

Abstract
The Ccr4-Not complex is a highly conserved regulator of mRNA metabolism. The transcription regulatory function of this complex in higher eukaryotes, however, is largely unexplored. Here we report that CNOT1, the large human subunit, represses the ligand-dependent transcriptional activation function of oestrogen receptor (ER) α. Promoter recruitment assays indicate that CNOT1 contains an intrinsic ability to mediate transcriptional repression. Furthermore, CNOT1 can interact with the ligand-binding domain of ERα in a hormone-dependent fashion and is recruited with other Ccr4-Not subunits to endogenous oestrogen-regulated promoters dependent on the presence of ligand. In addition, siRNA-mediated depletion of endogenous CNOT1 or other Ccr4-Not subunits in breast cancer cells results in deregulation of ERα target genes. Finally, CNOT1 interacts in a ligand-dependent manner with RXR and represses transcription mediated by several RXR heterodimers. These findings define a function for the human Ccr4-Not complex as a transcriptional repressor of nuclear receptor signalling that is relevant for the understanding of molecular pathways involved in cancer.
Keywords: Ccr4-Not, nuclear receptor, oestrogen, transcriptional repression
Introduction
Small lipophilic ligands such as steroid and thyroid hormones as well as fat-soluble vitamins exert their diverse biological effects through the action of nuclear receptors (NRs). The oestrogen receptors (ERs) α and β, for example, mediate the effects of oestrogens. These compounds, such as 17β-estradiol (E2), are steroid hormones that are important for the development and growth of the reproductive organs, bone density, and the function of the cardiovascular system (Nilsson et al, 2001; Hewitt et al, 2005). ERα can modulate gene expression directly as a ligand-activated transcription factor. Upon binding of E2 to the C-terminal ligand-binding domain (LBD), the receptor undergoes a conformational change resulting in dissociation from heat-shock chaperones, dimerisation, and binding to specific response elements. Alternatively, ligand-bound ERα can be recruited to target genes via other transcriptional activators, such as AP-1 (Nilsson et al, 2001; McKenna and O'Malley, 2002; Perissi and Rosenfeld, 2005). Upon binding of agonists, a rearrangement of α-helix 12 (H12) in the LBD creates an interaction surface for proteins containing an LXXLL motif (L leucine, X any amino acid; LXM) (Heery et al, 1997; Torchia et al, 1997; Nettles and Greene, 2005). Many positive cofactors containing this motif are identified and shown to be important for ERα activity. These coactivators function by recruitment of chromatin remodelling factors, including histone acetyltransferases and arginine methyltransferases (McKenna and O'Malley, 2002; Perissi and Rosenfeld, 2005). ERα can also directly bind to the TFTC complex, the mammalian counterpart of yeast coactivator SAGA, and Mediator, which acts as a bridge to the basal transcription machinery (Kang et al, 2002; Yanagisawa et al, 2002). This plethora of coactivators associates in a highly ordered and cyclical fashion with oestrogen-regulated promoters (Shang et al, 2000; Metivier et al, 2003). Surprisingly, although ligand-bound receptors are generally thought to activate transcription, several factors are described that associate with ERα and attenuate transcription in a ligand-dependent manner. These negative cofactors, which include NRIP1/RIP140 and LCoR, require an LXM for NR-interactions and contain potent repression domains when tethered to a heterologous DNA-binding domain (Cavailles et al, 1995; Fernandes et al, 2003; Christian et al, 2004). In addition, the human tumour antigen PRAME was recently identified as a ligand-dependent repressor of retinoic acid signalling (Epping et al, 2005).
The Ccr4-Not complex is a regulator of diverse functions in mRNA metabolism in yeast (Denis and Chen, 2003; Collart and Timmers, 2004). It was first discovered as a regulator of transcription by RNA polymerase II, but several subunits are also implicated in a cytoplasmic function in mRNA turnover apart from a nuclear function in transcription (Tucker et al, 2001; Yamashita et al, 2005).
Several human orthologues of subunits of this complex have been identified (Bogdan et al, 1998; Albert et al, 2000; Dupressoir et al, 2001). Analysis of the human subunits resulted in the identification of CNOT4 as a RING-type ubiquitin-protein ligase (Albert et al, 2002). In addition, a repression domain that requires histone deacetylase activity is present in the C-terminus of CNOT2 (Zwartjes et al, 2004). Interestingly, the CNOT7/CAF1 subunit interacts with Tob/Btg family members that are implicated with transcriptional regulation of NRs (Bogdan et al, 1998; Rouault et al, 1998; Ikematsu et al, 1999; Yoshida et al, 2001). Cnot7 knockout mice display testicular phenotypes and Cnot7 is proposed to be a positive coregulator of RXRβ (Berthet et al, 2004; Nakamura et al, 2004). In addition, it was put forward that the BTG1 and BTG2 proteins recruit CNOT7/CAF1 as a positive cofactor of ERα (Prevot et al, 2001).
In this report, we describe CNOT1, the human orthologue of the essential large subunit of the yeast Ccr4-Not complex. CNOT1 contains several LXMs, which are not present in any other subunit of the complex, and represses the ligand-dependent transcriptional activation function of ERα. Promoter recruitment assays indicate that CNOT1 contains an intrinsic ability to mediate transcriptional repression. Furthermore, CNOT1 can interact directly in a ligand-dependent fashion with ERα(LBD) and is recruited with other Ccr4-Not subunits to endogenous E2-regulated promoters in a ligand-dependent manner. In addition, depletion of endogenous CNOT1 or other components of the Ccr4-Not complex in breast cancer cells results in deregulation of endogenous ERα target genes. Finally, CNOT1 interacts in a ligand-dependent manner with RXR and represses transcription mediated by several RXR heterodimers.
Results
Identification of CNOT1, the human orthologue of yeast Not1p
We previously reported the identification of a partial cDNA encoding a human homologue of Not1p, the large subunit of the yeast Ccr4-Not complex (Albert et al, 2000). Subsequently, a cDNA was isolated exactly matching full-length cDNAs that became available during the course of this study. The gene is located on chromosome 16q21 and encompasses over 110 kb. The messenger is 8.4 kb long, in agreement with our previous analysis (Albert et al, 2000), and made up of 49 exons. Bioinformatic searches indicate that eukaryotic genomes harbour a single homologue of Not1p. The human homologue, CNOT1, encompasses 2376 amino acids and is 20% identical (32% similar) to its yeast counterpart, but regions with significantly higher homology to yeast Not1p were identified (Figure 1A). Antibodies were generated that recognized a high-molecular weight doublet of the expected size (predicted MW 267 kDa). The two isoforms may arise by alternative splicing (data not shown). To confirm that CNOT1 is a component of the human Ccr4-Not complex, we used these antibodies, and antibodies recognizing the CNOT2 and CNOT8/CALIF subunits, for immunoprecipitation analysis (Figure 1B). As expected, these proteins are part of a complex that also includes CNOT3 indicating that CNOT1 is the human orthologue of yeast Not1p. These immunocomplexes were specific, because they did not contain GAPDH and were not precipitated when a control rabbit antibody was used.
Figure 1.
