: 10 μm. (F) The number of cells with highly elongated and enriched mitochondria, filamentous-network, intermediate, or completely fragmented mitochondrial structures in (E) were counted and are shown as a percentage. More than 100 cells were counted for three different optical fields.
As for mitochondrial morphology, paraplegin knockdown induced a highly extended and connected tubulo-network mitochondrial morphology in approximately 30% of the cells, which is rarely observed in mock-transfected cells (Figure 8E, and Figure 8F, orange bars). Repression of PARL, on the other hand, did not affect mitochondrial morphology (Figure 8E and F).
Taken together, we concluded that m-AAA protease paraplegin stimulated OPA1 processing, which reflected in mitochondrial morphologic changes.
Discussion
We used two rat OPA1 splice variants, variants 1 and 7, and demonstrated that each variant is present as two (L and S1) and three (L, S1, and S2) proteolytically processed isoforms of distinct lengths, respectively. OPA1 proteins are synthesized as preproteins with a bipartite-type MTS at the N-termini. During mitochondrial import, MPP-induced cleavage occurred at residue 87–88 of the preproteins in the matrix to form the L-isoforms. Then, the second processing occurred at residues 194–195 to produce the S1 isoforms. In the case of variant 7, further proteolytic processing occurred at around 217–223, present in the 37 amino-acid residue segment derived from exon 5b to produce the S2 isoform. All these isoforms were present within the steady-state mitochondria, although their levels were distinct for each variant and probably depended on the condition of the cells.
In yeast, Mgm1 exists in two isoforms (l-Mgm1 and s-Mgm1) and both forms are required to rescue Δmgm1 cells. l-Mgm1 contains two putative TMs in the N-terminus (upstream TM1 and the more C-terminal TM2) and is anchored to the inner membrane through TM1. Processing of l-Mgm1 occurs within TM2 by Pcp1/Rbd1 when it is translocated into the inner membrane by the pulling force provided by ATP and the mitochondrial import motor complex (Herlan et al, 2004). We found that OPA1 processing was also significantly compromised in Pcp1-deficient yeast cells. Most importantly, however, OPA1 processing was almost completely blocked in m-AAA protease-deficient yeast cells, where Mgm1 processing was not affected, and its reintroduction efficiently restored the processing (Supplementary Figure S5B and C). It is known that yeast cytochrome c peroxidase is processed by novel two-step sequential mechanism involving Pcp1 and m-AAA protease (Esser et al, 2002). In case of OPA1 processing in yeast cells, Pcp1 and m-AAA protease might be involved independently but not sequentially, as the processing intermediate was not detected in Δpcp1 cells in contrast to the case for yeast cytochrome c peroxidase (Esser et al, 2002).
In marked contrast, in mammalian cells, repression or exogenous expression of mammalian rhomboid protease PARL had no effect on OPA1 processing. Confirming our results, OPA1 processing still proceeded in the PARL-deficient mouse cells (S Cipolat, T Rudka, L Scorrano and BD Strooper; personal communication). We showed in the present study that m-AAA-protease paraplegin is involved in OPA1 processing in mammalian cells. Exogenous expression of paraplegin efficiently stimulated OPA1 processing in a metal-binding site-dependent manner, although paraplegin knockdown only weakly affected the processing (Figure 8). This might be caused by incomplete repression of endogenous paraplegin. Or, another possibility might be that paraplegin cooperatively regulates OPA1 processing with PARL or other protease yet to be identified.
At present, we have no appropriate interpretation for the discrepancy in the processing reaction between yeast Mgm1 and mammalian OPA1. One explanation might be that PARL directly cleaved OPA1, and m-AAA protease is required for full activation of PARL in the inner membrane. If PARL is not rate limiting in the OPA1 processing reaction, expression of PARL should not stimulate the OPA1 processing. It is also possible that overexpression of paraplegin affects the mitochondrial morphology and OPA1 processing by indirect processing of unknown proteins, as m-AAA protease should have various substrates in inner membrane. It should be noted, however, that corepression of paraplegin and PARL, or paraplegin and AFG3L2 had no significant effect on OPA1 processing (data not shown). Clearly, further studies are needed to elucidate the exact mechanisms of OPA1 processing in the mitochondrial inner membrane.
