

Abstract
Packaging DNA in nucleosomes and higher-order chromatin structures restricts its accessibility and constitutes a barrier for all DNA transactions including gene regulation and DNA repair. How and how fast proteins find access to DNA buried in chromatin of living cells is poorly understood. To address this question in a real time in vivo approach, we investigated DNA repair by photolyase in yeast. We show that overexpressed photolyase, a light-dependent DNA-repair enzyme, recognizes and repairs UV-damaged DNA within seconds. Rapid repair was observed in various nucleosomal regions of the genome including inactive and active genes and repressed promoters. About 50% of cyclobutane pyrimidine dimers were removed in 5 s, >80% in 90 s. Heterochromatin was repaired within minutes, centromeres were not repaired. Consistent with fast conformational transitions of nucleosomes observed in vitro, this rapid repair strongly suggests that spontaneous unwrapping of nucleosomes rather than histone dissociation or chromatin remodeling provides DNA access. The data impact our view on the repressive and dynamic nature of chromatin and illustrate how proteins like photolyase can access DNA in structurally and functionally diverse chromatin regions.
Keywords: chromatin, DNA repair, nucleosome, photolyase, yeast
Introduction
How DNA can be accessed by proteins when it is packaged in chromatin of eukaryotic cells is a fundamental question concerning all DNA transactions from transcription to replication, recombination, and repair of DNA lesions (Kornberg and Lorch, 1999; Felsenfeld and Groudine, 2003; Downey and Durocher, 2006). Nucleosomes are the repeated structural units of eukaryotic chromosomes made of about two turns of DNA wrapped around an octamer of core histone proteins, two each of H2A, H2B, H3, and H4 (Richmond and Davey, 2003). They control the access of proteins to DNA, act as repressors by making recognition sites for proteins inaccessible, but they also present recognition sites on the surface. Occluded sites may become exposed and accessible to proteins by dynamic transitions involving transient dissociation of histones, unwrapping of DNA, or changing the position of histone octamers on the DNA sequence (nucleosome mobility). Those intrinsic properties may be modulated by chromatin remodeling activities that chemically modify the histones, exchange histone variants, and/or alter the structure and position of nucleosomes (Kornberg and Lorch, 1999; Felsenfeld and Groudine, 2003; Flaus and Owen-Hughes, 2004; Mellor, 2005).
While a role of chromatin remodeling in transcription is well established (see references above), increasing evidence supports a role of chromatin remodeling in DNA repair. Double-strand break repair involves recruitment and phosphorylation of H2A.X, acetylation of histones in the vicinity of the lesion, as well as a local chromatin expansion immediately after DNA damage induction (Downs et al, 2000, 2004; van Attikum and Gasser, 2005; Kruhlak et al, 2006). Chromatin remodeling plays also a role in nucleotide excision repair (NER) which is a multistep-multienzyme reaction that removes bulky lesions from double-stranded DNA including ultraviolet (UV) light-induced pyrimidine dimers (Gontijo et al, 2003; Gong et al, 2005; Yu et al, 2005). Remodeling includes rearrangement and/or reassembly of nucleosomes after DNA-repair synthesis (Smerdon and Lieberman, 1978; Green and Almouzni, 2003), acetylation of histones (Smerdon et al, 1982; Brand et al, 2001; Teng et al, 2002; Yu et al, 2005), as well as monoubiquitination of histone H2A by a ubiquitin E3 ligase that is recruited to the damage by UV-damaged DNA-binding protein complex (UV-DDB) (Kapetanaki et al, 2006). While histone modifications may destabilize chromatin and facilitate damage accessibility, the mechanisms of how DNA lesions are recognized in chromatin remain unclear.
Real time methods developed in the last few years provided a dynamic picture of nucleosomes and chromatin interactions of proteins involved in transcription and DNA repair (Hager et al, 2004; Mone et al, 2004; Mellor, 2005; Metivier et al, 2006). On one side, fluorescent energy transfer (FRET) studies with reconstituted nucleosomes in vitro suggested that nucleosomes exist in a dynamic equilibrium between fully wrapped and a set of partially unwrapped states. These conformational transitions occur in the second and subsecond time scale and provide a short window of opportunity for proteins to access DNA that is normally protected in nucleosomes (Li and Widom, 2004; Li et al, 2005; Tomschik et al, 2005). On the other hand, photobleaching studies in higher eukaryotic cells revealed that transcription and repair factors tagged with fluorescent proteins are highly mobile and exchange within seconds and minutes with target sites in the nucleus, whereas the core histones reside stably associated with chromatin (Hager et al, 2004; Mone et al, 2004; Kimura, 2005). Only histone H1 exchanges in minutes (Bustin et al, 2005). While these live-cell imaging techniques provide a time resolution in the seconds or even subseconds time scale, they are limited by a low resolution at the cytological level and cannot resolve how proteins interact with chromatin at the level of nucleosomes. In addition, they also sample unspecific and nonproductive transient interactions. Alternative kinetic data on protein–DNA interactions were obtained by chromatin immunoprecipitation (ChIP) (Hager et al, 2004; Mellor, 2005; Metivier et al, 2006). This technique depends on the efficiency of chemical protein–DNA crosslinking. It therefore samples rather productive interactions, yet the resolution is limited by the DNA fragment size of a few hundred base pairs and the time resolution is limited by the crosslinking time, which is in the range of several minutes. Moreover, crosslinking of proteins to the target DNA might shift the equilibrium towards the bound state. Thus, it remains largely unknown whether and how rapid structural transitions occur in nucleosomes that are imbedded in the chromosomal context or how fast proteins can access nucleosomal DNA in living cells.
To directly address those questions, we have studied DNA repair of UV lesions by photolyase in yeast as an alternative light-regulated real-time approach. In contrast to live-cell imaging and ChIPs, this approach samples a productive interaction of a protein with the target DNA at a resolution that spans from whole chromatin domains to individual dipyrimidines (e.g. Suter et al, 1997, 2000a; Suter and Thoma, 2002). UV light preferentially generates cyclobutane pyrimidine dimers (CPDs). CPDs are repaired by NER, and in many organisms including the yeast Saccharomyces cerevisiae by photolyase (Friedberg, 2003). Photolyase binds the damaged DNA, flips the pyrimidine dimer into the active site, and reverts it to the native bases in a light-dependent reaction (photoreactivation, PR) (Sancar, 2003; Mees et al, 2004). NER and PR are modulated by protein–DNA interactions in chromatin including nucleosomes, heterochromatin, and centromeres (Thoma, 1999, 2005). As dipyrimidines occur throughout the genome and as UV-damage formation and PR are light dependent, this system allows a direct and rapid assessment of DNA accessibility to photolyase in living cells. Here, we show that nucleosomal DNA can be repaired by photolyase in seconds (50% in 5 s, >80% in 90 s) under conditions where photolyase is not limiting, whereas heterochromatin is more slowly repaired and centromeres are not repaired. Those data directly demonstrate that dynamic transitions of nucleosomes allow proteins to access DNA in living cells within seconds and impact our view on the repressive role of nucleosomes in DNA recognition.
Results
In wild-type yeast, photolyase repairs 0.3 CPDs/kb UV lesions in nucleosomes in about 2 h and nucleosome-free DNA in about 15 min (Suter et al, 1997, 2000a, 2000b). Repair is slow in the center of nucleosomes and increases towards the ends, suggesting that transient unfolding, dissociation, or nucleosome mobility facilitate damage exposure (Suter and Thoma, 2002). To test whether the structural and dynamic properties of nucleosomes or the abundance of photolyase are rate limiting, we have overexpressed yeast photolyase. RAD1, an essential gene for NER, and the endogenous photolyase gene (PHR1) were deleted to avoid competitive repair by NER and damage-induced changes of photolyase levels (Sebastian et al, 1990), respectively. Moreover, the cells were transformed with a minichromosome (YRpTRURAP) as an internal standard that is independent of chromosomal position effects and allows comparison with previous studies (Thoma, 1986; Suter et al, 1997; Suter and Thoma, 2002).