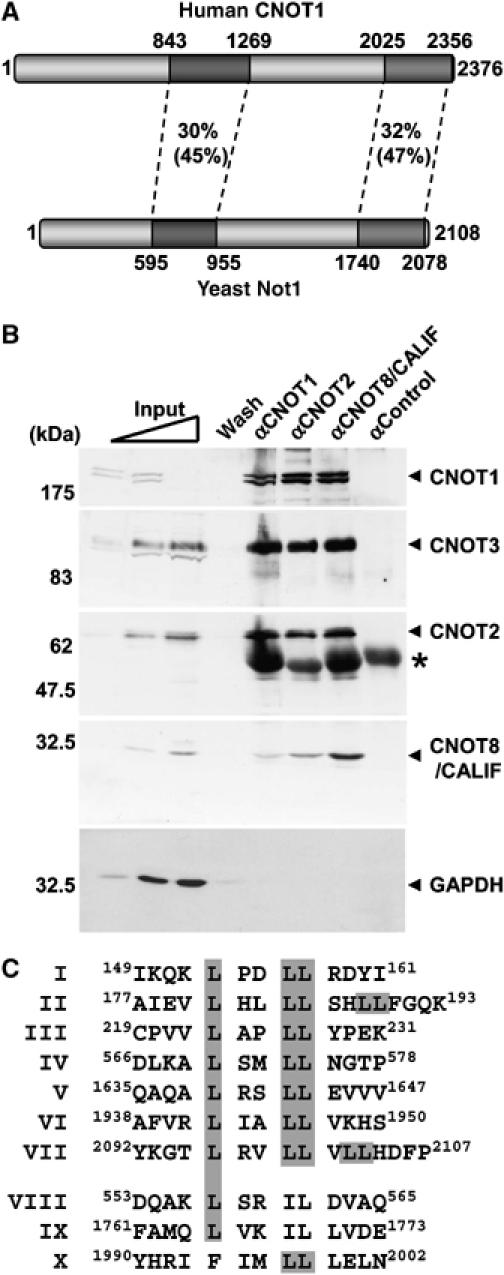
Schematic diagram of CNOT1 and interaction with components of the human Ccr4-Not complex. (A) Identification of CNOT1, the human orthologue of yeast Not1p. Regions of high identity (similarity) are shaded. Overall amino-acid identity (similarity) is 20% (32%). (B) Human CNOT1 interacts with several components of the human Ccr4-Not complex in a co-immunoprecipitation assay. Immunoprecipitation analysis was carried out using the indicated antibodies, and Ccr4-Not subunits and GAPDH were detected by immunoblotting. IgH bands are indicated by an asterisk. We consistently observed that the signal obtained using anti-CNOT1 antibodies was strongly reduced when high amounts of cell lysate were loaded. Note that all immunoblots were obtained using the same membrane. (C) LXXLL motifs (LXM) identified in CNOT1. Shaded in grey are leucine residues that are part of a consensus (I–VII) or variant LXM (VIII–X).
Several motif-searching routines did not identify conserved domains, but manual inspection of the amino-acid sequence identified nine consensus LXXLL motifs (LXMs) including two overlapping pairs and three variant LXM-like sequences (Figure 1C). These LXMs are not present in any other component of the human Ccr4-Not complex, and prompted us to investigate the function of CNOT1 in NR-mediated transcription.
CNOT1 represses ligand-dependent transcriptional activation by ER
Using the Cancer Profiling Database Oncomine version 3.0 (http://www.oncomine.org), we found that differential expression of several subunits of the Ccr4-Not complex (downregulation of CNOT2 and CNOT8/CALIF, and overexpression of CNOT9/RQCD1) correlates with ERα status in breast cancer. Therefore, we focused initially on ERα (NR3A1), a prototypical member of the NR superfamily to study the effect of CNOT1 on NR-mediated transcription. Expression of the Gal4 DNA-binding domain joined to the LBD of mouse ERα resulted in strong activation of reporter gene transcription in the presence of ligand. Remarkably, coexpression of full-length CNOT1 resulted in attenuation of ligand-dependent transcription by ERα(LBD) in a dose-dependent manner (Figure 2A). This effect was specific, as firefly luciferase activity was determined relative to Renilla luciferase driven by the constitutive CMV promoter, which was not affected. In addition, repression was not observed in the presence of the Gal4 DNA-binding domain alone, or when a Gal4 fusion with the E2F1 activation domain was used (data not shown).
Figure 2.
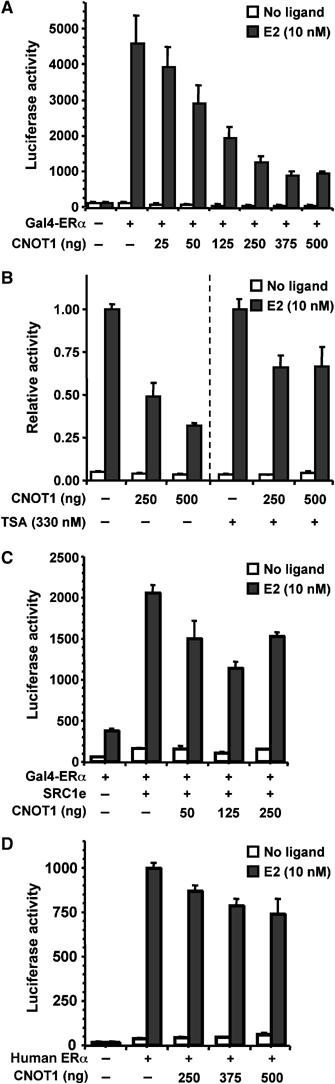
CNOT1 represses ligand-dependent transcriptional activation by ERα in COS-7 cells. (A) CNOT1 attenuates ligand-dependent transcription mediated by Gal4-mERα. Expression plasmid pSG424-mERα(AF2) was used encoding the Gal4 DNA-binding domain fused to the mouse ERα(LBD) domain (amino acids 313–599). A plasmid containing five GAL4-binding sites upstream of a TK minimal promoter and the firefly luciferase gene was used as a reporter. Luciferase activities were obtained for cells treated with vehicle or E2 (10 nM). (B) Repression by CNOT1 of Gal4-mERα ligand-dependent transcription is partially relieved by addition of the HDAC-inhibitor TSA. (C) Repression by CNOT1 of SRC1e-mediated coactivation of ligand-dependent transcription mediated by Gal4-mERα. (D) Attenuation of ligand-dependent transcription by full-length human ERα by CNOT1. A plasmid containing three EREs upstream of a TK minimal promoter and a firefly luciferase gene was used as a reporter. Error bars in panels (A–D) indicate standard deviations.
The repression of ligand-dependent transcription mediated by ERα(LBD) was partially relieved by addition of trichostatin A (TSA), an inhibitor of histone deacetylases (HDACs) (Figure 2B). This suggests that repression mediated by CNOT1 may function via both HDAC-dependent and -independent mechanisms. CNOT1 also inhibited ligand-dependent transcription when SRC1e, a member of the p160 family of coactivators (Kalkhoven et al, 1998), was coexpressed (Figure 2C). This is consistent with mechanisms in which SRC1e and CNOT1 compete for an overlapping binding site on ERα(LBD), or in which subsequent action of SRC1e and CNOT1 is required. Next, we tested whether CNOT1 could inhibit transcription by full-length human ERα. Indeed, ligand-dependent transcription was repressed in a dose-dependent manner (Figure 2D). The moderate repression observed in this case may be due to the influence of the N-terminal activation domain of ERα. Similar repression was observed with several other oestrogen-responsive element (ERE) reporters (data not shown). Taken together, these data show that expression of CNOT1 can result in repression of ligand-dependent transcription mediated by ERα.
Promoter recruitment of CNOT1 results in repression of transcription
To investigate whether CNOT1 has an intrinsic ability to repress transcription, we looked at the effect of promoter recruitment of CNOT1 on transcription. Full-length and two N-terminal and two C-terminal fragments of CNOT1 were fused to the Gal4 DNA-binding domain (Figure 3A). Expression of cDNAs encoding these proteins in COS-7 cells resulted in approximately equal levels of Gal4-CNOT1 fusion proteins, with slightly higher levels of the N-terminal fragments (Figure 3B). Expression of Gal4-CNOT1 in the presence of a luciferase reporter plasmid containing five GAL4 recognition sites resulted in a marked repression of transcription of both N-terminal and C-terminal fragments (Figure 3C and D). This effect was specific, because (i) the firefly luciferase was determined relative to Renilla luciferase driven by a constitutive promoter that was unaffected; (ii) no effect was observed when only the DNA-binding domain of Gal4 was expressed; and (iii) no or marginal repression was observed in the absence of GAL4 sites upstream of the reporter gene (data not shown). Addition of TSA partially relieved inhibition of transcription by Gal4-CNOT1 (Figure 3E and F). In agreement with the observations made using ligand-activated transcription by ERα (Figure 2), these data suggest that there are at least two independent repression domains located in the N- and C-terminal regions of CNOT1 that can both mediate repression via HDAC-dependent and -independent mechanisms.
Figure 3.
