

Abstract
Retinoic acid (RA) constitutes the major active ingredient of vitamin A and is required for various biological processes. The tissue RA level is maintained through a cascade of metabolic reactions where retinal dehydrogenases (RALDHs) catalyze the terminal reaction of RA biosynthesis from retinal, a rate-limiting step. We showed that dietary supplement of cholesterol enhanced the expression of RALDH1 and 2 genes and the cellular RA content in vital organs such as brain, kidney, liver and heart. Consistently, the cholesterol-lowering agent (pravastatin sodium) downregulated the expression of RALDH1 and 2 genes in several organs especially the liver and in cultured liver cells. Further, cholesterol metabolites, predominantly the oxysterols, the natural ligands for liver X receptor (LXR), induced these genes via upregulation of sterol regulatory element binding protein-1c (SREBP-1c) that bound to the regulatory regions of these genes. Knockdown of LXRα/β or SREBP-1c downregulated the expression of RALDH genes, which could be rescued by re-expressing SREBP-1c, suggesting SREBP-1c as a direct positive regulator for these genes. This study uncovered a novel crosstalk between cholesterol and RA biosynthesis.
Keywords: cholesterol, LXR, retinal dehydrogenase, retinoic acid, SREBP-1c
Introduction
Vitamin A is an essential nutrient for the vertebrate. Retinoic acid (RA), the major active metabolite of vitamin A, is critically important for embryogenesis and is involved in many cellular activities such as proliferation, differentiation and apoptosis (Gudas et al, 1994). RA exerts the biological effects, primarily, through its binding to nuclear receptors, RA receptors (RARs) and retinoid X receptors (RXRs) that act on the genomic targets (Chambon, 1996). In principle, it is vital for animals to maintain a homeostatic control of retinoid metabolism, especially the biosynthesis of RA (Napoli, 1996). A number of key enzymes and proteins involved in retinoid metabolism have been identified and functionally characterized. For instance, serum retinol binding protein (RBP), cellular retinol-binding proteins (CRBPs), retinol dehydrogenases (RoDHs), retinal dehydrogenases (RalDHs) and cellular retinoic acid binding proteins (CRABPs) constitute just a short list. In the cascade of metabolic reactions, the conversion of retinal to RA by RalDHs is known to be one terminal rate-limiting step of RA biosynthesis (Blomhoff et al, 1991; Napoli, 1996).
Four RalDH isozymes, RALDH1, RALDH2, RALDH3 and RALDH4, are known for the vertebrate (Niederreither et al, 2002a). The substrate specificity for these four isozymes differs (Lin et al, 2003) and they synthesize RA from retinal in a tissue-specific manner (Niederreither et al, 2002b). Transgenic and knockout animal studies have been conducted to address the physiological roles of RALDH1 and RALDH2 genes, primarily in organogenesis during embryonic development (Niederreither et al, 2002a). While many studies strongly support RALDH2 as the major RA-synthesizing enzyme during early embryogenesis (Napoli, 1996), it has been shown that both RALDH1 and RALDH2 are expressed in an organ- and tissue-specific manner during developmental stages and in adulthood (Wang et al, 2001; Niederreither et al, 2002a; Lin et al, 2003). This would suggest that both enzymes are probably important for the biosynthesis of RA throughout life. The physiological function of these two enzymes for RA biosynthesis has been well established, and the physiological role of RA is also well known. In addition to its effects in developmental stages, RA is also known to be important for adult animals such as for retinoid metabolism in the liver (Wolf, 2001; Ross, 2003). In gene chip studies, it was found that the expression of several ALDHs could be differentially affected in sterol regulatory element binding protein (SREBP)-1/SREBP-2 transgenic animals (Horton et al, 2003) and also by treating animals with a liver X receptor (LXR) agonist (Stulnig et al, 2002). However, it remains elusive as to the regulation of the biosynthetic machinery of RA in a physiological context. In particular, two important issues need to be addressed, that how the genes for these rate-limiting enzymes are regulated and that whether the regulation of these genes can reflect the level of endogenous RA in different tissues.
In this study, we attempted to address these two issues in a physiological context. Due to the potentially lethal, or teratogenic, effects of disturbing RA biosynthesis during embryogenesis, we initiated this study using adult animals. We uncovered a novel mechanism of control over the expression of RALDH1 and RALDH2 genes, as well as the endogenous tissue RA status, regulated by supplemented and endogenous cholesterol. We found that cholesterol metabolism generated LXR ligands to activate LXR. The activated LXR upregulated the expression of SREBP-1c via the LXR response element on its promoter. Finally, SREBP-1c directly upregulated the expression of RALDH1 and 2 genes through the sterolregulatory response elements (SREs) found to reside on their proximal promoters. Consequently, the level of endogenous tissue RA is elevated.
Results
Dietary cholesterol elevates the level of RAR ligands in different tissues
To determine if there existed a relationship between sterol metabolism and RA biosynthesis, mice were fed with a cholesterol-enriched diet for 2 weeks to elevate their cholesterol, and the level of endogenous RAR ligands was monitored. Total plasma cholesterol was compared between the cholesterol-fed and the control groups, confirming significant (two- to five-fold) increase of blood cholesterol (Supplementary Figure S1). Organic solvent extracts of tissues were then applied in an RAR trans-activation assay. Extracts from the vital organs (such as brain, kidney, liver and heart) of cholesterol-fed mice trans-activated RAR significantly higher as compared to the control (Figure 1A). Among these, liver extract induced the highest level of RAR activation, with a 12-fold increase as compared to the control.
Figure 1.
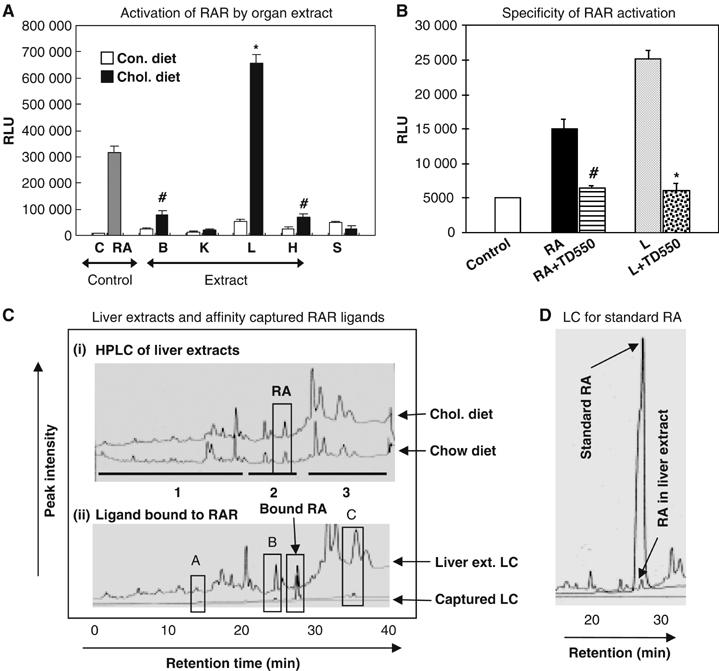
Elevation of tissue RA level in mice by enriched cholesterol diet. (A) Activation of Gal4RAR-LBD (relative luciferase unit, RLU) in COS-1 cells by organ extracts. The letters C, B, K, L, H stands for control, brain, kidney, liver and heart extracts (100 μg/ml), S for serum (50 μl/ml) and RA for 9-cis RA (1 μM). Brain and heart extracts from cholesterol-fed mice enhanced RAR activation by three-fold (#P<0.05) and liver extract by 12-fold (*P<0.01) compared to chow diet. (B) Evaluation of specificity of RAR ligands from liver extract (L) using RAR antagonist TD550 (5 μM). Suppression of Gal4RAR-LBD activation by RA (1 μM) by two-fold (# versus RA, P<0.05) and cholesterol fed liver extract (100 μg/ml) by four-fold (* versus L, P<0.001), suggesting elevation RAR specific ligands in cholesterol fed animal liver. (C) An overlay of HPLC charts of liver extracts from control (bottom) and cholesterol-fed (top) mice. Affinity captured RA from liver extract by His-tagged RAR protein (bottom). RAR selectively captured RA (overlay with RA in original LC) from liver extract. (D) An overlay of liquid chromatograms (LC) from reference at-RA and liver extract. Reporter assays were conducted in triplicate and binding assay was carried twice.