What is the functional significance of the L- and S-isoforms in mammalian mitochondrial morphogenesis? Considering that dissipation of ΔΨ or proapoptotic stimuli induced the S-isoform formation concomitant with mitochondrial fragmentation, and exogenous expression of the L-isoform complemented the OPA1-depleted phenotype to induce extension of mitochondrial networks, it seemed that mitochondrial fusion depended on the L-isoform. Indeed, we could demonstrate using CCCP-treated cells that the L-isoform, but not S-isoform, exhibited mitochondrial fusion-stimulating activity. As confirming these results, mitochondria in the S-isoform-expressing cells exhibited small fragmented morphology (see AIF-230 in Supplementary Figure S4), resembling that of the fusion-defect mitochondria in OPA1-depleted cells (Chen et al, 2005). These results seemed to be consistent with the notion that mitochondrial fusion reaction proceeds in Δpcp1 yeast cells (Sesaki et al, 2003), indicating that the s-isoform of Mgm1 is not essential for mitochondrial fusion reaction.
What is the mechanism underlying activation of OPA1 processing? Because mitochondrial AAA-proteases can extract TM from the membrane bilayer and translocate the unfolded hydrophilic domain in the trans-side across the membrane (Arlt et al, 1996; Leonhard et al, 1999), it is conceivable that paraplegin translocates the OPA1 L-isoforms across the membrane into the matrix side unless the IMS domain is tightly folded or firmly associated with other components. Conditions that dissipate ΔΨ or induce apoptosis stimulate processing at the downstream of TM1 as if the membrane-anchoring function of TM1 is compromised (Figure 9). We therefore examined if translocation of the L-isoform could be impeded by increasing TM1 hydrophobicity (average hydrophobicity increased from 0.65 to 1.23–2.35). However, the processing of OPA1 was not inhibited by this manipulation (Figure 5). These properties are in striking contrast to those of Mgm1 processing by Pcp1/Rbd1 in yeast; conversion of l-Mgm1 to s-isoform is strongly inhibited by increasing hydrophobicity of the TM1 (Herlan et al, 2004). This difference might reflect an intrinsic difference in the substrate driving system between mammalian and yeast mitochondria. m-AAA protease seems to extract substrate proteins irrespective of the hydrophobicity of their TMs.
Figure 9.
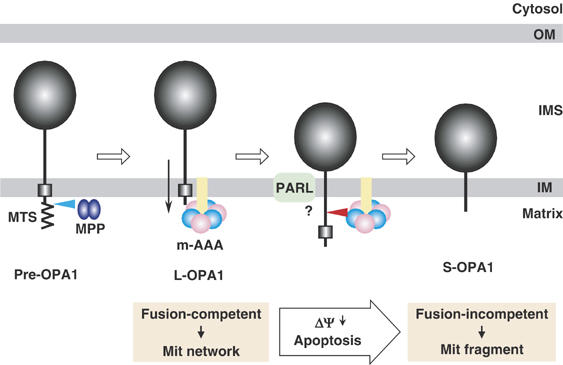
Model of proteolytic processing of OPA1. See Discussion for details.
In yeast, Mgm1 couples with Fzo1 of the outer membrane via Ugo1 to function in the mitochondrial fusion reaction (Sesaki and Jensen, 2004). Similarly, OPA1 requires Mfn1 of the outer membrane to promote mitochondrial fusion in mammalian cells (Cipolat et al, 2004), although the functional homologue of yeast Ugo1 is not known. If this is the case, ΔΨ might be required to maintain mechanical coupling of the fusion machinery of the outer membrane with the OPA1-containing inner membrane machinery, which prevents paraplegin-powered translocation and processing of OPA1 L-isoforms across the inner membrane. Once this mechanical coupling is broken by dissipation of ΔΨ, the L-isoforms would be translocated further into the matrix to form the S-isoforms.
Materials and methods
Cell culture and transfection
HeLa cells were cultured in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum under 5% CO2 at 37°C. DNA transfection was performed using lipofectamine (Invitrogen) as recommended by the manufacturer. SiRNAs were transfected to HeLa cells three times using oligofectamine (Invitrogen) as recommended by the manufacturer. The expression plasmids and the sequences for siRNA were described in supplemental section. The cells were fixed with 4% paraformaldehyde for fluorescent microscopy. Mitochondrial morphology was analyzed by 20 nM MitoTracker Red CMXRos (Molecular Probes) staining before fixation. All panels of microscopic images were 50 μm × 50 μm.