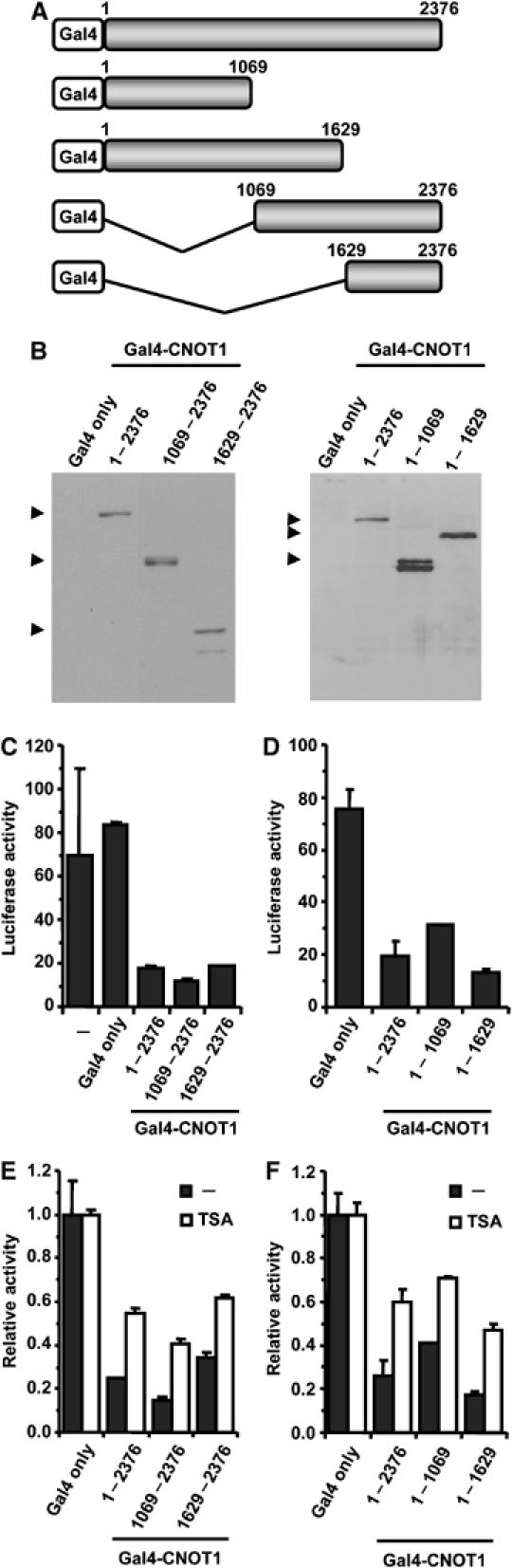
Repression of transcription by promoter recruitment of CNOT1. (A) Schematic representation of Gal4 (DNA-binding domain)-CNOT1 fusion proteins. (B) Immunoblot analysis of cells expressing the indicated Gal4-CNOT1 fusion. Fusion proteins were detected using antibodies recognizing the Gal4 DNA-binding domain. (C, D) Repression of transcription by promoter recruitment of CNOT1. Plasmids containing five GAL4-binding sites upstream of a TK minimal promoter and the firefly luciferase gene were used as a reporter. (E, F) Repression of transcription by promoter recruitment of (fragments of) CNOT1 is partially relieved by addition of TSA. Error bars in panels (C)–(F) indicate standard deviations.
Oestrogen-dependent interaction between CNOT1 and the ligand-binding domain of ER
Next, we addressed whether CNOT1 could interact directly in a ligand-dependent manner with ERα. To this end, we used a yeast two-hybrid assay that is sensitive and capable of identifying dynamic protein–protein interactions. A set of plasmids encoding LexA-CNOT1 fusion proteins (Figure 4A) were cotransformed in yeast with plasmids conditionally expressing the mouse ERα(LBD) fused to a B42 acidic activation domain. Appropriate expression of all fusion proteins was observed by immunoblotting (data not shown). As a measure for protein–protein interactions, β-galactosidase reporter activities were determined in lysates prepared from cells grown in the presence of E2 or vehicle. Strong β-galactosidase activities were measured with fragments that contain LXMs, but not in case of the fragment corresponding to amino acids 648–948 of CNOT1 that does not contain an LXM, or the LexA DNA-binding domain only (Figure 4B). The reporter activities are likely to represent bona fide protein–protein interactions, because no β-galactosidase activity was measured in the absence of ligand (Figure 4B). In addition, significantly reduced amounts of reporter activities were measured in a parallel experiment in which the B42 activation domain fused to a transcriptionally inactive ERα(LBD) was expressed (Figure 4C). This inactive ERα(LBD) contains the amino-acid substitutions M547A L548A in α-helix H12 that compromise the interaction with LXM motifs (Cavailles et al, 1994; Heery et al, 1997). Moreover, when antagonists of ERα were added to the growth medium, only background β-galactosidase activities were determined (Figure 4D). Finally, it was previously reported that extended LXMs (consisting of an LXM flanked on either side by four amino acids) display selectivity in NR interactions (Coulthard et al, 2003). Therefore, we fused the LXMs of CNOT1 and their flanking four amino acids (both N- and C-terminal; Figure 1C) to the LexA domain, and assayed for β-galactosidase activity in the presence of B42-ERα(LBD) and E2 or vehicle. Remarkably, the LXMs interact selectively with the ERα(LBD) in a strictly ligand-dependent manner. For example, LXM I (amino acids 149–161) and the variant LXXIL (LXM IX, amino acids 1761–1773) interacted very strongly in the presence of E2 with ERα(LBD), whereas LXM III (amino acids 219–231) or LXXIL (LXM VIII, amino acids 553–565) did not display any β-galactosidase activity (Figure 4E). Interestingly, the LexA-CNOT1 fusion proteins that interact with ERα(LBD) contain at least one interacting LXM (Figure 1C; Figure 4A and B). To understand whether these LXMs are also important for repression of ERα-mediated transcription by CNOT1, we constructed a series of mutants in which a single LXM was changed (LXXLL to LXXAA). However, these single LXM I-IX mutants, as well as a double LXM I/LXM IX mutant, did not relieve repression by CNOT1 (data not shown). Therefore, the interaction between CNOT1 and ERα(LBD) is complex and may be mediated by several LXMs in vivo. Co-immunoprecipitation between endogenous ERα and the Ccr4-Not complex was unsuccessful, indicating that the interaction might be highly dynamic (data not shown). Taken together, our data suggest that CNOT1 can interact with ERα in a strictly agonist-dependent manner via several of its (variant) LXM motifs.
Figure 4.
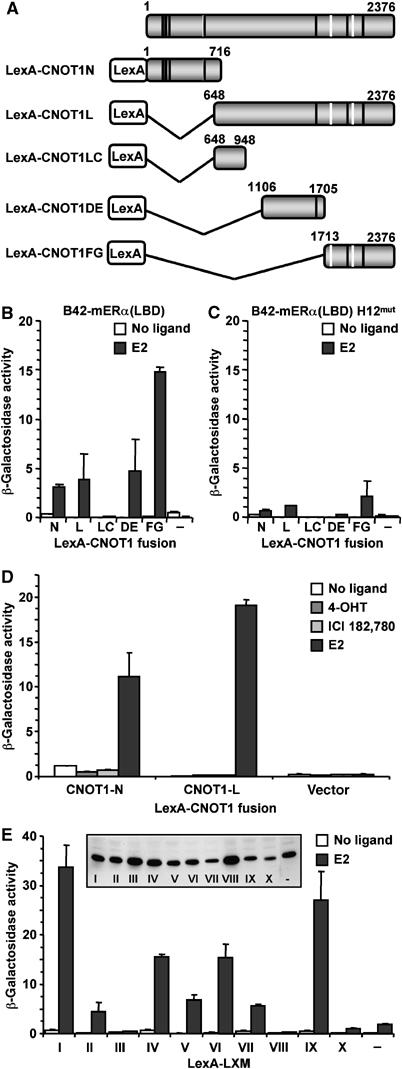
Ligand-dependent interaction between CNOT1 and the ligand-binding domain of ERα. (A) Schematic representation of full-length CNOT1 and LexA-CNOT1 constructs. Black bars indicate the presence of consensus LXXLL motifs (LXMs), whereas white bars indicate nonconsensus LXMs (see Figure 1C). (B) Human CNOT1 interacts in a hormone-dependent manner with the LBD of mouse (m) ERα in a yeast two-hybrid assay. Yeast cells were transformed with plasmids encoding LexA-CNOT1 fragments and B42-mERα and a β-galactosidase reporter plasmid containing eight upstream LexA-binding sites. Cells were grown in the presence of vehicle or E2 (1 μM), and β-galactosidase activities were determined. (C) Amino-acid substitutions M547A L548A in α-helix H12 of mERα(LBD) compromise binding to CNOT1. (D) Binding of CNOT1 to mERα(LBD) is not observed in the presence of antagonists 4-hydroxy tamoxifen (4-OHT) and ICI 182,780. (E) E2-dependent specific binding of CNOT1 LXMs to mERα(LBD). LexA-LXMs were expressed to similar levels in yeast as determined by immunoblotting using antibodies recognizing the LexA DNA-binding domain (inset). Error bars in panel (B–E) indicate errors of the mean.