The specificity of RAR activation by tissue extracts was verified by blocking with an RAR antagonist TD550 (Farooqui et al, 2003), supporting that cholesterol enhanced the level of RAR-specific ligands in these tissues (Figure 1B). We then compared the level of RA in liver extracts between the cholesterol-fed and control groups by HPLC analyses (Figure 1C, upper). The area under the curve of RA peak in the chromatogram of the treated liver extract was higher than that of the control. A standard RA liquid chromatogram overlay is shown in Figure 1D. Interestingly, the peak intensity in region 1 of the control extract was higher than that of the treated extract, whereas the peak intensities of regions 2 and 3 of the treated extract were higher than the control (Figure 1C), showing that cholesterol feeding in animals also generated some other hydrophobic metabolites.
To identify RAR ligand in liver extracts, in vitro affinity-capture experiments were conducted, followed by HPLC analyses of the captured compounds (Figure 1C, lower). RA was captured from the liver extract by RAR. The receptors also captured some other minor, or nonspecifically bound, components (blocked areas A, B and C). A GC-MS analysis showed that these components are fatty acids (area B) and their methyl esters (area C) (data not shown), probably due to nonspecific interaction. Together, these data suggested that cholesterol feeding in adult animals generated significant amounts of RAR ligand in various vital organs. Presumably, the activity or level of the rate-limiting enzymes for RA biosynthesis was enhanced.
Cholesterol supplement enhances the expression of RALDH1 and RALDH2 genes in animals and cells
Elevated levels of RA in animal tissues as a result of cholesterol feeding can be attributed to enhanced RalDHs (the rate-limiting enzymes) for RA biosynthesis and/or an inhibition of RA catabolism by downregulating Cyp26 enzyme family. We first examined mRNA levels of RALDH1 and 2 genes in different tissues. It appeared that RALDH1 and 2 genes were significantly, but differentially, induced (Figure 2A) in most of these organs. Thus, increased RA content in these tissues was probably due to locally elevated expression of these two rate-limiting, RA-synthesizing enzymes. The basal level of RALDH3 expression in these organs was very low and exhibited no detectable change in cholesterol-fed animals (data not shown).
Figure 2.
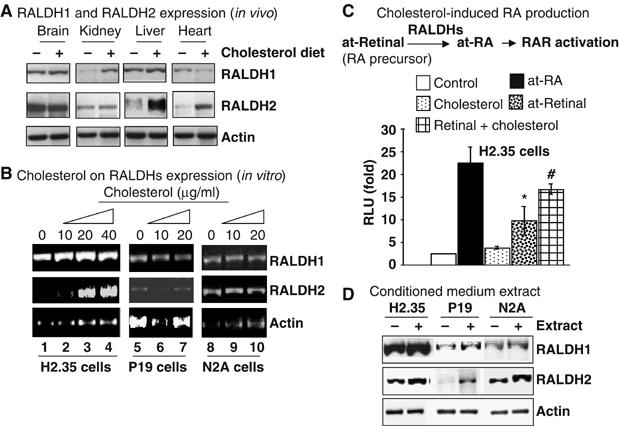
Effect of cholesterol on expression of RALDHs genes. (A) The mRNA level of RALDHs (RT–PCR) from the pulled mRNA of five mice. (B) Elevation of RALDH2 mRNA level in H2.35 cells by cholesterol (20 μM) and RALDHs showed no response to cholesterol in P19 and N2A cells. (C) Enhanced trans-activation of Gal4RAR-LBD (∼1.9-fold, # versus *, P<0.05) by at-retinal (10 μM) in the presence of cholesterol in H2.35 cells. (D) Get back responsiveness of RALDHs in N2A and P19 cells to concentrated conditioned medium extract prepared from cholesterol treated liver cells. All experiments were repeated at least twice.
We next determined the effect of cholesterol supplement using cyclodextrin-based water soluble cholesterol on the expression of RALDH1 and 2 genes in three different cell lines including a mouse hepatoma cell line H2.35, a mouse neuroblasotoma cell line N2A and a mouse embryonal carcinoma cell line P19. Surprisingly, RALDH2 mRNA was significantly elevated only in H2.35 cells dose dependently, but not in either N2A or P19 cells, and RALDH1 mRNA was elevated to a lesser extent (Figure 2B). We also conducted cholesterol supplement experiments using an isopropanolic solution of regular cholesterol containing no cyclodextrin to deliver cholesterol to H2.35 cells. Similar results were obtained as that using water soluble cholesterol, which confirmed that the effect on RALDH2 was contributed by cholesterol rather than by cyclodextrin. We also asked whether the expression pattern of these two genes in H2.35 cells reflected RA production in this cell line by conducting trans-activation assay using Gal4-fused ligand binding domain (LBD) of RAR provided with all-trans retinal as a precursor for RA biosynthesis. As expected, in the presence of cholesterol and retinal, the level of RAR activation was much higher (∼1.9-fold) than retinal alone, supporting the notion that cholesterol supplement enhanced the biosynthesis of endogenous RA from retinal in liver cells (Figure 2C).
Cholesterol metabolites induced RALDH expression
Since P19 and N2A cells did not to respond to cholesterol treatment in terms of elevating RALDH1 and 2 genes, we speculated that cholesterol might be metabolized to produce certain active metabolites only in H2.35 cells. To test this hypothesis, we prepared a concentrated condition medium of H2.35 cells with, or without, cholesterol treatment, which was supplemented to P19 and N2A cultures. The concentrated condition medium of the cholesterol-treated H2.35 cells significantly elevated RALDH2 mRNA, but to a lesser extent for RALDH1, in P19 and N2A cells (Figure 2D). This suggested that cholesterol metabolites, but not cholesterol itself, induced the expression of RALDH genes, but more significantly for the RALDH2 gene.
A wide range of cholesterol metabolites, especially the oxysterols, is known as the natural ligands of LXR (Liang et al, 2002). As predicted, 22(R)-hydroxy cholesterol significantly elevated RALDH2 mRNA level in H2.35 cells in a dose-dependent manner (Figure 3A). To determine whether the effect was dependent, specifically, upon LXR activation, a synthetic LXR ligand (TO901317) was applied, which also induced RALDH2 gene expression in a similar fashion (Figure 3A). Therefore, the activation of LXR by cholesterol metabolites resulted in the elevation of RALDH2 gene expression in liver cells. We also tested whether dexamethasone, a steroid ligand for glucocorticoid receptor, could affect RALDH expression. It was found that dexamethasone exerted little effect on the expression of RALDHs (not shown). Together, the data suggested a direct or indirect role for LXR in regulating the expression of RALDH genes.