Determination of N-termini of OPA1 isoforms
Mitochondria isolated from HeLa cells expressing OPA1-FLAG were sonicated and centrifuged to prepare the membrane fraction. After solubilization with 1% Triton X-100, the clarified supernatant was subjected to immunoisolation using M2-agarose beads (Sigma Chemical Co.). Samples were eluted by FLAG peptide, separated by SDS–PAGE, and blotted onto a polyvinylidene difluoride membrane. OPA1 bands were cut out and subjected to N-terminal sequencing by Edman degradation. N-terminal deletion constructs of OPA1 variants were synthesized in vitro using PURESYSTEM (Post Genome Institute Co.).
Analyses
Preparation of total cell lysates and subcellular fractionation were performed as described previously (Ishihara et al, 2004). For pulse-chase experiment, HeLa cells were cultured in DMEM without methionine and cysteine for 1 h, then labeled with 35S-labeling mix (Express, Pharmacia) for 30 min (pulse). These cells were further cultured in DMEM with 10% FCS. The cells were recovered, washed, and examined for immunoprecipitation using anti-FLAG or anti-OPA1 antibodies and subsequent digital autoradiography as described previously (Ishihara et al, 2001). The band intensities were quantified by BAS 2500 (Fuji Film). Co-immunoprecipitation experiments using digitonin-solubilized mitochondria were performed as described (Ishihara et al, 2004).
Supplementary Material
Supplementary Information
Acknowledgments
We thank W Neupert and A Reichert for providing anti-Mgm1 antibodies, M van der Heide and M Veenhuis for TEV protease cDNA, M Hamasaki and Y Ohsumi for yeast protease mutants, and H Otera for HeLa cells stably expressing Bcl-XL. We also thank EI Rugarli, A Bernacchia, and K Sakaki for helpful discussion. This work was supported by grants from the Ministry of Education, Science, and Culture of Japan, from the Human Frontier Science Program, Core Research from Evolutional Science and Technology, and Takeda Science Foundation.
References
- Alexander C, Votruba M, Pesch UE, Thiselton DL, Mayer S, Moore A, Rodriguez M, Kellner U, Leo-Kottler B, Auburger G, Bhattacharya SS, Wissinger B (2000) OPA1, encoding a dynamin-related GTPase, is mutated in autosomal dominant optic atrophy linked to chromosome 3q28. Nat Genet 26: 211–215 [DOI] [PubMed] [Google Scholar]
- Arlt H, Tauer R, Feldmann H, Neupert W, Langer T (1996) The YTA10-12 complex, an AAA protease with chaperone-like activity in the inner membrane of mitochondria. Cell 85: 875–885 [DOI] [PubMed] [Google Scholar]
- Atorino L, Silvestri L, Koppen M, Cassina L, Ballabio A, Marconi R, Langer T, Casari G (2003) Loss of m-AAA protease in mitochondria causes complex I deficiency and increased sensitivity to oxidative stress in hereditary spastic paraplegia. J Cell Biol 163: 777–787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casari G, De Fusco M, Ciarmatori S, Zeviani M, Mora M, Fernandez P, De Michele G, Filla A, Cocozza S, Marconi R, Durr A, Fontaine B, Ballabio A (1998) Spastic paraplegia and OXPHOS impairment caused by mutations in paraplegin, a nuclear-encoded mitochondrial metalloprotease. Cell 93: 973–983 [DOI] [PubMed] [Google Scholar]
- Chen H, Chan DC (2004) Mitochondrial dynamics in mammals. Curr Top Dev Biol 59: 119–144 [DOI] [PubMed] [Google Scholar]
- Chen H, Chomyn A, Chan DC (2005) Disruption of fusion results in mitochondrial heterogeneity and dysfunction. J Biol Chem 280: 26185–26192 [DOI] [PubMed] [Google Scholar]
- Cipolat S, Martins de Brito O, Dal Zilio B, Scorrano L (2004) OPA1 requires mitofusin 1 to promote mitochondrial fusion. Proc Natl Acad Sci USA 101: 15927–15932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delettre C, Griffoin JM, Kaplan J, Dollfus H, Lorenz B, Faivre L, Lenaers G, Belenguer P, Hamel CP (2001) Mutation spectrum and splicing variants in the OPA1 gene. Hum Genet 109: 584–591 [DOI] [PubMed] [Google Scholar]