DNA-repair experiments with the overexpressing strain were carried out in glucose media and at room temperature. Aliquots of the cell suspension were irradiated with UV light for 11 s (150 J/m2) and within seconds (<10 s) transferred to the PR chamber. Irradiation with photoreactivating light was under light saturation conditions. For CPD analysis, the DNA was isolated, digested with appropriate restriction enzymes and cut at CPDs with T4-endonuclease V. The cutting sites were visualized in different regions of the genome and minichromosome (Figures 1 and 2). The top bands represent the fraction of undamaged restriction fragments; the bands in the lower part represent DNA cut at CPDs. The damage generated with 150 J/m2 was approximately 0.35 CPDs/kb per DNA strand and varied from 0.30 to 0.45 CPDs/kb between individual experiments (see Materials and methods). This damage corresponds to approximately 8400 CPDs/genome (12 Mbp) and about one CPD per eight nucleosomes (assuming a nucleosome repeat length of 165 bp (Thomas and Furber, 1976)).
Figure 1.
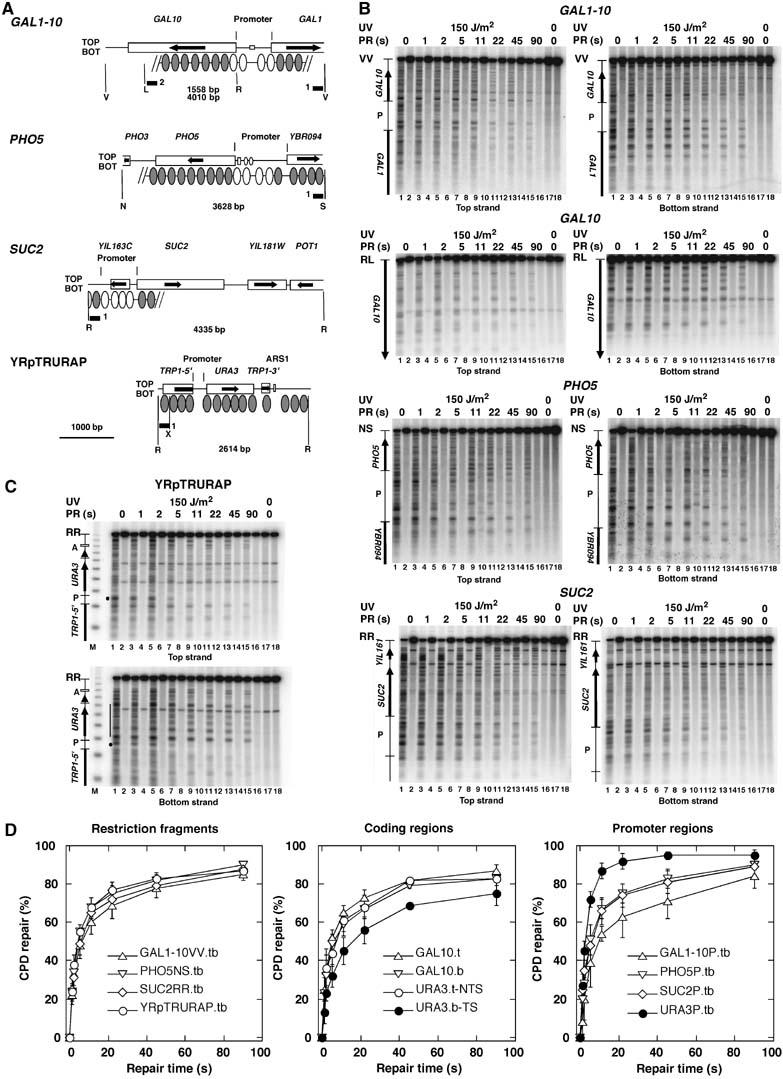
Photolyase repairs UV lesions in yeast chromatin in seconds. (A) Relevant features of the chromosomal GAL1–10-, PHO5-, and SUC2-loci and the minichromosome YRpTRURAP. Indicated are restriction sites (R, EcoRI; V, EcoRV; L, SalI; N, NciI; S, StuI; X, XbaI); the length of the restriction fragments (bp); the probes used for hybridization (black bars); the open reading frames of the genes (boxes with arrows); the promoter regions as used in this study with promoter elements (squares and dots); positioned nucleosomes where known (ovals); nucleosomes subjected to remodeling during gene activation (white ovals). (B, C) PhosphorImager scans of Southern blots visualizing the CPD distribution in the top and bottom strands. ABY19 cells were irradiated for 11 s with 150 J/m2 of UV light (lanes 1–16), exposed to photoreactivating light for 0–90 s (PR (s); lanes 3–16). The DNA was digested with restriction enzymes (indicated in A), cut at CPDs with T4-endonuclease V (odd lanes 1–17) or mock treated (even lanes 2–18), fractionated on alkaline agarose gels, blotted and hybridized to strand-specific probes (black bars in A). The top band represent the fraction of intact restriction fragments that contained no CPDs. Indicated are the relevant ORFs (arrows), promoter regions (P). M, a DNA size marker (multiples of 256 bp). (D) Repair curves showing the fraction of CPDs repaired as a function of time. The initial damage was set as 0% repair. Data are means and standard deviations from three independent UV experiments. Extensions t, b, and tb indicate that the top-, bottom-, or both strands were included in the calculation. Top and bottom strand were combined (tb), when the differences between individual curves were not significant.
Figure 2.
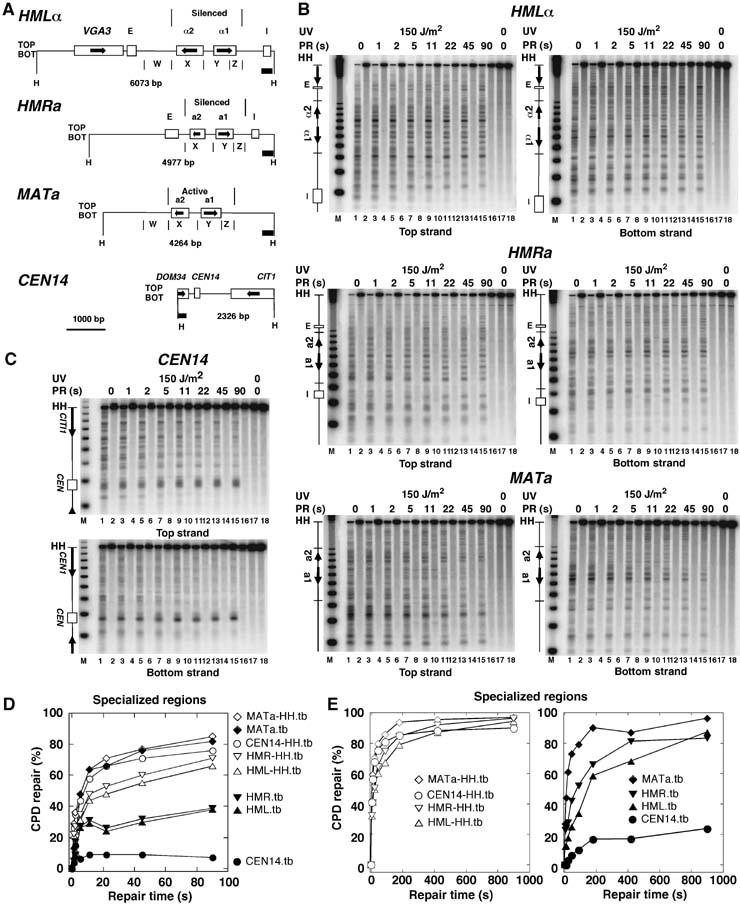
Heterogeneity of rapid repair in specialized chromatin. (A) Relevant features of the chromosomal silent mating type loci (HML and HMR), the active MATa locus, and the region surrounding the centromere of chromosome XIV (CEN14). (B, C) Repair analysis in the mating type loci and CEN14, respectively, as described in Figure 1. (D) Repair curves of restriction fragments (MATa-HH.tb, HML-HH.tb, HMR-HH.tb, CEN14-HH.tb) and the specialized loci (MATa.tb, HML.tb, HMR.tb, CEN14.tb) depicted in (A, B). Data are given as the average of the top and bottom strand (tb) of one of the experiments shown in Figure 1. (E) Repair of silent chromatin (HML, HMR) in minutes. ABY19 cells were irradiated for 11 s with 150 J/m2 of UV light, and exposed to photoreactivating light for 0–900 s.