Figure 3.
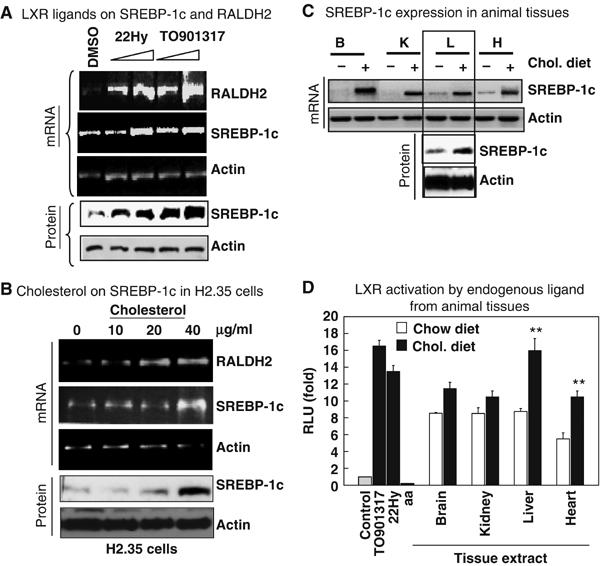
Effects of LXR ligands on RALDH2 gene. (A) LXR ligands, 22(R)-hydroxy cholesterol (22Hy) and a synthetic TO901317 dose dependently induced the mRNA of RALDH2 consistently with the mRNA level (top) and protein level (bottom) of SREBP-1c in H2.35 cells. (B) Cholesterol was shown to elevate the mRNA level and protein of SREBP-1c dose dependently in H2.35 cells similar to pure LXR ligands, suggesting oxysterol mediated activation of SREBP-1c via LXR. (C) Elevation of mRNA level of SREBP-1c in cholesterol fed animal tissue. (D) Activation of LXR by organ extracts from cholesterol fed mice similar to LXR ligands. Arachidonic acid (aa) was used as a control for LXR antagonist. Liver and heart extracts from cholesterol-fed animal activated LXR by two-fold (**P<0.01) compared to the extracts from control mice. The experiments for (A–C) were repeated twice, and for (D) thrice.
We then asked how LXR regulated the RALDH2 gene expression. A promoter analysis for transcription factor binding sites using the TESS (Transcription Element Search System) software, which are available online (http://www.cibl.upenn.edu/tess) revealed no LXR binding site, but a SREBP-1c binding site (TATCACCCAC) between −442 bp and −431 bp of RALDH2 gene. Since LXR activation induces SREBP-1c expression, we then determined the effect of synthetic and natural LXR ligands, as well as cholesterol itself, on the expression of SREBP-1c in H2.35 cells. As expected, SREBP-1c was induced at both mRNA and protein levels (Figure 3A and B). To further verify this link in a physiological setting, the endogenous level of SREBP-1c in the relevant organs was compared between the cholesterol-fed and control animals. It appeared that SREBP-1c expression at both mRNA and protein levels was significantly increased in most of these organs of cholesterol-fed animals (Figure 3C). By using an LXR trans-activation assay (Figure 3D), as predicted, most of the tissue extracts prepared from cholesterol-fed animals significantly trans-activated the ligand-binding domain of LXR fused to a Gal4BD vector. Taken together, these in vivo and in vitro data are consistent with the notion that high cholesterol activated SREBP-1c expression through the production of LXR ligands, which in turn elevated the expression of RALDH2.
SREBP-1c directly upregulated the expression of RALDH1 and RALDH2 gene
A promoter analysis revealed an sterol regulatory element (SRE) on the RALDH2 gene promoter. In the EMSA assay, it was found that SREBP-1c strongly bound to this cognate DNA sequence (Figure 4A, upper panel). A minimal mutation on this SRE abolished SREBP-1c binding. A transient transfection-based reporter assay was then conducted using a reporter containing the wild-type and the mutant SREs, which revealed that SREBP-1c indeed enhanced the activities of the reporter containing the wild-type SRE, but not the mutant SRE (Figure 4B, upper). Similarly, the effect of SREBP-1c on the reporters containing the endogenous promoters for both RALDH1 and 2, each spanning a 1.5 kb upstream region of the transcription initiation sites, was also tested. The data showed that SREBP-1c could also significantly activate the activities of the reporters driven by the natural promoters of RALDH1 and 2 (Figure 4B, lower panel). We also examined whether SREBP2 could bind to this SRE and activate the promoters. It appeared that SREBP2 could also bind to the same SRE site (Figure 4A, lower panel). SREBP2 also activated the reporter of either the SRE or the natural promoter (Figure 4B). However, in this system, SREBP-1c seemed to be more potent than SREBP2. Together, the data confirmed SREBP-1c and SREBP 2 as the direct positive regulators for RALDH2 gene.
Figure 4.
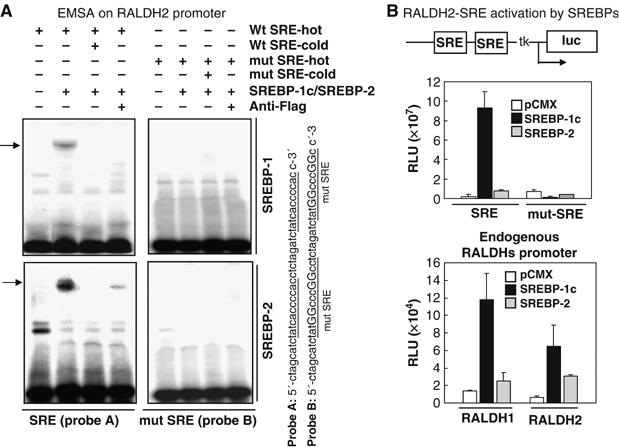
Role of SREBPs as transcription factors for RALDHs gene regulation. (A) In EMSA assay, both SREBP-1c/SREBP2 could bind to wild-type SRE derived from RALDH2 promoter, but not to the mutant SRE. (B) Activation of RALDH2 SRE and mutant-SRE luciferase reporters (top) and also reporters containing 1.5 kb endogenous promoters of both RALDH1 and RALDH2 by SREBP-1c/SREBP-2.
Enriched cholesterol diet also induced RALDH1 expression, but more significantly in different tissues such as the kidney (Figure 2A). Induction of RALDH1 gene expression by cholesterol also occurred, but less significantly, in H2.35 cells (Figure 2B). While no typical SRE (TATCACCCAC) site was predicted for the RALDH1 promoter, a cluster of two sequences resembling the modified SRE YCAY(C/A/G)YCAY, where Y can be C or T (Soccio et al, 2005) were found, TCATGCCCT (A) and TCTGCCCAT (A), located at −98 bp to −90 bp, and −81 bp to −73 bp, respectively. An EMSA assay and a reporter driven by this sequence were conducted, which confirmed that SREBP-1c could bind to this sequence (Supplementary Figure S2A) and was able to activate a reporter driven by this particular SRE cluster (Supplementary Figure S2B).