Recruitment of Ccr4-Not subunits to oestrogen-responsive promoters in MCF-7 breast cancer cells
To investigate whether the observed functional and two-hybrid interactions are important for the regulation of endogenous genes, we employed MCF-7 cells. These cells are derived from an ERα-positive breast carcinoma and require E2 for efficient proliferation. To assess whether CNOT1 and other Ccr4-Not subunits are directly recruited to endogenous promoters that are regulated by E2, we carried out chromatin immunoprecipitation (ChIP) analyses. MCF-7 cells were treated with E2 or vehicle, and subjected to in vivo formaldehyde crosslinking. Subsequently, cell lysates were prepared and used for ChIP analysis using antibodies against ERα, RNA polymerase II (RNAPII), CNOT1, CNOT2, and CNOT8/CALIF. Purified DNA was analysed by quantitative PCR using oligonucleotides specific for five different E2-regulated promoters. Background signal obtained using control rabbit IgG was subtracted. In agreement with the observation that these promoters are known to be hormone-regulated (Frasor et al, 2003; Bourdeau et al, 2004), E2-induced binding of ERα and RNAPII was observed on each promoter (Figure 5A and B). In addition, E2-induced binding was also observed for all examined Ccr4-Not subunits CNOT1, CNOT2, and CNOT8/CALIF on each promoter analysed (Figure 5C–E). Taken together, these results indicate that repression of transcription by CNOT1 likely occurs via direct recruitment of the Ccr4-Not complex to hormone-regulated promoters.
Figure 5.
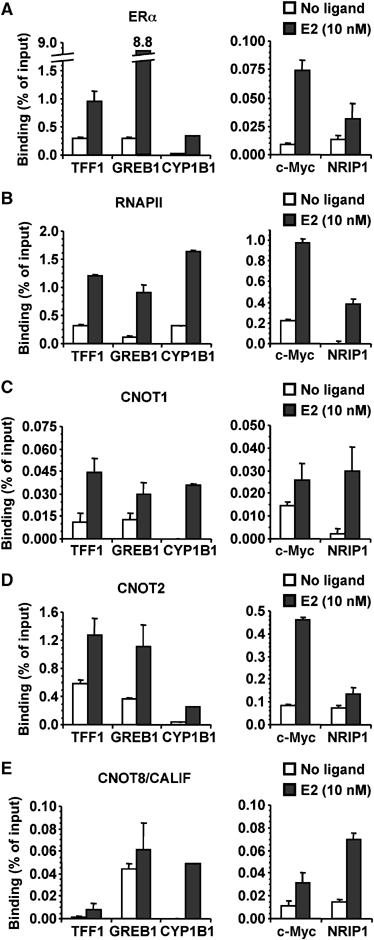
Recruitment of Ccr4-Not subunits to E2-responsive promoters in MCF-7 breast cancer cells. MCF-7 breast cancer cells were stimulated with E2 or vehicle and subjected to ChIP using antibodies recognizing (A) ERα, (B) RNA polymerase II, (C) CNOT1, (D) CNOT2, and (E) CNOT8/CALIF. Purified DNA was analysed by SYBR Green-based quantitative PCR using oligonucleotides amplifying promoter regions of the indicated genes. Background signals obtained using control IgG were subtracted from specific signals. Error bars indicate errors of the mean.
siRNA-mediated knockdown of CNOT1 results in induction of ERα target genes TFF1 and c-Myc
To analyse the effect of CNOT1 on endogenous ERα target genes in MCF-7 cells, we used RNAi to knockdown CNOT1 protein levels using two different siRNAs. These siRNAs specifically depleted CNOT1 protein levels, and did not affect protein levels of the Ccr4-Not components CNOT2 and CNOT3, or of ERα. α-Tubulin was used as a loading control (Figure 6A and data not shown). In addition, knockdown of CNOT1 did not influence a number of known cofactors of ligand-dependent transcription by ERα, including the histone acetyltransferases p300, CBP, and Tip60, the p160 family members SRC-1 and AIB1/SRC-3, and the ligand-dependent repressor RIP140 (Figure 6B). Subsequently, we isolated total RNA from cells treated with these siRNA molecules. Reverse-transcriptase quantitative PCR (RT–PCR) was carried out using intron-spanning primers specific for the well-characterised oestrogen target gene TFF1 (pS2) using GAPDH as a reference gene. Whereas little induction upon E2 treatment was observed in mock-treated cells, or cells treated with control siRNA, significantly higher induction was seen when cells were depleted for CNOT1 (Figure 6C). Similar observations were made when induction of c-Myc was analysed, which requires an AP-1 site to recruit ERα (Figure 6D). These results support the notion that CNOT1 is a transcriptional repressor of endogenous ERα target genes.
Figure 6.
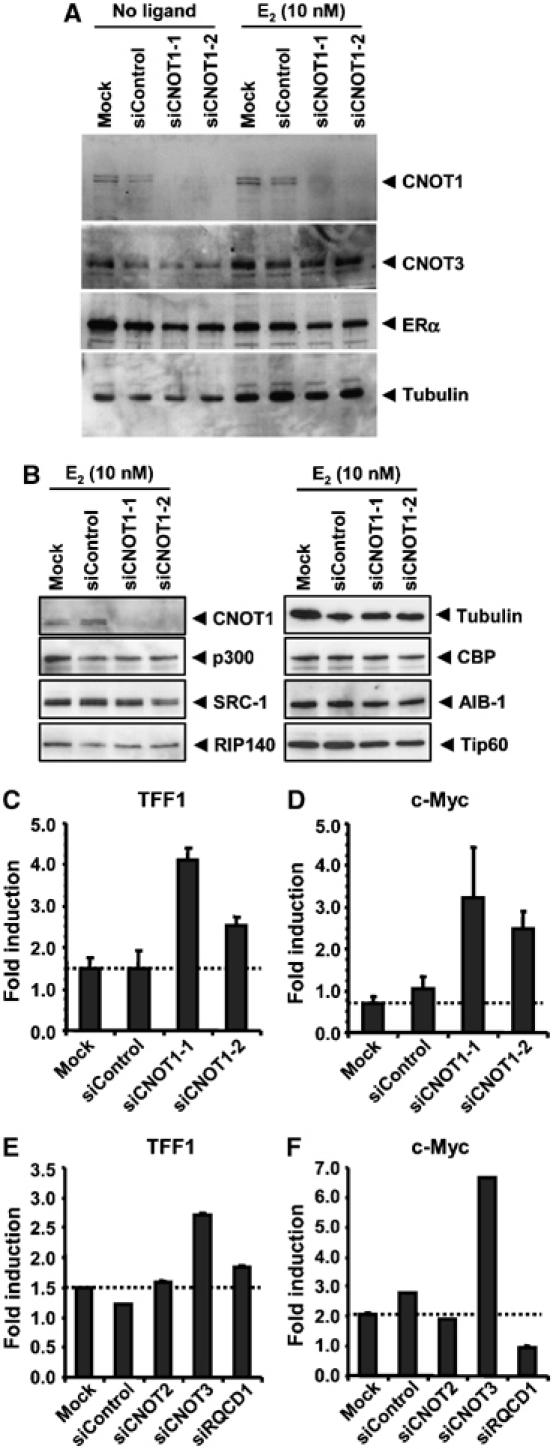
siRNA-mediated knockdown of CNOT1 results in increased induction of ERα target genes TTF1 and c-Myc. (A) Knockdown of CNOT1 by siRNA. Two different double-stranded RNA oligonucleotides were used. A nontargeting control was also included. Protein lysates were prepared 72 h after transfection and analysed by immunoblotting using the indicated antibodies. (B) Knockdown of CNOT1 does not affect protein levels of known ligand-dependent cofactors of ERα. (C) Increased TFF1 (pS2) mRNA induction in cells treated with siRNAs directed against CNOT1. mRNA levels were determined in MCF-7 breast cancer cells before and after treatment with vehicle or E2 by RT–PCR using GAPDH as a reference. Subsequently, the fold induction was calculated. (D) Increased c-Myc mRNA induction in cells treated with siRNAs directed against CNOT1. Levels of c-Myc mRNA were determined using GAPDH as a reference. (E) E2-mediated induction of TFF1 mRNA is increased in cells with depleted CNOT3 mRNA. MCF-7 cells were transfected with siRNAs against the Ccr4-Not subunits CNOT2, CNOT3, and CNOT9/RQCD1. (F) Differential effects on c-Myc mRNA induction caused by depletion of the Ccr4-Not subunits CNOT3 and CNOT9/RQCD1. Errors of the mean are indicated in panels (C)–(F).