Repair of nucleosomes in seconds
Figure 1 shows the repair data in the minichromosome YRpTRURAP and in three representative chromosomal regions (PHO5, SUC2, and GAL1–10). All of them are packaged in nucleosomes and show nucleosome eviction or remodeling in the promoter region upon activation of transcription (Hirschhorn et al, 1992; Cavalli and Thoma, 1993; Lohr, 1997; Boeger et al, 2004; Korber et al, 2004; Reinke and Horz, 2004). Repair was extremely fast in all DNA regions. This is obvious by the disappearance of bands and the increasing intensity of the top band (odd lanes 1–15). To allow direct comparison between different regions, repair curves were calculated as the fraction of CPDs removed versus repair time. About 50% of the lesions were removed in only 5 s from all chromatin domains shown in Figure 1D, whereas more than 80% of the lesions were repaired in 90 s. Thus, most CPDs were accessible to photolyase in less than 90 s. Given a nucleosome repeat length of about 165 bp (Thomas and Furber, 1976) and a nucleosome core size of 147 bp (Richmond and Davey, 2003), the major fraction of CPDs removed during rapid repair originates from nucleosome cores. Thus, photolyase gains access to nucleosomes within seconds.
There was no dramatic difference between repair rates in various nucleosomal regions, namely the restriction fragments containing the PHO5, SUC2, GAL1–10 regions (55±4, 49±6, 48±7%, respectively, in 5s; the numbers represent mean and standard deviation of three independent experiments, Figure 1B and D) and the coding region of the inactive GAL10 gene which is completely covered with nucleosomes and does not contain regulatory elements that might affect chromatin structures (51±4% in 5 s). Apparently, rapid accessibility of DNA reflects a general property of nucleosomal chromatin in yeast.
Rapid repair of nucleosomes in repressed promoters
Given the general concept of repressive nucleosomes and the fact that nucleosomes are involved in regulation of transcription, it is conceivable that promoter regions of repressed genes contain specialized nucleosomes with different composition and stability (Kornberg and Lorch, 1999; Mellor, 2005; Zhang et al, 2005). Indeed, the repressed PHO5 and GAL1–10 promoters contain the variant histone H2A.Z (Htz1) (Santisteban et al, 2000). It was proposed that Htz1-bearing nucleosomes at repressed/basal promoters facilitate activation through their susceptibility to loss, thereby helping to expose promoter DNA (Zhang et al, 2005). We therefore measured repair in the inactive PHO5-, GAL1–10-, and SUC2- promoters. All three promoter regions were almost as efficiently repaired as the other nucleosomal regions (80% in 90 s; in 5 s about 39±13% (GAL1–10), 51±8% (PHO5), and 48±11% (SUC2); Figure 1B and D). Thus, there is no indication that repressed promoters contain particularly stable or unstable nucleosomes that would resist or facilitate DNA accessibility to DNA-binding proteins like photolyase.
In contrast to the repressed promoters, the promoter of the URA3 gene represents a well-known example of a nucleosome-free open promoter (Thoma, 1986; Suter et al, 1997, 2000a). Indeed, this promoter was repaired much more rapidly (72±4% in 5s; Figure 1C and D) providing direct evidence for the different chromatin structures of the repressed and open promoters.
Differential repair in specialized chromatin regions
Deviations from those standard nucleosomal repair rates, however, were observed in functionally constrained regions such as transcribed genes, centromeres, and heterochromatin.
Transcription elongation affects chromatin structure and DNA repair. URA3 is a rarely transcribed gene containing six positioned nucleosomes flanked by two non-nucleosomal regions at the 5′-end (open promoter) and 3′-end, respectively (Thoma, 1986; Bedoyan et al, 1992). During transcription elongation RNA-polymerase II is transiently stalled at CPDs on the transcribed strand and inhibits repair of CPDs by photolyase which results in slow repair of the transcribed strand (Livingstone-Zatchej et al, 1997; Suter et al, 1997). Such a slow repair of the transcribed strand of URA3 was obvious even in seconds range (bottom strand, URA3b-TS; Figure 1C and D), indicating that some RNA-polymerases are stalled at the CPDs but released again within seconds. Previous experiments have shown that photoreactivation and NER of the nontranscribed strand are modulated by the nucleosomes (Wellinger and Thoma, 1997; Suter and Thoma, 2002). Here we show that repair of the nontranscribed strand of URA3 by overexpressed photolyase was similar to repair of the other nucleosomal regions (top strand, URA3t-NTS; 44±7%) indicating that transcription did not destabilize nucleosomes in the few seconds of the experiment.
No repair was detected in the centromere of chromosome XIV, whereas the centromere flanking regions were efficiently repaired (Figure 2). This supports our previous results that showed a strong inhibition of photoreactivation and nucleotide excision repair by kinetochore proteins (Capiaghi et al, 2004).
Heterochromatin refers to a fraction of eukaryotic genomes where transcription is silenced by a repressive chromatin structure that involves silencing proteins bound to nucleosomes, initiates at silencers and spreads over relatively large distances (Richards and Elgin, 2002). Silencing in yeast occurs in subtelomeric regions, in the rDNA locus, and in the silent mating-type loci HMLα and HMRa, whereas the active mating-type locus MATa (or MATα) is not silenced (Rusche et al, 2003). Reduced repair rates (in the time scale of hours) were previously observed in subtelomeric regions (NER and PR) (Livingstone-Zatchej et al, 2003) and in HMLα (NER) (Terleth et al, 1989). Figure 2 shows that with overexpressed photolyase, both heterochromatic HMLα and HMRa loci were more slowly repaired than the active MATa locus and the other chromosomal regions reaching about 40 and 80%, respectively, in 90 s (Figure 2B and D). A more extensive kinetic revealed that 80% repair of the silent loci required about 7–15 min (Figure 2E). Thus, despite silencing, chromatin remains dynamic and the DNA is accessible to photolyase.
Overexpression of photolyase and UV lesions do not disrupt chromatin
Having observed this rapid repair, one might wonder whether and to what extent repair is influenced by the overexpressed photolyase itself or by potential damage-induced disruption of nucleosomes. Photolyases strongly discriminate between pyrimidine dimers and nondamaged DNA. Comparison of the binding constants of the CPD photolyases to dimers versus nondamaged DNA indicates that the discrimination ratio is about 105 (Sancar, 2006). To test whether binding of overexpressed photolyase to undamaged DNA disturbs chromatin structures and thereby facilitates accessibility of CPD sites, we analyzed nucleosome positioning and stability by micrococcal nuclease digestion. The results did not show any obvious difference between the overexpression strain (ABY19) and a control strain (ABY07), neither on the minichromosome nor in the PH05 region (Figure 3). Therefore, rapid repair is not caused by chromatin disruption induced by interactions of photolyase with undamaged DNA. However, as photolyase bends DNA at the CPD site by about 50 degrees (Mees et al, 2004), it is conceivable that those specific interactions disturb the structure of nucleosomes. We cannot assess this topic in yeast by nuclease footprinting, as the fraction of nucleosomes containing a CPD at one particular site is very low (one out of eight nucleosomes contains one CPD located somewhere within 147 bp).