Physiological relevance of SREBP-1c to the expression of RALDH1 and RALDH2 genes
To further confirm the physiological relevance of RALDH1 and 2 genes regulation by SREBP-1c, RNA silencing of SREBP-1c targeting the C-terminus (Supplementary data) was performed in H2.35 cells (Figure 5A). It revealed significant knockdown of SREBP-1c, with approximately 68% reduction at RNA and 80% reduction at protein levels. As a result, RALDH2 and RALDH1 was significantly, 72 and 67%, respectively, downregulated. The direct action of SREBP-1c on these gene promoters in vivo was assessed using chromatin immunoprecipitation (ChIP) assays, which revealed an increased binding of SREBP-1c to the endogenous RALDH1 and 2 genes in the presence of natural and synthetic LXR ligands (Figure 5B). Finally, a cholesterol-lowering agent (HMG-CoA reductase inhibitor, pravastatin sodium) was applied in both animals and H2.35 cells. The total blood cholesterol was significantly (50–60% reduction) reduced (Supplementary Figure S1B), which effectively downregulated the expression of both RALDH1 and 2 genes in the liver (Figure 5C, right panel) and in liver cells (Figure 5C, left panel). We also tested whether cholesterol supplement could rescue the suppressive effect of pravastatin on RALDHs expression. As expected, cholesterol supplement could significantly rescue the effect of pravastatin on RALDH2 in cultured cells (Figure 5C, lower panel). We also compared the effects of pravastatin-treated and normal liver extracts on LXR activation (Figure 5D, lower panel). The data showed that pravastatin-treated liver extract was much weaker than the control liver extract in LXR activation, which was consistent with the reduced liver cholesterol level in pravastatin-treated mice (Figure 5D).
Figure 5.
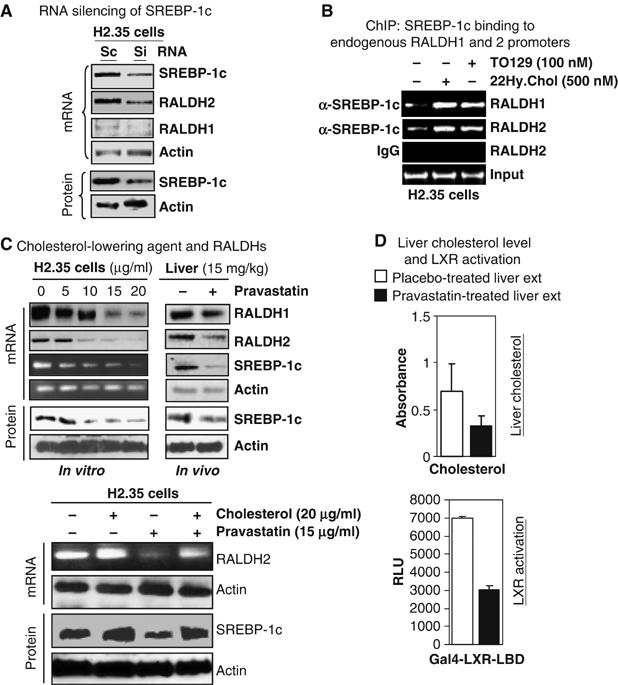
Regulation of RALDHs gene by SREBP-1c. (A) RNA silencing of SREBP-1c (knockdown 68% at mRNA and 80% at protein levels) could result in reduction of mRNA (RT–PCR) level of RALDH2 by 72% and RALDH1 by 67%. (B) ChIP assay on endogenous RALDH1 and RALDH2 genes. LXR ligands could enhance SREBP-1c binding to SRE sites on both RALDH1 and RALDH2 gene promoter in H2.35 cells. (C) Effect of HMG-CoA reductase inhibitor in H2.35 cells and animal. Pravastatin sodium could reduce the mRNA level of RALDHs genes in consistent with the reduction of mRNA and protein level of SREBP-1c in a dose-dependent fashion in liver cells (left panel) and liver (right panel, i.p. 15 mg/kg/day, n=5). Supplemental cholesterol could rescue the pravastatin effect on RALDH2 gene. (D) Pravastatin lowered the liver cholesterol level in mice (top). Activation of LXR by pravastatin treated mice liver extract was weaker than that of control mice. Experiments in all panels were conducted at least twice and RT–PCR was performed from pulled mRNA of five mice.
Together, these data confirm that metabolism of cholesterol enhances the level of LXR ligands in animal tissues, which in turn activates LXR for upregulating SREBP-1c expression, that ultimately directs signals for elevated expression of RALDH2 gene expression by binding to their promoter regions and, as a result, enhancing the biosynthesis of RA.
Effect of silencing LXRα/β or SREBP-1 and cholesterol supplement
The above data suggested that LXRs mediated the upregulation of RALDHs through SREBP-1c. We then conducted gene silencing of LXRs, followed by cholesterol supplement to determine whether cholesterol could rescue the expression of RALDHs in the absence of LXRs (Figure 6A). It appeared that cholesterol could efficiently activate the RALDH2 gene in the absence of LXRα, but it could hardly activate this gene in LXRβ-knockdown cells. This indicated that LXRβ might play a more prominent role in regulating RALDHs. In the case of LXRs double knockdown, cholesterol supplement could no longer activate RALDHs. As a result of LXRα/LXRβ knockdown, SREBP-1c was nearly complete absent (Figure 6A). These data suggested that activation of RALDH genes by cholesterol indeed involve LXRs. Further, to examine the effect of SREBP-1, we ectopically expressed SREBP-1c in the LXRα/β knockdown cells. The results showed that ectopic expression of SREBP-1c could rescue the LXR-silencing phenotype in terms of RALDH2 gene expression (Figure 6C). This suggested a direct role for SREBP-1c in regulating RALDH2 gene. We then silenced SREBP-1c, followed by cholesterol supplement. The result showed that cholesterol could no longer activate RALDH2 in SREBP-1c knockdown cells (Figure 6B). Similar results were obtained by silencing of SREBP cleavage-activating protein (SCAP), which blocked the production of SREBPs (Figure 6B). In this experiment, cholesterol was also unable to rescue the phenotype of SCAP knockdown. SREBPs cleavage system was thought to be strictly regulated by sterols. In high cholesterol-fed animal, SREBPs cleavage system could be interrupted. Therefore, elevation of SREBP-1c by oxysterol-activated LXR should no longer activate the RALDH genes. However, our results showed an upregulation of RALDH1 and RALDH2 genes in high cholesterol-fed animals. It was possible that SREBP-1c could be cleaved by a sterol-independent mechanism as reported (Shimano, 2002), to exert its regulatory effect on RALDH genes in high cholesterol-fed animals.
Figure 6.