To examine whether other Ccr4-Not components are also involved in repression of ligand-mediated transcription by ERα, we used siRNAs against several other Ccr4-Not components, in particular against CNOT2, CNOT3, and CNOT9/RQCD1. The siRNAs used depleted protein levels in the case of CNOT2 and CNOT3, whereas all siRNAs tested typically depleted 80–90% of the mRNA levels of the respective genes (data not shown). Interestingly, whereas CNOT2 and CNOT9/RQCD1 siRNAs had only marginal effects on the induction of the TFF1 (pS2) gene, depletion of CNOT3 significantly increased the induction of this gene in response to E2 (Figure 6E). In agreement with these observations, depletion of CNOT3 strongly increased the induction by E2 of the c-Myc gene, whereas the knockdown of CNOT2 had no effect. Intriguingly, the use of siRNAs against CNOT9/RQCD1 abolished E2-dependent induction of the c-Myc gene (Figure 6F). Together, these results indicate that the repressive function of the Ccr4-Not complex on E2-dependent transcription relies on at least two subunits, CNOT1 and CNOT3. Furthermore, it suggests that—in addition—the complex may also exert positive effects on oestrogen-mediated transcription via the CNOT9/RQCD1 subunit.
Interaction and repression of RXR-mediated transcription
To study whether the Ccr4-Not complex also interacts with other members of the NR superfamily, we tested whether CNOT1 displayed ligand-dependent interactions with RARα, TRβ, PPARγ, and RXRα. Interestingly, CNOT1 did not display such interactions with RARα, TRβ, and PPARγ (Figure 7A–C), but did interact in a ligand-dependent fashion with RXRα (Figure 7D). Surprisingly, a ligand-independent interaction was observed between the N-terminal region of CNOT1 and full-length TRβ (Figure 7B). Intriguingly, this may be mediated by the N-terminal activation region of TRβ. Regardless of this, however, the above results indicate specificity in the ligand-dependent interactions between CNOT1 and NRs.
Figure 7.
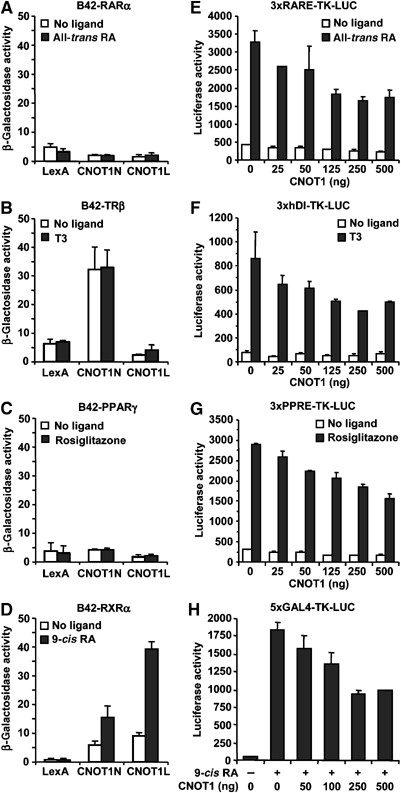
Interaction and repression of RXR-mediated transcription. (A–C) Human CNOT1 does not interact in a ligand-dependent manner with RARα (A), TRβ (B), or PPARγ (C) in a yeast two-hybrid assay. Yeast cells were transformed with plasmids encoding LexA-CNOT1 fragments, the indicated B42-NR construct, and a β-galactosidase reporter plasmid containing eight upstream LexA-binding sites. Cells were grown in the presence of vehicle or ligand (1 μM) as indicated. (D) Ligand-dependent interaction between CNOT1 and RXRα in a yeast two-hybrid assay. Cells were grown in the presence of vehicle of 9-cis retinoic acid. (E–G) Repression by CNOT1 of ligand-dependent transcription mediated by RA/RXR (E), TRβ/RXR (F), and PPARγ/RXR (G) heterodimers. COS-7 cells were transfected with the indicated amount of a CNOT1 expression plasmid, a plasmid expressing the TRβ or PPARγ, the indicated firefly luciferase reporter plasmid, and grown in the presence of the indicated ligand (1 μM). (H) Repression of ligand-dependent transcription mediated by Gal4-RXRγ by overexpression of CNOT1. A plasmid containing five GAL4-binding sites upstream of a TK minimal promoter and the firefly luciferase gene was used as a reporter. COS-7 cells were grown in the absence or presence of 9-cis retinoic acid (1 μM) as indicated.
Subsequently, we tested whether CNOT1 affects the transcriptional transactivation activity of these NRs by transfecting COS-7 cells with reporter constructs carrying response elements for RAR (RARE), TRβ (hDI), and PPARγ (PPRE). Remarkably, although no ligand-dependent interaction with the corresponding NRs was observed, CNOT1 repressed transcription of all reporters tested (Figure 7E–G). However, these NRs tested are functional as heterodimers with RXR. Therefore, the repressive effect by CNOT1 might be mediated via the interaction with the RXR partner. To directly test whether CNOT1 can repress the ligand-dependent activation function of RXR, we used a fusion of the Gal4 DNA-binding domain with RXR. Indeed, ligand-mediated transcription of the Gal4-RXRγ hybrid receptor was repressed by CNOT1 in a dose-dependent fashion (Figure 7H). Similar results were obtained using Gal4-RXRβ (data not shown). Together, these results indicate that CNOT1 can specifically interact with some members of the NR family, including ERα and RXR, and repress their ligand-dependent transcription activity.
Discussion
The regulation of specific transcriptional and cellular programmes by the mammalian Ccr4-Not complex is poorly understood. Cnot7 knockout mice display a defect in spermatogenesis (Berthet et al, 2004; Nakamura et al, 2004). In addition, interactions with human Tob/Btg family members point to a role in transcriptional regulation, but physiological target genes remained obscure. Here we show that human Ccr4-Not acts as a ligand-dependent repressor of ERα-mediated transcription of both ectopic and endogenous genes. These effects are likely direct, because CNOT1 can interact in an agonist-dependent manner with the LBD of ERα, and several Ccr4-Not components are recruited in an E2-dependent manner to known ERα target genes. In addition, CNOT1 interacts in a ligand-dependent manner with RXR and can repress the transcriptional activity of RXR/NR heterodimers.
Regulation of ligand-bound ER by the Ccr4-Not complex
The function of the Ccr4-Not complex in ERα-mediated transcription is clearly different from the well-characterised NCoR and SMRT repressor complexes whose interaction with members of the NR family is lost upon binding of ligand (McKenna and O'Malley, 2002; Perissi and Rosenfeld, 2005). In contrast, the Ccr4-Not complex resembles repressors of liganded NRs, such as LCoR, NRIP1/RIP140, and PRAME (Cavailles et al, 1995; Fernandes et al, 2003; Christian et al, 2004; Epping et al, 2005). How these factors contribute to ligand-regulated transcription has not been clarified yet. Although some genes are clearly downregulated upon exposure of cells to E2, ligand-dependent negative cofactors appear to have a more general role during activation. A possible explanation may relate to the highly dynamic behaviour of transcription factors. Fluorescence microscopy of living cells shows that the glucocorticoid receptor rapidly exchanges on promoter-binding sites in the presence of ligand (McNally et al, 2000). In addition, analysis of protein–DNA complexes indicates that activation and recruitment of cofactors by ERα is a cyclic process that involves the ordered assembly and disassembly of a plethora of high-molecular weight factors (Shang et al, 2000; Metivier et al, 2003). It can be envisaged that negative cofactors may play a role in the exchange of positive cofactors.
An alternative, not mutually exclusive mechanism, may be that transcriptional activation by ERα is regulated by a balance of positive and negative cofactors. Thus, the relative contribution of coactivators and corepressors may determine the induction of a given E2-target gene resulting in differential gene expression. This may be analogous to observations made in yeast. A temperature-sensitive allele of SRB4, an essential component of the Mediator complex associated with activation, can be suppressed by inactivating mutations in several Ccr4-Not components, including the large subunit NOT1 (Lee et al, 1998). Thus, equilibrium between Mediator and the Ccr4-Not complex is required for yeast viability. Interestingly, mammalian Mediator is implicated with a positive function in ERα-mediated transcription (Kang et al, 2002), whereas we show in this report a negative function for the CNOT1 subunit of the Ccr4-Not complex. This model would also be compatible with a role for negative cofactors in the prevention of oestrogen-induced transcription of certain genes, which may contribute to tissue-specific gene expression.