Figure 3.
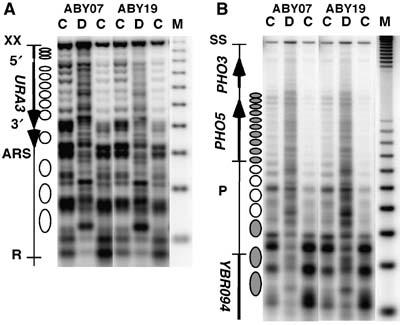
Overexpression of photolyase does not disrupt nucleosome positions and stability. Chromatin structure of strains with (ABY19) and without photolyase overexpression (ABY07) in YRpTRURAP (A) and in the PHO5 locus (B). DNA (D) and chromatin (C) were digested with different amounts of micrococcal nuclease. The DNA was purified, cut with XbaI (YRpTRURAP) or StuI and NciI (PHO5) before loading on an agarose gel. After blotting, the membranes were hybridized with probe 1 generated by random priming (YRpTRURAP) and probe 1, bottom strand probe generated by primer extension (PHO5), indicated in Figure 1A. No differences were observed using the top stand probe (not shown). M, size marker (multiples of 256 bp). Indicated are the nuclease-sensitive regions (URA3-3′, URA3-5′, ARS), the open reading frames of the genes (arrows), and the positioned nucleosomes (ellipses). White ellipses represent nucleosomes in PHO5 that are evicted during transcription activation (Reinke and Horz, 2004).
With respect to damage-induced nucleosome disruption, it was previously shown that nucleosomes resist UV irradiation and that UV-damaged nucleosomes can be purified from genomic chromatin (Gale et al, 1987; Schieferstein and Thoma, 1998; Gaillard et al, 2003). It seems that the inherent flexibility and distortions in nucleosomal DNA support the accommodation of DNA lesions (Richmond and Davey, 2003; Thoma, 2005). One might, however, argue that UV lesions disrupt or destabilize nucleosomes in living cells thereby facilitating damage recognition and repair. Assuming that CPDs are transiently exposed in a dynamic equilibrium, it is also conceivable that specific binding of photolyase to exposed CPDs shifts the equilibrium. To test those hypotheses, ABY19 cells were UV irradiated and incubated in the dark for 0–20 min before PR (11 s). An increase of repair is expected, if UV lesions disrupt chromatin and/or if photolyase binds to the exposed CPDs in the dark. As shown for a standard nucleosomal region (coding region of GAL10), repair increased slightly by about 10% as a consequence of dark incubation (Figure 4). Thus, UV lesions and binding of photolyase in the dark exert only a mild effect on site exposure of DNA lesions in living cells.
Figure 4.
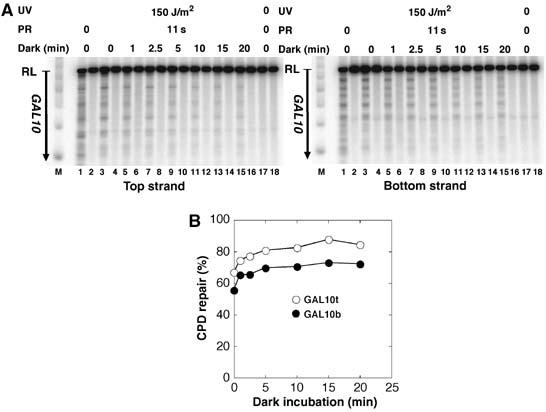
Repair after dark incubation. ABY19 cells were UV irradiated with 150 J/m2, incubated in the dark for 0, 1, 2.5, 5, 10, and 20 min and photoreactivated for 11 s. CPDs were displayed (A) and measured (B) in the GAL10 coding region (LR) as described in Figure 1.
Overexpressed photolyase is not rate limiting
A central question is whether the amount of photolyase and the exposure of CPDs in chromatin are rate-limiting factors in chromatin repair under our conditions. As yeast photolyase was not only found in the nucleus but also in the cytoplasm and mitochondria (Yasui et al, 1992) and as its activity depends on two chromophore cofactors, which may be present only in a fraction of the overexpressed enzymes, an accurate quantification of active and inactive enzymes in the nucleus is not possible. We therefore have addressed this problem by exploring the repair capacity of the overexpressed photolyase pool (Figure 5). Cells were irradiated with an increasing UV dose from 75 to 1000 J/m2 and PR was carried out for a constant time of 45 s. DNA damage and repair was measured in the minichromosome YRpTRURAP and in the nucleosomal GAL10 region (GAL10-LR as in Figure 1). The initial damage ranged between 0.2 and 0.8 CPDs/kb (Figure 5A). The number of CPDs repaired in 45 s increased with the increased initial damage demonstrating that photolyase was not limiting (Figure 5B). Moreover, the fraction of CPDs repaired in 45 s was fairly constant (Figure 5C), indicating that a similar fraction of CPDs was exposed within 45 s repair time irrespective of the initial damage. Thus, it is the exposure of CPDs that limits repair and not the amount of active photolyase. We also noticed a slight decrease of the repaired fraction in the samples with high amounts of DNA lesions (Figure 5C). We think that this decrease is due to a potential inactivation of photolyase by the high UV dose applied to those cells, rather than by structural changes of chromatin. Finally, those data show that fluctuations of CPDs/kb in our standard conditions with relative low levels of DNA lesions do not significantly affect CPD repair.
Figure 5.
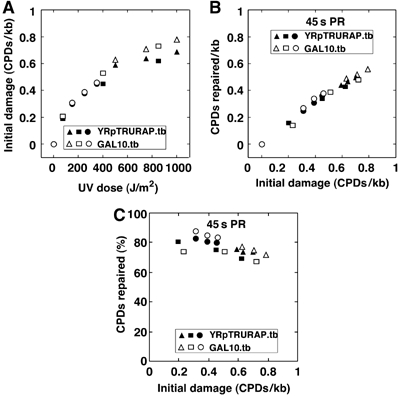
UV-dose-dependent CPD formation and PR. (A) Initial damage. (B) CPD repair in 45 s PR. (C) Fraction of CPDs repaired in 45 s PR. ABY19 cells were UV irradiated with 75–1000 J/m2, and photoreactivated for 45 s (PR). CPDs were measured in the GAL10 coding region (GAL10.tb, average of top and bottom strand) and YRpTRURAP (YRpTRURAP.tb, average of top and bottom strand) as described in Figure 1. Triangles, circles, and squares are data from three independent experiments.
Discussion
All chromosomal regions with the exception of some engaged in specific functions were repaired on a time scale of seconds. As photolyase is inhibited by nucleosomes in vitro (Schieferstein and Thoma, 1998; Gaillard et al, 2003), repair in vivo reflects dynamic properties of chromatin that make DNA accessible to proteins. This is to our knowledge the first direct evidence demonstrating that proteins can access DNA buried in nucleosomes of living cells within seconds, and therefore impacts both our understanding of nucleosome dynamics and our view of how similar proteins involved in other DNA transactions can access DNA.