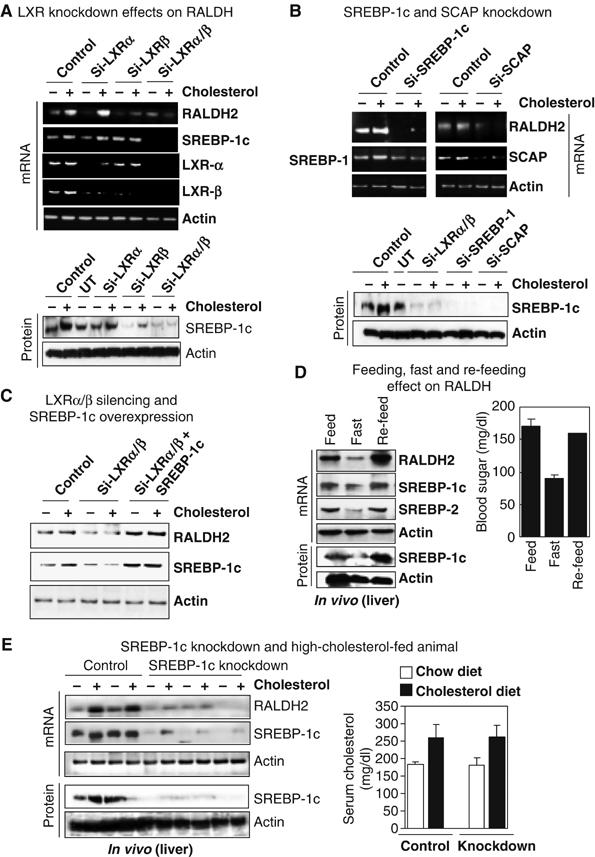
Physiological relevance of RALDHs gene regulation by SREBPs. (A) Double knockdown of LXRα and LXRβ strongly downregulated RALDH2 expression in liver cells, which was not rescued by supplemental cholesterol. (B) RALDH2 was also downregulated by knockdown of either SREBP-1c or SCAP. Supplemental cholesterol could not reverse their effect on RALDH2. (C) Overexpression of SREBP-1c in LXRα/β-null cells could rescue the LXRs double knockdown effect on RALDH2 gene. (D) Re-feeding of fasting animals could robustly increase RALDH2 transcription, suggesting SREBP-1c as a strong positive regulator of RALDH2 gene. (E) Knockdown of mouse liver SREBP-1c by hydrodynamics-based delivery of si-RNA (short hairpin RNAi vector, 100 μg/mice/1.6 ml in 5 s) of SREBP-1c downregulated the RALDH2 expression, which was not rescued by high cholesterol feeding.
To distinguish the roles of SREBP-1 and SREBP2 in terms of regulation of RALDH genes, we conducted feed, fasting and re-feed experiments in mice (Figure 6D). It is known that fasting downregulates the transcription as well as the formation of both SREBP 1 and 2 (Horton et al, 1998). Re-feeding animals with a fat-free high-carbohydrate diet usually enhanced the transcription, as well as activation of SREBP-1c protein, but not SREBP2, which should come back to a normal level. Our results showed that, fasting for 18 h dramatically reduced both SREBPs, in consistence with the downregulation of RALDH2 gene (Figure 6D). An 8-h re-feed significantly enhanced the level of SREBP-1c and RALDH2 expression in mouse liver. This suggested that, compared to SREBP-2, SREBP-1c is more potent for RALDH2 gene regulation. Finally, we conducted liver-specific knockdown of SREBP-1c by hydrodynamics-based siRNA delivery using RNAi vector containing the U6 promoter to generate short-hairpin siRNA to silence SREBP-1c in vivo. The mice were also fed high cholesterol diet. The results showed that SREBP-1c knockdown indeed significantly downregulated RALDH2 gene in liver (Figure 6E). The high cholesterol diet could no longer rescue the effect of SREBP-1 knockdown on RALDH2 gene expression.
Discussion
In this report, we present, for the first time, a novel mechanism of cross-regulation of RA biosynthesis by cholesterol metabolites. A regulatory pathway of RALDH1 and 2 genes by cholesterol metabolites is uncovered, that is mediated by SREBP-1c's direct upregulation of these two genes through cholesterol metabolic shunt in animals and cell lines. This finding suggests a need for careful investigation of the cross-regulation of metabolic machineries for cholesterol and for vitamin A.
The expression of RA catabolic enzymes (Cyp26a1, Cyp26b1 and Cyp26c1) in cholesterol-fed animals reacted differently. For instance, Cyp26a1 mRNA in liver was slightly induced (data not shown), probably due to the feed back regulation of Cyp26a1 by the elevated RA content in the liver for a detoxification purpose. In contrast, mRNA of Cyp26b1 was significantly reduced in the testis of cholesterol-fed animals (data not shown). In H2.35 cells, cholesterol feeding had no effects on the mRNA level of Cyp26a1 and Cyp26c1, but reduced that of Cyp26b1 (data not shown). Therefore, the effects of cholesterol on the endogenous tissue RA content occurred primarily by the induction of RALDH genes; RA catabolism could be secondary. Further studies are needed to gain insight into RA catabolism affected by cholesterol.
RA is involved in a wide spectrum of biological processes, many of which are affected by sterol metabolic shunt (Peet et al, 1998; Costet et al, 2000; Venkateswaran et al, 2000; Joseph et al, 2002). Therefore, our findings of upregulation of RALDHs genes and RA synthesis by oxysterols reveals a direct role of cholesterol in regulating RA biosynthesis and, possibly, crucial biological processes regulated by RA. However, the regulation of RA biosynthesis in the brain can be more complex because the brain cholesterol pool is separated from circulation by blood–brain barrier and the brain has an independent cholesterol metabolic pathway (Levi et al, 2005). This is consistent with the observation that both RALDH1 and RALDH2, while expressed in the brain, are only moderately affected by the cholesterol-enriched diet (Figure 2A). However, the cholesterol lowering agent, pravastatin, appeared to significantly reduce RALDH2 expression in the brain (data not shown) and the level of brain RA is significantly elevated by dietary cholesterol (Figure 1A). It is possible that other RalDHs may be involved in brain RA synthesis (Niederreither et al, 2002a). A gene chip study conducted for the LXR agonist-treated animals and LXR-knockout mice revealed various degrees of alteration in the expression of several ALDHs (Stulnig et al, 2002). Although cholesterol transport and metabolism in the brain is separated from the whole body circulation, brain RA biosynthesis remains vulnerable to a disturbance in the dietary cholesterol and cellular cholesterol. This should involve a highly coordinated network of regulatory pathways that remain to be determined.
Cholesterol metabolism and fatty acid synthesis are interrelated. Oxysterol activates LXR to upregulate fatty acid synthase (FAS) via SREBP-1c (Yoshikawa et al, 2001). SREBPs are transcription factors cleaved from 125 kDa precursors and are required for the maintenance of lipid homeostasis (Eberle et al, 2004). At least three members are found in the SREBP family: SREBP-1a, SREBP-1c and SREBP-2 isoforms. SREBP-1a and SREBP-2 isoforms, involved in cholesterol synthesis, are suppressed by a high serum cholesterol status through a feedback mechanism. Active cleavage of their precursors occurs by sterol depletion (Eberle et al, 2004). Our findings that both SREBP-1 and 2 can regulate the expression of RALDH genes suggest the physiological relevance of a highly coordinated, intricate regulation of RALDH genes by high and low cholesterol diets. Potentially, a high cholesterol diet where SREBP2 is downregulated, SREBP-1c would take over the control over RA synthesis; whereas a low sterol condition, when SREBP2 is upregulated, SREBP-2 might be more important for RA synthesis. This type of physiological coordination of RA synthesis by different SREBPs presumably evolved in accordance with the physiological demands for a homeostatic supply of RA under various dietary conditions. However, SREBP-1c is expressed in most tissues, especially in the liver and the white adipose tissue (Shimomura et al, 1997) and its cleavage is triggered by other cellular factors such as insulin (Eberle et al, 2004). Further, SREBP-1c gene is known to be directly induced by LXRs through an RXR/LXR binding site on the SREBP-1c gene promoter (Repa et al, 2000; Yoshikawa et al, 2001). While both SREBP-1 and SREBP-2 could bind to the RALDH gene promoters and activate their reporters, our data would suggest that SREBP-1c is more relevant to the regulation of RALDH genes in the liver under a high-cholesterol dietary condition.