What is the mechanism used by the Ccr4-Not complex to repress ERα-mediated transcription? The observation that the repressive effects of CNOT1 are partially relieved by TSA suggests that histone deacetylases can be recruited. In addition, the CNOT4 RING-type ubiquitin ligase that interacts with the ubiquitin-conjugating enzyme UbcH5b (Albert et al, 2002; Dominguez et al, 2004) may also contribute to ERα-mediated transcription. Overexpression of CNOT1 did not affect protein levels of Gal4-ERα(LBD) in transient transfection assays (data not shown). However, as no target of the CNOT4/UbcH5b enzyme pair has been identified yet, it will be of interest whether ubiquitylation of liganded ERα and/or of other cofactors by CNOT4 can influence transcriptional activation of E2 target genes. Interestingly, the Ccr4-Not complex has a modular architecture in that subunits can mediate different functions of the complex. For example, CNOT3 is required for repression of ERα, whereas CNOT2 is expendable. In addition, the CNOT9/RQCD1 subunit appears to act gene-specifically, as it is required for oestrogen-dependent activation of the c-Myc target gene, but not of TFF1.
Is repression of ERα by the Ccr4-Not complex mediated via mRNA turnover?
The Ccr4-Caf1 subunits of the Ccr4-Not complex are part of a physically and functionally separable submodule of the complex (Bai et al, 1999) and are implicated with a cytoplasmic function in mRNA decay (Tucker et al, 2001; Yamashita et al, 2005). This raises the possibility that the repressive effects of CNOT1 on transcription are due to defects in mRNA turnover. However, several lines of evidence contradict this notion. The observations that repression of reporter activity depended specifically on promoter recruitment of the ligand-binding domain of ERα and could be partially relieved by the addition of the HDAC inhibitor TSA together suggest that CNOT1 acts at the stage of transcription initiation (Figure 2). In agreement with these observations, promoter recruitment of CNOT1 (Figure 3) and of CNOT2 and CNOT9/RQCD1 also inhibited reporter activity (Zwartjes et al, 2004). Finally, several Ccr4-Not components were directly recruited to E2-responsive promoters in the presence of ligand (Figure 5). These observations are difficult to reconcile with an exclusive function for the Ccr4-Not complex in cytoplasmic mRNA decay and indicate that the Ccr4-Not complex can—in addition—act as a repressor of transcription.
Repression of RXR-mediated transactivation by CNOT1
The observations that CNOT1 interacts in a ligand-dependent fashion with RXR and represses the transactivation function of RXR and several heterodimers of RXR suggest that the Ccr4-Not complex may affect transcription mediated by a large group of NRs. Interestingly, mouse Cnot7 was shown to interact with the N-terminal activation domain of RXR that does not require the presence of ligand (Berthet et al, 2004; Nakamura et al, 2004). In agreement with the observation that several subunits affect transcription of ERα target genes in different ways, this is a further indication that subunits of the Ccr4-Not complex differentially contribute to NR-mediated transcription.
Anti-oestrogenic compounds or selective ER modulators (SERMs) are widely used in the treatment and prevention of breast cancer and severe postmenopausal symptoms. In addition, selective RXR ligands are tested for their potential as anticancer drugs (Gronemeyer et al, 2004). The findings described in this report define a function for the human Ccr4-Not complex as a transcriptional repressor of oestrogen and RXR signalling that is relevant for the understanding of molecular pathways involved in cancer and the pharmacology of selective ER and RXR ligands.
Materials and methods
Plasmids
pCMV-FLAG-CNOT1 contains the complete open-reading frame (amino acids 1–2376) of human CNOT1 fused to an N-terminal FLAG epitope tag in pcDNA3.1 (Invitrogen). The pGAL4-CNOT1 plasmids are based on pCMV-GAL4 (Zwartjes et al, 2004). pLexA-CNOT1 derivatives are based on plasmid pEG202 (Albert et al, 2000). The pLexA-LXM plasmids were constructed by ligation of synthetic oligonucleotides into the BamHI and XhoI sites of pEG202-NLS. Plasmids pLexA-RARα, -RXRα, -TRβ, and -PPARγ were derived from the corresponding pASV3-mod plasmids (Coulthard et al, 2003). Plasmid pFLAG-hERα was a kind gift of Dr WL Kraus (Cornell University, USA). Plasmids pB42-mERα(AF2) and pB42-mERα(AF2mut) were generated by transferring the EcoRI fragments from pSG424-mERα(AF2) and pSG424-mERα(AF2mut), respectively, to plasmid pJG4-5 (Cavailles et al, 1994). The expression plasmid pSG5-SRC1e, and the reporter plasmids p(ERE)3-TK-Luc, p(GAL4)5-TK-Luc, and pCMV-Renilla were described previously (Kalkhoven et al, 1998; Zwartjes et al, 2004).
Antibodies and protein immunoprecipitation analysis
Rabbit polyclonal antibodies directed against CNOT3 and CNOT8/CALIF were described previously (Albert et al, 2000; Aoki et al, 2002). Fusion proteins with Gal4 (amino acids 1–147) and LexA were detected using mouse monoclonal antibodies RK5C1 and 2-12, respectively (Santa Cruz), recognizing the DNA-binding domains. Rabbit polyclonal antibodies recognizing ERα were described previously (Acevedo et al, 2004) or are available from commercial sources (HC-20, Santa Cruz). Antibodies against a C-terminal fragment of CNOT1 (a kind gift of Dr MA Collart, CMU Geneva, CH) and full-length CNOT2 were developed by immunizing rabbits with His-tagged proteins expressed in E. coli. Antibodies recognizing RNA polymerase II, SRC-1, GAPDH, and α-tubulin were mouse monoclonal 8WG16, clone 1135 (Upstate), MAB374 (Chemicon), and DM1A (Calbiochem), respectively. Antibodies C-20, N-17, H-300, A-22, and N-15 (Santa Cruz) recognised AIB-1, Tip60, RIP-140, CBP, and p300, respectively.
For immunoprecipitation analysis of the Ccr4-Not complex, human embryonic kidney 293T cells (approximately 0.4 ml cell pellet volume) were lysed in 4.0 ml Lysis buffer (50 mM Tris–HCl pH 8.0, 150 mM NaCl, 5 mM MgCl2, 0.5 mM EDTA, 0.2% Nonidet P-40, 5% (v/v) glycerol, 0.5 mM dithiotreitol, and protease inhibitors). The cell suspension was left on ice for 5–10 min, passed 10 times through a 27-gauge needle, and spun for 10 min in a micro centrifuge (full speed, 4°C). Each immunoprecipitate contained 300 μl soluble lysate, 25 μl protein A-agarose, and 20 μl serum and was incubated o/n at 4°C. After washing three times with 500 μl Lysis buffer, bound proteins were analysed by 8% SDS–PAGE and immunoblotting.
Yeast two-hybrid analysis
Yeast two-hybrid analysis was carried out as described with minor modifications (Winkler et al, 2004). Yeast transformants were directly used to inoculate 4 ml synthetic complete medium containing sucrose and galactose as carbon sources and 1 μM ligand or vehicle. A portion of this culture (1 ml) was used to prepare protein lysates, whereas the remaining part was used for β-galactosidase reporter assays. Ligands were dissolved in ethanol (17β-estradiol and 4-hydroxy tamoxifen; Sigma), DMSO (ICI 182,780; Tocris; all-trans and 9-cis retinoic acid; Sigma; Rosiglitazone) or 1 M NaOH (T3).
DNA transfection
COS-7 cells were routinely maintained in DMEM containing 10% foetal bovine serum (FBS) supplemented with glutamine and antibiotics. For reporter assays, DNA transfections were carried out in 12-well plates. Approximately 10 000 cells were plated in each well. Medium was replaced with DMEM without phenol-red containing 5% dextran-coated charcoal stripped (DCC) FBS 24 h before transfection. Cells were transfected using Fugene 6 transfection reagent as suggested by the manufacturer (Roche) using a total amount of 750 ng DNA per well (5 ng ERα, 10 ng TRβ-, 10 ng PPARγ-, or 2.5 ng Gal4-RXRγ expression plasmid; 2.5 ng pCMV-Renilla; 200 ng luciferase reporter; and pCMV-FLAG-CNOT1 expression plasmid as indicated made up to a total of 550 ng using empty vector). Where indicated, 150 ng pSG5-SRC1e or empty vector was used. At 24 h after transfection, medium was replaced with DMEM without phenol-red containing 5% DCC-FBS and 10 nM 17β-estradiol (E2) or vehicle (ethanol). Cell lysates were prepared 48 h after transfection and reporter activities were measured using the Dual Luciferase Assay (Promega) as described by the manufacturer. Where indicated, TSA was added during the final 24 h before cell lysis.