With removal of half of the lesions in 5 s and >80% in 90 s, we observed an extremely fast damage recognition and repair in living cells. In other words, approximately 4200 CPDs and >6700 CPDs were removed from the genome in the first 5 and 90 s, respectively. We do not know whether this is the fastest possible access to chromatin at room temperature. However, we examined the repair capacity of overexpressed photolyase and found that photolyase was in excess and CPD exposure in chromatin was rate limiting in our standard conditions (Figure 5). Moreover, we showed that overexpression of photolyase per se does not disturb or disrupt undamaged chromatin by unspecific interactions with DNA (Figure 3). In addition, the dark incubation experiment revealed that nucleosomes are not significantly disrupted by UV lesions and that photolyase does not remarkably affect the equilibrium of damage exposure by specific binding to CPDs in the dark (Figure 4). Finally, reduced repair rates in nucleosomal regions compared with open promoters (URA3P, Figure 1) are direct evidence for the presence of nucleosomes during the repair time.
Given a nucleosome repeat length of 165 bp and a core size of 147 bp, the major fraction of CPDs removed during rapid repair of nucleosomal regions originates from nucleosome cores. We therefore can evaluate how structural and dynamic properties of nucleosomes allow damage recognition and repair. One question to be discussed is whether the location of CPDs on the nucleosome surface affects repair. As nucleosomes consist of two turns of DNA wrapped around the histone octamer, damage accessibility and repair might depend on the rotational setting. CPDs located on the helical turns that face the histone octamer or the adjacent superhelical turn of DNA may be shielded from recognition by photolyase, whereas CPDs located on helical turns that face the solvent might be directly accessible (Thoma, 2005). However, when the damage distribution was analyzed in nucleosome cores isolated from UV-irradiated chromatin, CPDs were found preferentially but not exclusively at sites where the phosphate backbone of the DNA was farthest from the core histone surface (Gale et al, 1987). Moreover, irradiation of nucleosomes with defined DNA sequences revealed that CPDs are generated irrespective of their position in the nucleosome, including sites that face the histone octamer (Schieferstein and Thoma, 1996, 1998; Gaillard et al, 2003). Thus, a preferential orientation of CPDs per se cannot account for rapid and complete repair.
Can photolyase recognize and repair UV lesions on the nucleosome surface? CPD repair in nucleosomes has been tested in vitro using Escherichia coli photolyase. The data showed a severe inhibition as well as inefficient and incomplete repair at some sites, but no correlation with the rotational setting (Schieferstein and Thoma, 1998). In contrast to the in vitro experiments, high-resolution analysis of PR in yeast nucleosomes revealed slow repair in the center, increased repair rates towards the ends, and differential repair at individual sites that are only a few base pairs apart, indicating that the ends of nucleosomes become more readily repaired than the center and that some repair may occur on the nucleosome surface. As all lesions were repaired in about 2 h, we assumed that the nucleosomal DNA becomes accessible by dynamic structural changes (Suter and Thoma, 2002).
Does rapid repair measure intrinsic properties of chromatin or does it require chromatin remodeling? ATP-dependent remodeling activities (SWI/SNF and ISW2) can act on UV-damaged nucleosomes in vitro and facilitate PR (Gaillard et al, 2003). In principle, nucleosome remodeling complexes can be recruited to specific sites via interactions with DNA-binding proteins (e.g. transcription factors). For PR, such a recruitment process is highly unlikely, as photolyase is a monomeric enzyme that recognizes and repairs the damage. Moreover, there are no indications by genome-wide interaction studies that photolyase contacts nuclear proteins or remodeling complexes (Ho et al, 2002). Alternatively, remodeling complexes may interact nonspecifically and globally with the genome and enhance dynamics of chromatin at random. We cannot formally exclude this possibility. Global remodeling, however, would imply that the abundance of a few hundred to a few thousand copies of remodeling complexes (SWI/SNF about 200; ISW2 and RSC, about 2000 copies/cell (Ghaemmaghami et al, 2003)) have to act on about 72 000 nucleosomes within a few seconds. In conclusion, rapid repair within seconds most likely reflects intrinsic and spontaneous dynamic properties of nucleosomes in the budding yeast and elucidates a new time scale with respect to DNA exposure in chromatin of living cells.
Several mechanisms that expose nucleosomal DNA to proteins are currently discussed and tested (Flaus and Owen-Hughes, 2004; Mellor, 2005; Thoma, 2005). DNA sites in nucleosomes may become accessible to proteins by transient dissociation of histones or unwrapping of DNA from the histone octamer. In both cases, the ends of nucleosomal DNA become preferentially exposed. Based on the in vivo photobleaching experiments with histones (Kimura, 2005), the dissociation of core histones is far too slow to account for rapid repair. However, in vitro FRET experiments with fluorescently labeled DNA and histones revealed that conformational transitions consistent with unwrapping occur on the subsecond to second time scale (Li et al, 2005; Tomschik et al, 2005). Thus, DNA accessibility to photolyase within seconds is most consistent with such an unwrapping mechanism. In addition, complete repair implies that also the central region of nucleosomal DNA becomes accessible and suggests that nucleosome mobility is somehow involved. A favored model suggests that nucleosomes move as a result of unwrapping and rebinding of nucleosomal DNA at different positions (references in Flaus and Owen-Hughes, 2004; Mellor, 2005). This generates a DNA bulge, which can eventually diffuse around the histone octamer by subsequent steps of unwrapping and rewrapping. Therefore, mobility as a multistep process must be slow compared with individual unwrapping transitions, but it might be that some repair takes place when lesions are exposed in the bulge, whereas another fraction of lesions is repaired after the histone octamer has moved.
We have observed similar repair rates in several chromosomal loci and in a minichromosome as well as reduced repair rates in a centromere, in silenced chromatin and in transcribed DNA, confirming that photolyase is a sensitive tool to study chromatin structure and dynamics in living cells. Interestingly, however, nucleosomes in repressed promoters showed no obvious indication of an altered stability that would prevent or facilitate DNA accessibility, although different histone composition and modification states of histones may suggest so (Santisteban et al, 2000; Mellor, 2005; Zhang et al, 2005). To provide a more detailed picture on differential accessibility and chromatin dynamics in the yeast genome, it will be important to extend our studies on other regions that differ with respect to composition and function.
How fast proteins can bind to DNA in chromatin depends on several parameters, including their size and conformation, their affinity for DNA, and whether or not they are complexed with other proteins before binding to DNA. Yeast photolyase is a monomeric protein with a molecular weight of 66 291 and contacts a 6–8-bp region around the dimer and strongly discriminates between binding of undamaged and damaged DNA (Baer and Sancar, 1989; Sancar, 2006). From this perspective, photolyase results can help to evaluate how similar proteins involved in other DNA-dependent processes could access target sites in chromatin. Extrapolating from our observations, it is conceivable that live-cell imaging experiments with various transcription factors, hormone receptors, and DNA-repair enzymes indeed report productive interactions with nucleosomal target sites (Hager et al, 2004; Mone et al, 2004).
In contrast to overexpressed photolyase, natural amounts of photolyase (about 272–688 molecules/cell (Yasui and Laskowski, 1975; Ghaemmaghami et al, 2003)) and other less-abundant proteins or protein complexes might profit from remodeling events in the genome. A few repair studies on histone acetylation point in that direction. Treatment of human cells with butyrate, a histone deacetylase inhibitor, resulted in hyperacetylation of histones and a stimulation of NER (Smerdon et al, 1982). In yeast, an increase of histone H3 acetylation several minutes after UV irradiation correlated with an enhanced accessibility of nucleosomal DNA for a restriction enzyme in the repressed MFA2 promoter (Yu et al, 2005). Deletion of the Gcn5 histone acetyl transferase resulted in a marked reduction of photoreactivation and NER in the MFA2 gene, but less so in RPB2, whereas no detectable defect was seen for repair of the genome overall (Teng et al, 2002). Gcn5 was also required for efficient NER in the repressed MFA2 promoter (Yu et al, 2005). Thus, chromatin modifications appear to play a role at some loci under conditions where repair proteins are limiting, but it remains unclear how remodeling activities are targeted to the damage site or to particular regions of the genome. Interestingly, we noticed a slightly enhanced photoreactivation when cells were incubated in the dark after UV-damage formation (Figure 4). It is conceivable that this experiment detects enhanced accessibility of chromatin owing to damage-induced chromatin modifications.