SREBP-1c is involved in many biological processes. In adipogenesis, it elevates fatty acid synthesis to supply natural ligands of peroxisome proliferator-activated receptors (PPARs) for adipocyte differentiation and fat accumulation. Saturated fatty acids also enhance the expression of SREBP-1c through elevated expression of PGC-1α that coactivates LXR (Lin et al, 2005). Therefore, upregulation of SREBP-1c by oxysterols could result in the production of both RAR/RXR and PPAR-γ ligands. Activation of PPAR δ results in the enhanced hydrolysis of retinyl ester (the storage form of vitamin A in liver) to retinol for RA synthesis (Hellmans et al, 2003). Further, SREBP-1c gene is upregulated by other steroids like androgen and progesterone, which are synthesized from cholesterol (Heemers et al, 2001; Lacasa et al, 2001) and estrogen induces RALDH2 and RA synthesis in rat uterus (Li et al, 2004). Therefore, in addition to oxysterols, cholesterol may upregulate RALDH2 through SREBP-1c by other steroid metabolites generated from cholesterol. It needs to be determined how other steroid hormones can play roles in the regulation of RALDH genes.
Numerous studies have revealed the regulation of cholesterol and fatty acid metabolism by RA signaling through RXR (Shulman and Mangelsdorf, 2005). An LXR target gene, apolipoprotein E (apoE) was also reported to be affected by 9-cis-RA during the secretion process (Ishida et al, 2004; Liang et al, 2004; Ripolles et al, 2004). Our current study presents the first evidence for the regulation of tissue RA biosynthesis by cholesterol metabolites. Accordingly, it is tempting to speculate crosstalk between cholesterol and retinoid metabolic pathways through crossregulation of their metabolic machineries by the reciprocal hormones. Of particular significance is the implication of the impact of cholesterol status on the maintenance of local RA biosynthesis in different tissues, as well as the overall vitamin A homeostasis in animals.
Materials and methods
Reagents and drugs
Detection reagents for estimation of serum/plasma triglyceride (Thermo DMA), cholesterol (Thermo DMA), free fatty acids (FFA) (Wako, USA) and protein (Bradford assay, Bio-Rad) were procured and used according to the supplier protocol. Regular cholesterol and water soluble cholesterol (Sigma), pravastatin sodium (Sigma), LXR ligands, TO901317 and 22(R)-hydroxy cholesterol from Cayman Chemical, Anarbor, MI. RAR antagonist TD550 was a kind gift from Dr Hiroyuki Kagechika, Tokyo University, Tokyo, Japan.
Plasmid constructs
The cDNA of mouse active SREBP-1c (p68) and SREBP-2 were PCR amplified from mouse (C57BL/6) liver cDNA, and then cloned SREBP-1c at BamHI and XbaI and SREBP-2 at EcoRI and HindIII sites into pCMX vector. Luciferase reporter plasmids containing two SREBP-1c response elements from RALDH1 and RALDH2 promoter were synthesized in vitro, and cloned into BamHI/NheI sites of pGL3-promoter vector (Promega, USA). Endogenous promoters both of RALDH1 and RALDH2 containing 1.5 Kb upstream of transcription initiation sites were cloned into pGL3-basic vector (Promega) at NheI and BglII sites. The detailed procedures are available in the Supplementary data.
Cell culture, cholesterol and drug treatment
COS-1, and N2A cells were maintained in DMEM containing 10% FBS, P19 cells was in MEM supplemented with 7.5% CS and 2.5% FBS, and H2.35 cells were cultured in DMEM low glucose (Gibco, USA) supplemented with 4% FBS and 20 nM dexamethasone. Water-soluble cholesterol (Sigma) and LXR ligands were added to 60–70% confluent cells in 6 cm plate and harvested after 24–36 h treatment except otherwise noted.
RT–PCR
Total RNA either from tissues or cells was isolated using Trizol™ reagent (Invitrogen, USA). The mRNA from five mice was pulled and RT reaction was conducted using Omniscript™ (Qiagene, USA) reverse-transcriptase enzyme. The cDNAs of RALDH1 (Aldh1a1, Accession No: NM_013467), RALDH2 (Aldh1a2, Accession No: NM_009022), SREBP-1c (Srebf1, Accession No: NM_011480), SREBP-2 (Srebf2, Accession No: NM_033218), LXRα (Nr1h3, Accession No: NM_013839), LXRβ (Nr1h2, Accession No: NM_009473), and β-actin were amplified by PCR. The information for primers and PCR condition is available in the Supplementary data.
In vivo experiment
ICR male mice (6-week old) were used for in vivo experiment. The control and treated groups containing five mice each was put in a case under a day night cycle. The animal was treated according to the Institutional Research Animal Resource (RAR) guidelines of the University of Minnesota. Regular cholesterol (Sigma) containing no cyclodextrin was supplied as aqueous suspension in a bottle instead of drinking water, while the control mice were treated with a placebo drink containing the all the excipients but no cholesterol for 2 weeks. On an average, 20 mg cholesterol/mice/day was fed. For cholesterol lowering experiment, mice were treated with 200 μl of pravastatin sodium (15 mg/kg/day) in PBS by intra-peritoneal (i.p.) route for 2 weeks. The control mice were treated with PBS. On day 15, the mice were killed, and total plasma cholesterol, FFA and triglyceride was estimated with detection reagents. Tissues from different organs were collected, homogenized, and processed for extraction and RNA isolation. For feed, fast and reefed experiment, the mice were fasted for 18 h and reefed with fat-free high-carbohydrate diet before killing the animal.
Preparation of organic extracts of animal tissues
Fresh homogenized tissues from different organs from control and treated mice were extracted with an equal mixture of ethyl acetate and diethyl ether for 24 h in the dark. The solvent was evaporated under reduced pressure using a rotary evaporator to dryness. The dried extract was dissolved in isopropanol, and employed for the reporter assay and biochemical analysis.
Affinity capture of RAR ligands and HPLC analysis
His-tagged RAR-LBD (250 μg) purified protein from bacteria was incubated with 25 μl of liver extract (500 μg) for 6 h at 4°C in 2.5 ml of 50 mM HEPES (pH 8.0) binding buffer containing 150 mM NaCl, 10% glycerol and 0.1% NP-40. The receptor was affinity bound to Nickel-agarose beads (Qiagen), unbound ligand was washed out, and the bound ligand was extracted by 100% isopropanol and run over an HPLC. The conditions for HPLC and affinity purification of receptor bound ligand are described in the Supplementary data.
Reporter assay
The 5 × 105 cells were plated on 24-well plates. After 12 h, regular medium were exchanged with medium supplemented with charcoal stripped FBS, and the cells were transfected with the reporter (SRE-tk-Luc and Gal4-tk-Luc) and expression (SREBP-1c, Gal4LXR-LBD, Gal4RAR-LBD, Gal4LXR-LBD) plasmids using Lipofectamine™-2000 (Invitrogene) transfection reagents. The LacZ gene expression plasmid was used as an internal control. At 12 h after transfection, the cells were treated with 100 μg/ml of isopropanolic tissue extract for 16–24 h except otherwise noted.