For promoter recruitment assays, COS-7 cells were plated in 12-well plates (10 000 cells/well). The next day, a mixture of 2.5 ng pCMV-Renilla, 200 ng luciferase reporter, 50 ng pCMV-FLAG, and 500 ng pCMV-Gal4 expression plasmids were transfected using Fugene 6 transfection reagent. Cell lysates were prepared 48 h after transfection and reporter activities were determined as described above.
Chromatin immunoprecipitation
MCF-7 NKI cells were routinely maintained in DMEM containing 10% FBS. ChIP was carried out as described with minor modifications (Acevedo et al, 2004). Before ChIP analysis, cells were grown for 2 days in DMEM without phenol-red containing 5% DCC-FBS. After addition of 10−8 M E2 or vehicle for 3 h, approximately 1–2 × 107 cells (for six ChIP assays) were crosslinked with 1% formaldehyde (in PBS) for 10 min at 37°C. Subsequently, cells were rinsed twice with ice-cold PBS, harvested using a rubber policeman, and transferred to two eppendorf tubes. After resuspension in Lysis buffer (each tube 350 μl; 50 mM Tris–HCl pH 8.0, 1% SDS, 10 mM EDTA, 1 mM dithiotreitol, and protease inhibitors), sonication was carried out for 3 × 30 s (on/off cycles, medium power) using a Diagenode BioRuptor. Subsequently, soluble chromatin lysates were diluted, precipitated, washed, incubated at 65°C to reverse crosslinking, and treated with proteinase K. DNA was purified using Qiaquick PCR purification spin columns (Qiagen) and analysed by SYBR Green-based quantitative PCR using a Chromo4 real-time fluorescence detector and the MJ Opticon Monitor 3.00 package (MJ Research/BioRad). Oligonucleotide sequences amplifying fragments of the TFF1 (pS2), c-Myc, GREB1, CYP1B1, and NRIP1 promoters were described elsewhere (Acevedo et al, 2004; Labhart et al, 2005).
siRNA transfection and RT–PCR
siRNAs directed against CNOT1, CNOT2, CNOT3, CNOT9/RQCD1, and a nontargeting control (siControl #1) were obtained from Dharmacon. Approximately 6 × 105 MCF-7 cells were resuspended in EPB buffer (2 mM HEPES–NaOH pH 7.2, 15 mM Na-phosphate pH 7.2, 250 mM mannitol, 1 mM MgCl2), mixed with 2 μg siRNA, transferred to an electroporation cuvette (0.1 cm gap, BioRad) and subjected to 16 pulses (1.5 ms, 1.5 s intervals) at 140 V using a Gene Pulser Xcell eukaryotic system (BioRad). Cells were resuspended in 4 ml medium, and plated in four wells of a 12-well plate. The next day, medium was replaced with DMEM without phenol-red containing 5% DCC-FBS. At 3 days post transfection, cells were stimulated with 10−8 M E2 or vehicle for 3 h. Total RNA was isolated using RNeasy mini columns (Qiagen) and converted into cDNA using Superscript II and oligo(dT) according to the manusfacturer's instructions (Invitrogen). Oligonucleotide sequences amplifying fragments of the TFF1 (pS2) and c-Myc cDNAs were described before (Acevedo et al, 2004). For protein analysis, cell pellets were collected 72 h post transfection, resuspended in 150 μl SDS-sample buffer and subjected to analysis by 8% SDS–PAGE.
Acknowledgments
We thank Dr MA Collart for generous gift of antibodies; Drs DM Heery, J Wong, G Folkers, and VKK Chatterjee for gift of plasmids; and Dr WL Kraus for advice on ChIP and quantitative PCR and kind gift of reagents. FM Jacobs, M Gijzen, I Gerritsen, and N Hamers are acknowledged for their contributions. Jimin Gao and Connie M Corcoran are thanked for technical assistance. GSW and HThMT were supported by grants from the Netherlands Organisation for Scientific Research (NWO-MW Pionier 900-98-142) and the European Union (6FP LSHG-CT-2004-502950); VJB by grant NCI R01-CA71540-06; and EK by a fellowship from the Royal Netherlands Academy of Arts and Sciences.
References
- Acevedo ML, Lee KC, Stender JD, Katzenellenbogen BS, Kraus WL (2004) Selective recognition of distinct classes of coactivators by a ligand-inducible activation domain. Mol Cell 13: 725–738 [DOI] [PubMed] [Google Scholar]
- Albert TK, Hanzawa H, Legtenberg YI, de Ruwe MJ, van den Heuvel FA, Collart MA, Boelens R, Timmers HT (2002) Identification of a ubiquitin-protein ligase subunit within the CCR4-NOT transcription repressor complex. EMBO J 21: 355–364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albert TK, Lemaire M, van Berkum NL, Gentz R, Collart MA, Timmers HT (2000) Isolation and characterization of human orthologs of yeast CCR4-NOT complex subunits. Nucleic Acids Res 28: 809–817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aoki T, Okada N, Wakamatsu T, Tamura TA (2002) TBP-interacting protein 120B, which is induced in relation to myogenesis, binds to NOT3. Biochem Biophys Res Commun 296: 1097–1103 [DOI] [PubMed] [Google Scholar]
- Bai Y, Salvadore C, Chiang YC, Collart MA, Liu HY, Denis CL (1999) The CCR4 and CAF1 proteins of the CCR4-NOT complex are physically and functionally separated from NOT2, NOT4, and NOT5. Mol Cell Biol 19: 6642–6651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berthet C, Morera AM, Asensio MJ, Chauvin MA, Morel AP, Dijoud F, Magaud JP, Durand P, Rouault JP (2004) CCR4-associated factor CAF1 is an essential factor for spermatogenesis. Mol Cell Biol 24: 5808–5820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogdan JA, Adams-Burton C, Pedicord DL, Sukovich DA, Benfield PA, Corjay MH, Stoltenborg JK, Dicker IB (1998) Human carbon catabolite repressor protein (CCR4)-associative factor 1: cloning, expression and characterization of its interaction with the B-cell translocation protein BTG1. Biochem J 336 (Part 2): 471–481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourdeau V, Deschenes J, Metivier R, Nagai Y, Nguyen D, Bretschneider N, Gannon F, White JH, Mader S (2004) Genome-wide identification of high-affinity estrogen response elements in human and mouse. Mol Endocrinol 18: 1411–1427 [DOI] [PubMed] [Google Scholar]
- Cavailles V, Dauvois S, Danielian PS, Parker MG (1994) Interaction of proteins with transcriptionally active estrogen-receptors. Proc Natl Acad Sci USA 91: 10009–10013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavailles V, Dauvois S, L'Horset F, Lopez G, Hoare S, Kushner PJ, Parker MG (1995) Nuclear factor RIP140 modulates transcriptional activation by the estrogen receptor. EMBO J 14: 3741–3751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christian M, Tullet JM, Parker MG (2004) Characterization of four autonomous repression domains in the corepressor receptor interacting protein 140. J Biol Chem 279: 15645–15651 [DOI] [PubMed] [Google Scholar]
- Collart MA, Timmers HT (2004) The eukaryotic Ccr4-Not complex: a regulatory platform integrating mRNA metabolism with cellular signaling pathways? Prog Nucleic Acid Res Mol Biol 77: 289–322 [DOI] [PubMed] [Google Scholar]
- Coulthard VH, Matsuda S, Heery DM (2003) An extended LXXLL motif sequence determines the nuclear receptor binding specificity of TRAP220. J Biol Chem 278: 10942–10951 [DOI] [PubMed] [Google Scholar]
- Denis CL, Chen J (2003) The CCR4-Not complex plays diverse roles in mRNA metabolism. Prog Nucleic Acid Res Mol Biol 73: 221–250 [DOI] [PubMed] [Google Scholar]
- Dominguez C, Bonvin AM, Winkler GS, van Schaik FM, Timmers HT, Boelens R (2004) Structural model of the UbcH5B/CNOT4 complex revealed by combining NMR, mutagenesis, and docking approaches. Structure (Camb) 12: 633–644 [DOI] [PubMed] [Google Scholar]