In conclusion, the rapid repair of UV lesions by photolyase sheds light on the intrinsic properties of nucleosomes that regulate DNA accessibility and impacts our view on the dynamic nature of structurally and functionally diverse chromatin regions in living cells.
Materials and methods
Yeast strains
The strains were constructed in a W303.1a background. ABY19 (MATa ade2-1 ura3-1 his3-11,15 trp1-1 leu2-3,112 can1-100 phr1Δ∷kanMX rad1Δ∷hisG YRpTRURAP YEp423GPDYPHR), ABY07 (MATa ade2-1 ura3-1 his3-11,15 trp1-1 leu2-3,112 can1-100 phr1Δ∷kanMX rad1Δ∷hisG YRpTRURAP). PHR1 and RAD1 were deleted using standard procedures. YRpTRURAP was described (Thoma, 1986). The overexpression plasmid YEp423GPDYPHR was generated by PCR amplification of the yeast PHR1 ORF and insertion in an overexpression vector containing the GPD promoter (p423GPD, ATCC No. 87355).
UV irradiation and repair
Irradiation and repair was carried out as described (Suter et al, 1997) with modifications. Cells were grown in 6 l of SD (synthetic minimal medium, 0.67% yeast nitrogen base without amino acids, 2% dextrose) supplemented with 20 mg/l adenine, 20 mg/l tryptophane, 30 mg/l leucine (and for ABY07 20 mg/l histidine) to a density of about 1.5 × 107 cells/ml, harvested, and resuspended in SD to a density of about 3 × 107 cells/ml. For each data point, 250 ml aliquots were (i) irradiated with 150 J/m2 UV light using germicidal lamps (Sylvania G15 T8 bulbs) for 11 s, (ii) adenine and the appropriate amino acids were added and (iii) the samples were transferred to the PR chamber and (iv) photoreactivated using Sylvania F15 T8/BLB bulbs (emission peak, 366 nm) at 1.5 mW/cm2 for different times. (v) The cells were chilled on ice. (vi) DNA was purified using QIAGEN Genomic Tips 500/G. All steps from UV irradiation (i) to DNA extraction (vi) were carried out at room temperature and in yellow light (Sylvania GE Gold fluorescent light). Dark incubation, which refers to the time between the end of damage formation and the beginning of PR (iv), was <10 s.
Analysis of CPDs by indirect end labeling
CPDs were mapped by indirect end labeling and quantified as described (Suter et al, 1997; Capiaghi et al, 2004). This method allows correlating CPD distribution with positioned nucleosomes, linker DNA, and nuclease-sensitive regions (compare Figures 1, 2 and 3). Individual bands are resolved within 10–50 bp at the lower and upper part of the gel, respectively (Thoma et al, 1984). DNA was cut with the appropriate restriction enzymes (see figure legends) and aliquots were cut at CPDs with T4 endonuclease V (T4-EndoV; Epicentre) or mock treated. The DNA was electrophoresed in 1.5% alkaline agarose gels, blotted to Zeta-Probe GT Genomic membranes, and hybridized with radioactively labeled strand-specific DNA probes (figure legends). Primers to generate DNA templates and probes that hybridize to the top- and bottom strand, respectively, were: YRpTRURAP probe 1 (1036-5′GCA AGC CGC AAA CTT TCA CCA ATG GAC CAG; 1037-5′GAG GGC CAA GAG GGA GGG CAT TGG TGA C); GAL1–10 probe 1 (1165-5′GAT TAC CTT TAT TCG TGC TCG; 1164-5′GCG GTG AGG AAG ATC ATG C); GAL1–10 probe 2 (954-5′CCG CCG AGT ACA TGC TGA TAG ATA ATG A; 953-5′CGC ACC ATA ATC TCC GTA CCC TCA ATA G); PHO5 probe 1 (980-5′tatc gaattc GGA ACT CCA GCA TCG TGC GCA AAT ATC, 981-5′tagt ggatcc CAA TCC ATT AAA ATA GGC CCA AGA AAT AGC); SUC2-probe 1 (977-5′tatc gaattc GTA GCA TAC CGA CTG GTA ATT TGC TCA C; 976-5′tagt ggatcc CGC AAC AAC CTA TAA TTG AGT TAA GTG CC); CEN14 (989-5′gctgGG AAG AAG TAA AGA GAA TAA TCC; 988-5′ccgcGC TTG GTA TGG TGA AAA AGA GG); HMLα (1001-5′gtccgaattcAT AAG GTA CAG TGT TCA TG; 1002-5′gtacggatccTC AAC ATG AAA GCC CGA C); HMRa (999-5′gtccgaattcAC CGG TTG AAT AAA CCT GG; 1064-5′GTC AAG CGC AAA TCC GA); MATa (997-5′gtccgaattcAA TCA TGC TTT TAG AAG GTG G: 998-5′gta cgg atccCA ACT CAA TAT CAT CCT CAC T). Small letters indicate extensions used for subcloning.
Quantification
The membranes were analyzed and quantified with a PhosphorImager (Molecular Dynamics). The positions of DNA lesions were determined using a molecular size marker (256-bp intervals; Thoma et al, 1984). The CPD content was calculated using the Poisson expression, –ln (RFa/RFb), where RFa and RFb represent the signal intensities of the intact restriction fragment of the T4-EndoV- and mock-treated DNA, respectively. Region-specific damage was calculated as the signal of that region in the T4-EndoV-treated DNA and divided by the signal of the whole lane. The corresponding signal of the mock-treated DNA was subtracted as background. Strand-specific probes were generated by primer extension using genomic DNA fragments as templates. The initial damage varied in individual UV experiments between 0.30±0.04 (mean and standard deviation of six restriction fragments: top and bottom strand of GAL10-RL, YRpTRURAP, GAL1–10-VV), 0.32±0.06 (mean and standard deviation of 10 restriction fragments: top and bottom strand of GAL10-RL, YRpTRURAP, GAL1–10-VV, PHO5-NS, SUC2-RR), 0.33±0.06 (mean and standard deviation of 10 restriction fragments: top and bottom strand of GAL10-RL, YRpTRURAP, GAL1–10-VV, PHO5-NS, SUC2-RR) and 0.45±0.06 CPDs/kb (mean and standard deviation of 14 restriction fragments: top and bottom strand of GAL1–10-VV, PHO5-NS, SUC2-RR, CEN14-HH, MATa-HH, HML-HH, HMR-HH). The overall average of the initial damage generated with 150 J/m2 was approximately 0.35 CPDs/kb (mean of the four experiments listed above). To allow direct comparison between different regions, repair curves were calculated as the fraction of CPDs removed versus repair time. The initial damage was set as 0% repair.
Chromatin analysis
Chromatin isolation and digestion with micrococcal nuclease digestion was carried out as described (Capiaghi et al, 2004). The cutting sites were analyzed by indirect end labeling as described for mapping CPDs, except that a neutral agarose gel was used for Figure 3A.
Acknowledgments
We thank U Suter for continuous support, S Schalbetter and S Wunderlin for assistance, C Capiaghi, M Fink, R Hahn, M Lopes, N Mantei, and J Sogo for discussions and comments on the manuscript. This research was supported by Grant No. 3100AO-102152 of the Swiss National Science Foundation and the ETH-Research Grants TH37/00-4 and TH41/05-2 (to FT).