EMSA assay
SREBP-1c N-terminal was generated by in vitro transcription/translation (TNT) coupled system (Promega). Synthetic oligonucleotide probes containing SRE sites were labeled with [α-32P]dCTP by RadPrime DNA labeling system (Gibco). Binding reactions were performed in 20 μl of 10 mM HEPES buffer (pH 7.4) conatining 75 mM KCl, 2.5 mM MgCl2, 0.1 mM EDTA, 4% Glycerol, 1 μg poly-dI/dC, 2 μl of TNT SREBP-1c/SREBP-2, and 10 ng probe DNA. For competition assay, 10-fold excess cold probe DNA was used. The mixtures were incubated at room temperature for 30 min, followed by adding with or without 1 μl anti-Flag antibody for further 30 min for super-shift. Samples were run onto a 6% polyacrylamide gel in TBE buffer at 4°C. After electrophoresis, the gel was fixed, dried and exposed to PhosphorImager screen (Molecular Dynamics).
Chromatin immunoprecipitation
ChIP assay were conducted as described previously (Park et al, 2005). Briefly, H2.35 cells cultured in DMEM containing DCC-FBS were treated with a gradient concentration of LXR ligands for 24 h. The cells were fixed with 10% formaldehyde and the reaction was quenched with 1 M glycine. Whole-cell extract was prepared in SDS-lysis buffer by ultra-sonication. The extracts were equilibrated according to protein concentration, and 500 μg protein was used for immunoprecipation using anti-SREBP-1c antibody. Precipitated DNA was amplified by PCR covering the SRE site on RALDH1 and RALDH2 using specific primers. The primer information is available in Supplementary data.
RNA silencing and Western blot
To knockdown SREBP-1 individual siGENOME duplex (catalog No. D-040814-01-0010, sequence: 5′-GCAAGGCCAUCGACUACAU-3′) targeting N-terminal region and siGENOME duplex targeting C-terminal region (Catalog No. D-0040814-04-0010, sequence: 5′-GGGCAGCUCUGUACUCCUU-3′) of SREBP-1c were purchased from Dharmacon and also an si-RNA duplex from Santa Cruz Biotechnology, Inc. (Catalog No. sc-35558) were used, and scramble RNA was from Dharmacon. Similarly, siGENOME duplex (Catalog No. D-040649-01-0010, sequence: 5′-GCCUCAAUGCCUGAUGUUU-3′) to knockdown LXRα, siGENOME duplex (Catalog No. D-042839-01-0010, sequence: 5′-CUACAUCGUGGUCAUCUUA-3′) to knockdown LXRβ, siGENOME smart pool reagent (Catalog No. M-040322-00-0005) to knockdown SCAP were procured from Dharmacon. The H2.35 cells in six-well plates were transfected with siRNA duplex using Lipofectamine-2000 (Invitrogen) transfection reagent to a final concentration of 100 nM siRNA. Mouse liver SREBP-1c was knocked down by hydrodynamics-based delivery of RNAi vector to generate target short hairpin siRNA for SREBP-1c following the established procedures (Liu et al, 1999; Kobayashi et al, 2004). Details are described in the Supplementary data. Expression of the target genes was monitored at the mRNA level by RT–PCR and the SREBP-1c protein level was monitored by Western blot using anti-SREBP-1 anti-body (Santa Cruz Biotechnology Inc, CA, Catalog No. Sc-336) to detect mature SREBP-1c (p68).
Statistical analysis
All values are expressed as mean±s.e.m. Comparsison of results between different groups was performed by paired t-test (Student's t-test). A P-value ⩽0.05 was considered to be statistically significant.
Supplementary Material
Supplementary Data
Acknowledgments
This work was supported by NIH Grants DK54733, DK60521, DA11190 and K02-DA13926 to LNW. We thank Dr CH Lee for his critical reading of this manuscript.
References
- Blomhoff R, Green MH, Green JB, Berg T, Norum KR (1991) Vitamin A metabolism: new perspectives on absorption, transport, and storage. Physiol Rev 71: 951–990 [DOI] [PubMed] [Google Scholar]
- Chambon P (1996) A decade of molecular biology of retinoic acid receptors. FASEB J 10: 940–954 [PubMed] [Google Scholar]
- Costet P, Luo Y, Wang N, Tall AR (2000) Sterol-dependent transactivation of the ABC1 promoter by the liver X receptor/retinoid X receptor. J Biol Chem 275: 28240–28245 [DOI] [PubMed] [Google Scholar]
- Eberle D, Hegarty B, Bossard P, Ferre P, Foufelle F (2004) SREBP transcription factors: master regulators of lipid homeostasis. Biochimie 86: 839–848 [DOI] [PubMed] [Google Scholar]
- Farooqui M, Franco PJ, Thompson J, Kagechika H, Chandraratna RA, Banaszak L, Wei LN (2003) Effects of retinoid ligands on RIP140: molecular interaction with retinoid receptors and biological activity. Biochemistry 42: 971–979 [DOI] [PubMed] [Google Scholar]
- Gudas LJ, Sporn MB, Roberts AB (1994) Cellular biology and biochemistry of retinoids. In The Retinoids, Sporn MB, Roberts AB, Goodman DS (eds), pp 443–520. New York: Raven Press [Google Scholar]
- Heemers H, Maes B, Foufelle F, Heyns W, Verhoeven G, Swingen JV (2001) Androgens stimulate lipogenic gene expression in prostate cancer cells by activation of the sterol regulatory element-binding protein cleavage activating protein/sterol regulatory element-binding protein pathway. Mol Endocrinol 15: 1817–1828 [DOI] [PubMed] [Google Scholar]
- Hellmans K, Rombotus K, Quarter E, Dittie AS, Knorr A, Michalik L, Rogiers V, Schuit F, Wahli W, Geerts A (2003) PPAR-beta regulates vitamin A metabolism-related gene expression in hepatic stellate cells undergoing activation. J Lipid Res 44: 280–295 [DOI] [PubMed] [Google Scholar]
- Horton JD, Bashmakov Y, Shimomura I, Shimano H (1998) Regulation of sterol regulatory element binding proteins in livers of fasted and refed mice. Proc Natl Acad Sci USA 95: 5987–5992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horton JD, Shah NA, Warrington JA, Anderson NN, Park SW, Brown MS, Goldstein JL (2003) Combined analysis of oligonucleotide microarray data from transgenic and knockout mice identifies direct SREBP target genes. Proc Natl Acad Sci USA 100: 12027–12032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishida BY, Bailey KR, Duncan KG, Chalkley RJ, Burlingame AL, Kane JP, Schwartz DM (2004) Regulated expression of apolipoprotein E by human retinal pigment epithelial cells. J Lipid Res 45: 263–271 [DOI] [PubMed] [Google Scholar]
- Joseph SB, Laffitte BA, Patel PH, Watson MA, Matsukuma KE, Walczak R, Collins JL, Osborne TF, Tontonoz P (2002) Direct and indirect mechanisms for regulation of fatty acid synthase gene expression by liver X receptors. J Biol Chem 277: 11019–11025 [DOI] [PubMed] [Google Scholar]