- Delettre C, Lenaers G, Griffoin JM, Gigarel N, Lorenzo C, Belenguer P, Pelloquin L, Grosgeorge J, Turc-Carel C, Perret E, Astarie-Dequeker C, Lasquellec L, Arnaud B, Ducommun B, Kaplan J, Hamel CP (2000) Nuclear gene OPA1, encoding a mitochondrial dynamin-related protein, is mutated in dominant optic atrophy. Nat Genet 26: 207–210 [DOI] [PubMed] [Google Scholar]
- Esser K, Tursun B, Ingenhoven M, Michaelis G, Pratje E (2002) A novel two-step mechanism for removal of a mitochondrial signal sequence involves the mAAA complex and the putative rhomboid protease Pcp1. J Mol Biol 323: 835–843 [DOI] [PubMed] [Google Scholar]
- Faber KN, Kram AM, Ehrmann M, Veenhuis M (2001) A novel method to determine the topology of peroxisomal membrane proteins in vivo using the tobacco etch virus protease. J Biol Chem 276: 36501–36507 [DOI] [PubMed] [Google Scholar]
- Ferreirinha F, Quattrini A, Pirozzi M, Valsecchi V, Dina G, Broccoli V, Auricchio A, Piemonte F, Tozzi G, Gaeta L, Casari G, Ballabio A, Rugarli EI (2004) Axonal degeneration in paraplegin-deficient mice is associated with abnormal mitochondria and impairment of axonal transport. J Clin Invest 113: 231–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank S, Gaume B, Bergmann-Leitner ES, Leitner WW, Robert EG, Catez F, Smith CL, Youle RJ (2001) The role of dynamin-related protein 1, a mediator of mitochondrial fission, in apoptosis. Dev Cell 1: 515–525 [DOI] [PubMed] [Google Scholar]
- Griparic L, van der Bliek AM (2001) The many shapes of mitochondrial membranes. Traffic 2: 235–244 [DOI] [PubMed] [Google Scholar]
- Griparic L, van der Wel NN, Orozco IJ, Peters PJ, van der Bliek AM (2004) Loss of the intermembrane space protein Mgm1/OPA1 induces swelling and localized constrictions along the lengths of mitochondria. J Biol Chem 279: 18792–18798 [DOI] [PubMed] [Google Scholar]
- Herlan M, Bornhovd C, Hell K, Neupert W, Reichert AS (2004) Alternative topogenesis of Mgm1 and mitochondrial morphology depend on ATP and a functional import motor. J Cell Biol 165: 167–173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herlan M, Vogel F, Bornhovd C, Neupert W, Reichert AS (2003) Processing of Mgm1 by the rhomboid-type protease Pcp1 is required for maintenance of mitochondrial morphology and of mitochondrial DNA. J Biol Chem 278: 27781–27788 [DOI] [PubMed] [Google Scholar]
- Ishihara N, Eura Y, Mihara K (2004) Mitofusin 1 and 2 play distinct roles in mitochondrial fusion reactions via GTPase activity. J Cell Sci 117: 6535–6546 [DOI] [PubMed] [Google Scholar]
- Ishihara N, Hamasaki M, Yokota S, Suzuki K, Kamada Y, Kihara A, Yoshimori T, Noda T, Ohsumi Y (2001) Autophagosome requires specific early Sec proteins for its formation and NSF/SNARE for vacuolar fusion. Mol Biol Cell 12: 3690–3702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishihara N, Jofuku A, Eura Y, Mihara K (2003) Regulation of mitochondrial morphology by membrane potential, and DRP1-dependent division and FZO1-dependent fusion reaction in mammalian cells. Biochem Biophys Res Commun 301: 891–898 [DOI] [PubMed] [Google Scholar]
- Jensen RE, Hobbs AE, Cerveny KL, Sesaki H (2000) Yeast mitochondrial dynamics: fusion, division, segregation, and shape. Microsc Res Tech 51: 573–583 [DOI] [PubMed] [Google Scholar]
- Karbowski M, Youle RJ (2003) Dynamics of mitochondrial morphology in healthy cells and during apoptosis. Cell Death Differ 10: 870–880 [DOI] [PubMed] [Google Scholar]
- Langer T, Kaser M, Klanner C, Leonhard K (2001) AAA proteases of mitochondria: quality control of membrane proteins and regulatory functions during mitochondrial biogenesis. Biochem Soc Trans 29: 431–436 [DOI] [PubMed] [Google Scholar]
- Lee YJ, Jeong SY, Karbowski M, Smith CL, Youle RJ (2004) Roles of the mammalian mitochondrial fission and fusion mediators Fis1, Drp1, and Opa1 in apoptosis. Mol Biol Cell 15: 5001–5011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Legros F, Lombes A, Frachon P, Rojo M (2002) Mitochondrial fusion in human cells is efficient, requires the inner membrane potential, and is mediated by mitofusins. Mol Biol Cell 13: 4343–4354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonhard K, Stiegler A, Neupert W, Langer T (1999) Chaperone-like activity of the AAA domain of the yeast Yme1 AAA protease. Nature 398: 348–351 [DOI] [PubMed] [Google Scholar]