References
- Baer M, Sancar GB (1989) Photolyases from Saccharomyces cerevisiae and Escherichia coli recognize common binding determinants in DNA containing pyrimidine dimers. Mol Cell Biol 9: 4777–4788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bedoyan J, Gupta R, Thoma F, Smerdon MJ (1992) Transcription, nucleosome stability, and DNA repair in a yeast minichromosome. J Biol Chem 267: 5996–6005 [PubMed] [Google Scholar]
- Boeger H, Griesenbeck J, Strattan JS, Kornberg RD (2004) Removal of promoter nucleosomes by disassembly rather than sliding in vivo. Mol Cell 14: 667–673 [DOI] [PubMed] [Google Scholar]
- Brand M, Moggs JG, Oulad-Abdelghani M, Lejeune F, Dilworth FJ, Stevenin J, Almouzni G, Tora L (2001) UV-damaged DNA-binding protein in the TFTC complex links DNA damage recognition to nucleosome acetylation. EMBO J 20: 3187–3196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bustin M, Catez F, Lim JH (2005) The dynamics of histone H1 function in chromatin. Mol Cell 17: 617–620 [DOI] [PubMed] [Google Scholar]
- Capiaghi C, Ho TV, Thoma F (2004) Kinetochores prevent repair of UV damage in Saccharomyces cerevisiae centromeres. Mol Cell Biol 24: 6907–6918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavalli G, Thoma F (1993) Chromatin transitions during activation and repression of galactose-regulated genes in yeast. EMBO J 12: 4603–4613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Downey M, Durocher D (2006) Chromatin and DNA repair: the benefits of relaxation. Nat Cell Biol 8: 9–10 [DOI] [PubMed] [Google Scholar]
- Downs JA, Allard S, Jobin-Robitaille O, Javaheri A, Auger A, Bouchard N, Kron SJ, Jackson SP, Cote J (2004) Binding of chromatin-modifying activities to phosphorylated histone H2A at DNA damage sites. Mol Cell 16: 979–990 [DOI] [PubMed] [Google Scholar]
- Downs JA, Lowndes NF, Jackson SP (2000) A role for Saccharomyces cerevisiae histone H2A in DNA repair. Nature 408: 1001–1004 [DOI] [PubMed] [Google Scholar]
- Felsenfeld G, Groudine M (2003) Controlling the double helix. Nature 421: 448–453 [DOI] [PubMed] [Google Scholar]
- Flaus A, Owen-Hughes T (2004) Mechanisms for ATP-dependent chromatin remodelling: farewell to the tuna-can octamer? Curr Opin Genet Dev 14: 165–173 [DOI] [PubMed] [Google Scholar]
- Friedberg EC (2003) DNA damage and repair. Nature 421: 436–440 [DOI] [PubMed] [Google Scholar]
- Gaillard H, Fitzgerald DJ, Smith CL, Peterson CL, Richmond TJ, Thoma F (2003) Chromatin remodeling activities act on UV-damaged nucleosomes and modulate DNA damage accessibility to photolyase. J Biol Chem 278: 17655–17663 [DOI] [PubMed] [Google Scholar]
- Gale JM, Nissen KA, Smerdon MJ (1987) UV-induced formation of pyrimidine dimers in nucleosome core DNA is strongly modulated with a period of 10.3 bases. Proc Natl Acad Sci USA 84: 6644–6648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghaemmaghami S, Huh WK, Bower K, Howson RW, Belle A, Dephoure N, O'Shea EK, Weissman JS (2003) Global analysis of protein expression in yeast. Nature 425: 737–741 [DOI] [PubMed] [Google Scholar]
- Gong F, Kwon Y, Smerdon MJ (2005) Nucleotide excision repair in chromatin and the right of entry. DNA Repair (Amst) 4: 884–896 [DOI] [PubMed] [Google Scholar]
- Gontijo AM, Green CM, Almouzni G (2003) Repairing DNA damage in chromatin. Biochimie 85: 1133–1147 [DOI] [PubMed] [Google Scholar]
- Green CM, Almouzni G (2003) Local action of the chromatin assembly factor CAF-1 at sites of nucleotide excision repair in vivo. EMBO J 22: 5163–5174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hager GL, Nagaich AK, Johnson TA, Walker DA, John S (2004) Dynamics of nuclear receptor movement and transcription. Biochim Biophys Acta 1677: 46–51 [DOI] [PubMed] [Google Scholar]
- Hirschhorn JN, Brown SA, Clark CD, Winston F (1992) Evidence that SNF2/SWI2 and SNF5 activate transcription in yeast by altering chromatin structure. Gene Dev 6: 2288–2298 [DOI] [PubMed] [Google Scholar]
- Ho Y, Gruhler A, Heilbut A, Bader GD, Moore L, Adams SL, Millar A, Taylor P, Bennett K, Boutilier K, Yang L, Wolting C, Donaldson I, Schandorff S, Shewnarane J, Vo M, Taggart J, Goudreault M, Muskat B, Alfarano C, Dewar D, Lin Z, Michalickova K, Willems AR, Sassi H, Nielsen PA, Rasmussen KJ, Andersen JR, Johansen LE, Hansen LH, Jespersen H, Podtelejnikov A, Nielsen E, Crawford J, Poulsen V, Sorensen BD, Matthiesen J, Hendrickson RC, Gleeson F, Pawson T, Moran MF, Durocher D, Mann M, Hogue CW, Figeys D, Tyers M (2002) Systematic identification of protein complexes in Saccharomyces cerevisiae by mass spectrometry. Nature 415: 180–183 [DOI] [PubMed] [Google Scholar]
- Kapetanaki MG, Guerrero-Santoro J, Bisi DC, Hsieh CL, Rapic-Otrin V, Levine AS (2006) The DDB1-CUL4ADDB2 ubiquitin ligase is deficient in xeroderma pigmentosum group E and targets histone H2A at UV-damaged DNA sites. Proc Natl Acad Sci USA 103: 2588–2593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura H (2005) Histone dynamics in living cells revealed by photobleaching. DNA Repair (Amst) 4: 939–950 [DOI] [PubMed] [Google Scholar]
- Korber P, Luckenbach T, Blaschke D, Horz W (2004) Evidence for histone eviction in trans upon induction of the yeast PHO5 promoter. Mol Cell Biol 24: 10965–10974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kornberg RD, Lorch Y (1999) Twenty-five years of the nucleosome, fundamental particle of the eukaryote chromosome. Cell 98: 285–294 [DOI] [PubMed] [Google Scholar]
- Kruhlak MJ, Celeste A, Dellaire G, Fernandez-Capetillo O, Muller WG, McNally JG, Bazett-Jones DP, Nussenzweig A (2006) Changes in chromatin structure and mobility in living cells at sites of DNA double-strand breaks. J Cell Biol 172: 823–834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li G, Levitus M, Bustamante C, Widom J (2005) Rapid spontaneous accessibility of nucleosomal DNA. Nat Struct Mol Biol 12: 46–53 [DOI] [PubMed] [Google Scholar]
- Li G, Widom J (2004) Nucleosomes facilitate their own invasion. Nat Struct Mol Biol 11: 763–769 [DOI] [PubMed] [Google Scholar]
- Livingstone-Zatchej M, Marcionelli R, Moller K, De Pril R, Thoma F (2003) Repair of UV lesions in silenced chromatin provides in vivo evidence for a compact chromatin structure. J Biol Chem 278: 37471–37479 [DOI] [PubMed] [Google Scholar]
- Livingstone-Zatchej M, Meier A, Suter B, Thoma F (1997) RNA polymerase II transcription inhibits DNA repair by photolyase in the transcribed strand of active yeast genes. Nucleic Acids Res 25: 3795–3800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lohr D (1997) Nucleosome transactions on the promoters of the yeast GAL and PHO genes. J Biol Chem 272: 26795–26798 [DOI] [PubMed] [Google Scholar]