- Kobayashi N, Matsui Y, Kawase A, Hirata K, Miyagishi M, Taira K, Nishikawa M, Takakura Y (2004) Vector-based in vivo RNA interference: dose- and time-dependent suppression of transgene expression. J Pharmacol Exp Ther 308: 688–693 [DOI] [PubMed] [Google Scholar]
- Lacasa D, Liepvre XL, Ferre P, Dugail I (2001) Progesterone stimulates adipocyte determination and differentiation 1/sterol regulatory element-binding protein 1c gene expression potential mechanism for the lipogenic effect of progesterone in adipose tissue. J Biol Chem 276: 11512–11516 [DOI] [PubMed] [Google Scholar]
- Levi O, Lutjohann D, Devir A, Bergmann KV, Hartman T, Michaelson DM (2005) Regulation of hippocampal cholesterol metabolism by apoE and environmental stimulation. J Neurochem 95: 987–997 [DOI] [PubMed] [Google Scholar]
- Li XH, Kakkad B, Ong DE (2004) Estrogen directly induces expression of retinoic acid biosynthetic enzymes, compartmentalized between the epithelium and underlying stromal cells in rat uterus. Endocrinology 145: 4756–4762 [DOI] [PubMed] [Google Scholar]
- Liang Y, Lin S, Beyer TP, Zhang Y, Wu X, Bales KR, DeMattos RB, May PC, Li SD, Jiang XC, Eacho PI, Cao G, Paul SM (2004) A liver X receptor and retinoid X receptor heterodimer mediates apolipoprotein E expression, secretion and cholesterol homeostasis in astrocytes. J Neurochem 88: 623–634 [DOI] [PubMed] [Google Scholar]
- Liang G, Yang J, Horton JD, Hammer RE, Goldstein JL, Brown MS (2002) Diminished hepatic response to fasting/refeeding and liver X receptor agonists in mice with selective deficiency of sterol regulatory element-binding protein-1c. J Biol Chem 277: 9520–9528 [DOI] [PubMed] [Google Scholar]
- Lin J, Yang R, Tarr PT, Wu PH, Handschin C, Li S, Yang W, Pei L, Uldry M, Tontonoz P, Newgard CB, Spiegelman BM (2005) Hyperlipidemic effects of dietary saturated fats mediated through PGC-1beta coactivation of SREBP. Cell 120: 261–273 [DOI] [PubMed] [Google Scholar]
- Lin M, Zhang M, Abraham M, Smith SM, Napoli JL (2003) Mouse retinal dehydrogenase 4 (RALDH4), molecular cloning, cellular expression, and activity in 9-cis-retinoic acid biosynthesis in intact cells. J Biol Chem 278: 9856–9861 [DOI] [PubMed] [Google Scholar]
- Liu F, Song Y, Liu D (1999) Hydrodynamics-based transfection in animals by systemic administration of plasmid DNA. Gene Therapy 6: 1258–1266 [DOI] [PubMed] [Google Scholar]
- Napoli JL (1996) Biochemical pathways of retinoid transport, metabolism, and signal transduction. Clin Immunol Immunopathol 80: S52–S62 [DOI] [PubMed] [Google Scholar]
- Niederreither K, Fraulob V, Garnier JM, Chambon P, Dolle P (2002b) Differential expression of retinoic acid-synthesizing (RALDH) enzymes during fetal development and organ differentiation in the mouse. Mech Dev 110: 165–171 [DOI] [PubMed] [Google Scholar]
- Niederreither K, Vermot J, Fraulob V, Chambon P, Dolle P (2002a) Retinaldehyde dehydrogenase 2 (RALDH2)- independent patterns of retinoic acid synthesis in the mouse embryo. Proc Natl Acad Sci USA 99: 16111–16116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park SW, Huq MM, Loh HH, Wei LN (2005) Retinoic acid-induced chromatin remodeling of mouse kappa opioid receptor gene. J Neurosci 25: 3350–3357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peet DJ, Turley SD, Ma W, Janowski BA, Lobaccaro JM, Hammer RE, Mangelsdorf DJ (1998) Cholesterol and bile acid metabolism are impaired in mice lacking the nuclear oxysterol receptor LXR alpha. Cell 93: 693–704 [DOI] [PubMed] [Google Scholar]
- Repa JJ, Liang G, Ou J, Bashmakov Y, Lobaccaro JM, Shimomura I, Shan B, Brown MS, Goldstein JL, Mangelsdorf DJ (2000) Regulation of mouse sterol regulatory element-binding protein-1c gene (SREBP-1c) by oxysterol receptors, LXRalpha and LXRbeta. Genes Dev 14: 2819–2830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ripolles PB, Nazih H, Neunlist M, Huvelin JM, Bard JM (2004) Effect of LPS on basal and induced apo E secretion by 25-OH chol and 9cRA in differentiated CaCo-2. J Cell Biochem 91: 786–795 [DOI] [PubMed] [Google Scholar]
- Ross AC (2003) Retinoid production and catabolism: role of diet in regulating retinol esterification and retinoid acid oxidation. J Nutr 133: 291S–296S [DOI] [PubMed] [Google Scholar]
- Shimano H (2002) Sterol regulatory element-binding protein family as global regulators of lipid synthetic genes in energy metabolism. Vitam Horm 65: 167–194 [DOI] [PubMed] [Google Scholar]
- Shimomura I, Shimano H, Horton JD, Goldstein JL, Brown MS (1997) Differential expression of exons 1a and 1c in mRNAs for sterol regulatory element binding protein-1 in human and mouse organs and cultured cells. J Clin Invest 99: 838–845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shulman AI, Mangelsdorf DJ (2005) Retinoid X receptor heterodimers in the metabolic syndrome. N Engl J Med 353: 604–615 [DOI] [PubMed] [Google Scholar]
- Soccio RE, Adams RM, Maxwell KN, Breslow JL (2005) Differential gene regulation of StarD4 and StarD5 cholesterol transfer proteins. Activation of StarD4 by sterol regulatory element-binding protein-2 and StarD5 by endoplasmic reticulum stress. J Biol Chem 280: 19410–19418 [DOI] [PubMed] [Google Scholar]
- Stulnig TM, Steffensen KR, Gao H, Reimers M, Dahlman-Wright K, Schuster GU, Gustafsson JA (2002) Novel roles of liver X receptors exposed by gene expression profiling in liver and adipose tissue. Mol Pharmacol 62: 1299–1305 [DOI] [PubMed] [Google Scholar]
- Venkateswaran A, Laffitte BA, Joseph SB, Mak PA, Wilpitz DC, Edwards PA, Tontonoz P (2000) Control of cellular cholesterol efflux by the nuclear oxysterol receptor LXR alpha. Proc Natl Acad Sci USA 97: 12097–12102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Sperkova Z, Napoli JL (2001) Analysis of mouse retinal dehydrogenase type 2 promoter and expression. Genomics 74: 245–250 [DOI] [PubMed] [Google Scholar]
- Wolf G (2001) Retinoic acid homeostasis: retinoic acid regulates liver retinol-esterifying enzyme as well as its own catabolic oxidation in liver. Nutr Rev 59: 391–394 [DOI] [PubMed] [Google Scholar]
- Yoshikawa T, Shimano H, Amemiya-Kudo M, Yahagi N, Hasty AH, Matsuzaka T, Okazaki H, Tamura Y, Iizuka Y, Ohashi K, Osuga J, Harada K, Gotoda T, Kimura S, Ishibashi S, Yamada N (2001) Identification of liver X receptor-retinoid X receptor as an activator of the sterol regulatory element-binding protein 1c gene promoter. Mol Cell Biol 21: 2991–3000 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Data