- Dupressoir A, Morel AP, Barbot W, Loireau MP, Corbo L, Heidmann T (2001) Identification of four families of yCCR4- and Mg2+dependent endonuclease-related proteins in higher eukaryotes, and characterization of orthologs of yCCR4 with a conserved leucine-rich repeat essential for hCAF1/hPOP2 binding. BMC Genomics 2: 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Epping MT, Wang L, Edel MJ, Carlee L, Hernandez M, Bernards R (2005) The human tumor antigen PRAME is a dominant repressor of retinoic acid receptor signaling. Cell 122: 835. [DOI] [PubMed] [Google Scholar]
- Fernandes I, Bastien Y, Wai T, Nygard K, Lin R, Cormier O, Lee HS, Eng F, Bertos NR, Pelletier N, Mader S, Han VK, Yang XJ, White JH (2003) Ligand-dependent nuclear receptor corepressor LCoR functions by histone deacetylase-dependent and -independent mechanisms. Mol Cell 11: 139–150 [DOI] [PubMed] [Google Scholar]
- Frasor J, Danes JM, Komm B, Chang KC, Lyttle CR, Katzenellenbogen BS (2003) Profiling of estrogen up- and down-regulated gene expression in human breast cancer cells: insights into gene networks and pathways underlying estrogenic control of proliferation and cell phenotype. Endocrinology 144: 4562–4574 [DOI] [PubMed] [Google Scholar]
- Gronemeyer H, Gustafsson JA, Laudet V (2004) Principles for modulation of the nuclear receptor superfamily. Nat Rev Drug Discov 3: 950–964 [DOI] [PubMed] [Google Scholar]
- Heery DM, Kalkhoven E, Hoare S, Parker MG (1997) A signature motif in transcriptional co-activators mediates binding to nuclear receptors. Nature 387: 733–736 [DOI] [PubMed] [Google Scholar]
- Hewitt SC, Harrell JC, Korach KS (2005) Lessons in estrogen biology from knockout and transgenic animals. Annu Rev Physiol 67: 285–308 [DOI] [PubMed] [Google Scholar]
- Ikematsu N, Yoshida Y, Kawamura-Tsuzuku J, Ohsugi M, Onda M, Hirai M, Fujimoto J, Yamamoto T (1999) Tob2, a novel anti-proliferative Tob/BTG1 family member, associates with a component of the CCR4 transcriptional regulatory complex capable of binding cyclin-dependent kinases. Oncogene 18: 7432–7441 [DOI] [PubMed] [Google Scholar]
- Kalkhoven E, Valentine JE, Heery DM, Parker MG (1998) Isoforms of steroid receptor co-activator 1 differ in their ability to potentiate transcription by the oestrogen receptor. EMBO J 17: 232–243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang YK, Guermah M, Yuan CX, Roeder RG (2002) The TRAP/Mediator coactivator complex interacts directly with estrogen receptors alpha and beta through the TRAP220 subunit and directly enhances estrogen receptor function in vitro. Proc Natl Acad Sci USA 99: 2642–2647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labhart P, Karmakar S, Salicru EM, Egan BS, Alexiadis V, O'Malley BW, Smith CL (2005) Identification of target genes in breast cancer cells directly regulated by the SRC-3/AIB1 coactivator. Proc Natl Acad Sci USA 102: 1339–1344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee TI, Wyrick JJ, Koh SS, Jennings EG, Gadbois EL, Young RA (1998) Interplay of positive and negative regulators in transcription initiation by RNA polymerase II holoenzyme. Mol Cell Biol 18: 4455–4462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenna NJ, O'Malley BW (2002) Combinatorial control of gene expression by nuclear receptors and coregulators. Cell 108: 465–474 [DOI] [PubMed] [Google Scholar]
- McNally JG, Muller WG, Walker D, Wolford R, Hager GL (2000) The glucocorticoid receptor: rapid exchange with regulatory sites in living cells. Science 287: 1262–1265 [DOI] [PubMed] [Google Scholar]
- Metivier R, Penot G, Hubner MR, Reid G, Brand H, Kos M, Gannon F (2003) Estrogen receptor-alpha directs ordered, cyclical, and combinatorial recruitment of cofactors on a natural target promoter. Cell 115: 751–763 [DOI] [PubMed] [Google Scholar]
- Nakamura T, Yao R, Ogawa T, Suzuki T, Ito C, Tsunekawa N, Inoue K, Ajima R, Miyasaka T, Yoshida Y, Ogura A, Toshimori K, Noce T, Yamamoto T, Noda T (2004) Oligo-astheno-teratozoospermia in mice lacking Cnot7, a regulator of retinoid X receptor beta. Nat Genet 36: 528–533 [DOI] [PubMed] [Google Scholar]
- Nettles KW, Greene GL (2005) Ligand control of coregulator recruitment to nuclear receptors. Annu Rev Physiol 67: 309–333 [DOI] [PubMed] [Google Scholar]
- Nilsson S, Makela S, Treuter E, Tujague M, Thomsen J, Andersson G, Enmark E, Pettersson K, Warner M, Gustafsson JA (2001) Mechanisms of estrogen action. Physiol Rev 81: 1535–1565 [DOI] [PubMed] [Google Scholar]
- Perissi V, Rosenfeld MG (2005) Controlling nuclear receptors: the circular logic of cofactor cycles. Nat Rev Mol Cell Biol 6: 542–554 [DOI] [PubMed] [Google Scholar]
- Prevot D, Morel AP, Voeltzel T, Rostan MC, Rimokh R, Magaud JP, Corbo L (2001) Relationships of the antiproliferative proteins BTG1 and BTG2 with CAF1, the human homolog of a component of the yeast CCR4 transcriptional complex: involvement in estrogen receptor alpha signaling pathway. J Biol Chem 276: 9640–9648 [DOI] [PubMed] [Google Scholar]
- Rouault JP, Prevot D, Berthet C, Birot AM, Billaud M, Magaud JP, Corbo L (1998) Interaction of BTG1 and p53-regulated BTG2 gene products with mCaf1, the murine homolog of a component of the yeast CCR4 transcriptional regulatory complex. J Biol Chem 273: 22563–22569 [DOI] [PubMed] [Google Scholar]
- Shang Y, Hu X, DiRenzo J, Lazar MA, Brown M (2000) Cofactor dynamics and sufficiency in estrogen receptor-regulated transcription. Cell 103: 843–852 [DOI] [PubMed] [Google Scholar]
- Torchia J, Rose DW, Inostroza J, Kamei Y, Westin S, Glass CK, Rosenfeld MG (1997) The transcriptional co-activator p/CIP binds CBP and mediates nuclear-receptor function. Nature 387: 677–6849192892 [Google Scholar]
- Tucker M, Valencia-Sanchez MA, Staples RR, Chen J, Denis CL, Parker R (2001) The transcription factor associated Ccr4 and Caf1 proteins are components of the major cytoplasmic mRNA deadenylase in Saccharomyces cerevisiae. Cell 104: 377–386 [DOI] [PubMed] [Google Scholar]
- Winkler GS, Albert TK, Dominguez C, Legtenberg YI, Boelens R, Timmers HT (2004) An altered-specificity ubiquitin-conjugating enzyme/ubiquitin-protein ligase pair. J Mol Biol 337: 157–165 [DOI] [PubMed] [Google Scholar]
- Yamashita A, Chang TC, Yamashita Y, Zhu W, Zhong Z, Chen CY, Shyu AB (2005) Concerted action of poly(A) nucleases and decapping enzyme in mammalian mRNA turnover. Nat Struct Mol Biol 12: 1054–1063 [DOI] [PubMed] [Google Scholar]
- Yanagisawa J, Kitagawa H, Yanagida M, Wada O, Ogawa S, Nakagomi M, Oishi H, Yamamoto Y, Nagasawa H, McMahon SB, Cole MD, Tora L, Takahashi N, Kato S (2002) Nuclear receptor function requires a TFTC-type histone acetyl transferase complex. Mol Cell 9: 553–562 [DOI] [PubMed] [Google Scholar]
- Yoshida Y, Hosoda E, Nakamura T, Yamamoto T (2001) Association of ANA, a member of the antiproliferative Tob family proteins, with a Caf1 component of the CCR4 transcriptional regulatory complex. Jpn J Cancer Res 92: 592–596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zwartjes CG, Jayne S, van den Berg DL, Timmers HT (2004) Repression of promoter activity by CNOT2, a subunit of the transcription regulatory Ccr4-not complex. J Biol Chem 279: 10848–10854 [DOI] [PubMed] [Google Scholar]