- Mees A, Klar T, Gnau P, Hennecke U, Eker AP, Carell T, Essen LO (2004) Crystal structure of a photolyase bound to a CPD-like DNA lesion after in situ repair. Science 306: 1789–1793 [DOI] [PubMed] [Google Scholar]
- Mellor J (2005) The dynamics of chromatin remodeling at promoters. Mol Cell 19: 147–157 [DOI] [PubMed] [Google Scholar]
- Metivier R, Reid G, Gannon F (2006) Transcription in four dimensions: nuclear receptor-directed initiation of gene expression. EMBO Rep 7: 161–167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mone MJ, Bernas T, Dinant C, Goedvree FA, Manders EM, Volker M, Houtsmuller AB, Hoeijmakers JH, Vermeulen W, van Driel R (2004) In vivo dynamics of chromatin-associated complex formation in mammalian nucleotide excision repair. Proc Natl Acad Sci USA 101: 15933–15937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinke H, Horz W (2004) Anatomy of a hypersensitive site. Biochim Biophys Acta 1677: 24–29 [DOI] [PubMed] [Google Scholar]
- Richards EJ, Elgin SC (2002) Epigenetic codes for heterochromatin formation and silencing: rounding up the usual suspects. Cell 108: 489–500 [DOI] [PubMed] [Google Scholar]
- Richmond TJ, Davey CA (2003) The structure of DNA in the nucleosome core. Nature 423: 145–150 [DOI] [PubMed] [Google Scholar]
- Rusche LN, Kirchmaier AL, Rine J (2003) The establishment, inheritance, and function of silenced chromatin in Saccharomyces cerevisiae. Annu Rev Biochem 72: 481–516 [DOI] [PubMed] [Google Scholar]
- Sancar A (2003) Structure and function of DNA photolyase and cryptochrome blue-light photoreceptors. Chem Rev 103: 2203–2238 [DOI] [PubMed] [Google Scholar]
- Sancar GB (2006) Damage recognition by DNA photolyase. In: Damage Recognition DNA, Siede W, Kow YW, Doetsch PW (ed), pp. 95–109. New York: Taylor and Francis Group [Google Scholar]
- Santisteban MS, Kalashnikova T, Smith MM (2000) Histone H2A. Z regulates transcription and is partially redundant with nucleosome remodeling complexes. Cell 103: 411–422 [DOI] [PubMed] [Google Scholar]
- Schieferstein U, Thoma F (1996) Modulation of cyclobutane pyrimidine dimer formation in a positioned nucleosome containing poly(dA.dT) tracts. Biochemistry 35: 7705–7714 [DOI] [PubMed] [Google Scholar]
- Schieferstein U, Thoma F (1998) Site-specific repair of cyclobutane pyrimidine dimers in a positioned nucleosome by photolyase and T4 endonuclease V in vitro. EMBO J 17: 306–316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sebastian J, Kraus B, Sancar GB (1990) Expression of the yeast PHR1 gene is induced by DNA-damaging agents. Mol Cell Biol 10: 4630–4637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smerdon MJ, Lan SY, Calza RE, Reeves R (1982) Sodium butyrate stimulates DNA repair in UV-irradiated normal and xeroderma pigmentosum human fibroblasts. J Biol Chem 257: 13441–13447 [PubMed] [Google Scholar]
- Smerdon MJ, Lieberman MW (1978) Nucleosome rearrangement in human chromatin during UV-induced DNA-repair synthesis. Proc Natl Acad Sci USA 75: 4238–4241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suter B, Livingstone-Zatchej M, Thoma F (1997) Chromatin structure modulates DNA repair by photolyase in vivo. EMBO J 16: 2150–2160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suter B, Schnappauf G, Thoma F (2000a) Poly(dA.dT) sequences exist as rigid DNA structures in nucleosome-free yeast promoters in vivo. Nucleic Acids Res 28: 4083–4089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suter B, Thoma F (2002) DNA-repair by photolyase reveals dynamic properties of nucleosome positioning in vivo. J Mol Biol 319: 395–406 [DOI] [PubMed] [Google Scholar]
- Suter B, Wellinger RE, Thoma F (2000b) DNA repair in a yeast origin of replication: contributions of photolyase and nucleotide excision repair. Nucleic Acids Res 28: 2060–2068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teng Y, Yu Y, Waters R (2002) The Saccharomyces cerevisiae histone acetyltransferase Gcn5 has a role in the photoreactivation and nucleotide excision repair of UV-induced cyclobutane pyrimidine dimers in the MFA2 Gene. J Mol Biol 316: 489–499 [DOI] [PubMed] [Google Scholar]
- Terleth C, van Sluis CA, van de Putte P (1989) Differential repair of UV damage in Saccharomyces cerevisiae. Nucleic Acids Res 17: 4433–4439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thoma F (1986) Protein–DNA interactions and nuclease-sensitive regions determine nucleosome positions on yeast plasmid chromatin. J Mol Biol 190: 177–190 [DOI] [PubMed] [Google Scholar]
- Thoma F (1999) Light and dark in chromatin repair: repair of UV-induced DNA lesions by photolyase and nucleotide excision repair. EMBO J 18: 6585–6598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thoma F (2005) Repair of UV lesions in nucleosomes-intrinsic properties and remodeling. DNA Repair (Amst) 4: 855–869 [DOI] [PubMed] [Google Scholar]
- Thoma F, Bergman LW, Simpson RT (1984) Nuclease digestion of circular TRP1ARS1 chromatin reveals positioned nucleosomes separated by nuclease-sensitive regions. J Mol Biol 177: 715–733 [DOI] [PubMed] [Google Scholar]
- Thomas JO, Furber V (1976) Yeast chromatin structure. FEBS Lett 66: 274–280 [DOI] [PubMed] [Google Scholar]
- Tomschik M, Zheng H, van Holde K, Zlatanova J, Leuba SH (2005) Fast, long-range, reversible conformational fluctuations in nucleosomes revealed by single-pair fluorescence resonance energy transfer. Proc Natl Acad Sci USA 102: 3278–3283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Attikum H, Gasser SM (2005) The histone code at DNA breaks: a guide to repair? Nat Rev Mol Cell Biol 6: 757–765 [DOI] [PubMed] [Google Scholar]
- Wellinger RE, Thoma F (1997) Nucleosome structure and positioning modulate nucleotide excision repair in the non-transcribed strand of an active gene. EMBO J 16: 5046–5056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasui A, Laskowski W (1975) Determination of the number of photoreactivating enzyme molecules per haploid Saccharomyces cells. Int J Radiat Biol Relat Stud Phys Chem Med 28: 511–518 [DOI] [PubMed] [Google Scholar]
- Yasui A, Yajima H, Kobayashi T, Eker AP, Oikawa A (1992) Mitochondrial DNA repair by photolyase. Mutat Res 273: 231–236 [DOI] [PubMed] [Google Scholar]
- Yu Y, Teng Y, Liu H, Reed SH, Waters R (2005) UV irradiation stimulates histone acetylation and chromatin remodeling at a repressed yeast locus. Proc Natl Acad Sci USA 102: 8650–8655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Roberts DN, Cairns BR (2005) Genome-wide dynamics of Htz1, a histone H2A variant that poises repressed/basal promoters for activation through histone loss. Cell 123: 219–231 [DOI] [PMC free article] [PubMed] [Google Scholar]