- McQuibban GA, Saurya S, Freeman M (2003) Mitochondrial membrane remodelling regulated by a conserved rhomboid protease. Nature 423: 537–541 [DOI] [PubMed] [Google Scholar]
- Misaka T, Miyashita T, Kubo Y (2002) Primary structure of a dynamin-related mouse mitochondrial GTPase and its distribution in brain, subcellular localization, and effect on mitochondrial morphology. J Biol Chem 277: 15834–15842 [DOI] [PubMed] [Google Scholar]
- Mozdy AD, Shaw JM (2003) A fuzzy mitochondrial fusion apparatus comes into focus. Nat Rev Mol Cell Biol 4: 468–478 [DOI] [PubMed] [Google Scholar]
- Nolden M, Ehses S, Koppen M, Bernacchia A, Rugarli EI, Langer T (2005) The m-AAA protease defective in hereditary spastic paraplegia controls ribosome assembly in mitochondria. Cell 123: 277–289 [DOI] [PubMed] [Google Scholar]
- Olichon A, Baricault L, Gas N, Guillou E, Valette A, Belenguer P, Lenaers G (2003) Loss of OPA1 perturbates the mitochondrial inner membrane structure and integrity, leading to cytochrome c release and apoptosis. J Biol Chem 278: 7743–7746 [DOI] [PubMed] [Google Scholar]
- Otera H, Ohsakaya S, Nagaura Z, Ishihara N, Mihara K (2005) Export of mitochondrial AIF in response to proapoptotic stimuli depends on processing at the intermembrane space. EMBO J 24: 1375–1386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellegrini L, Passer BJ, Canelles M, Lefterov I, Ganjei JK, Fowlkes BJ, Koonin EV, D'Adamio L (2001) PAMP and PARL, two novel putative metalloproteases interacting with the COOH-terminus of Presenilin-1 and -2. J Alzheimers Dis 3: 181–190 [DOI] [PubMed] [Google Scholar]
- Satoh M, Hamamoto T, Seo N, Kagawa Y, Endo H (2003) Differential sublocalization of the dynamin-related protein OPA1 isoforms in mitochondria. Biochem Biophys Res Commun 300: 482–493 [DOI] [PubMed] [Google Scholar]
- Sesaki H, Jensen RE (2004) Ugo1p links the Fzo1p and Mgm1p GTPases for mitochondrial fusion. J Biol Chem 279: 28298–28303 [DOI] [PubMed] [Google Scholar]
- Sesaki H, Southard SM, Hobbs AE, Jensen RE (2003) Cells lacking Pcp1p/Ugo2p, a rhomboid-like protease required for Mgm1p processing, lose mtDNA and mitochondrial structure in a Dnm1p-dependent manner, but remain competent for mitochondrial fusion. Biochem Biophys Res Commun 308: 276–283 [DOI] [PubMed] [Google Scholar]
- Shaw JM, Nunnari J (2002) Mitochondrial dynamics and division in budding yeast. Trends Cell Biol 12: 178–184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shepard KA, Yaffe MP (1999) The yeast dynamin-like protein, Mgm1p, functions on the mitochondrial outer membrane to mediate mitochondrial inheritance. J Cell Biol 144: 711–720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugioka R, Shimizu S, Tsujimoto Y (2004) Fzo1, a protein involved in mitochondrial fusion, inhibits apoptosis. J Biol Chem 279: 52726–52734 [DOI] [PubMed] [Google Scholar]
- Taylor AB, Smith BS, Kitada S, Kojima K, Miyaura H, Otwinowski Z, Ito A, Deisenhofer J (2001) Crystal structures of mitochondrial processing peptidase reveal the mode for specific cleavage of import signal sequences. Structure (Camb) 9: 615–625 [DOI] [PubMed] [Google Scholar]
- Westermann B (2003) Mitochondrial membrane fusion. Biochim Biophys Acta 1641: 195–202 [DOI] [PubMed] [Google Scholar]
- Wong ED, Wagner JA, Scott SV, Okreglak V, Holewinske TJ, Cassidy-Stone A, Nunnari J (2003) The intramitochondrial dynamin-related GTPase, Mgm1p, is a component of a protein complex that mediates mitochondrial fusion. J Cell Biol 160: 303–311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yaffe MP (1999) The machinery of mitochondrial inheritance and behavior. Science 283: 1493–1497 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Information