

Abstract
Escherichia coli is generally resistant to H2O2, with >75% of cells surviving a 3-min challenge with 2.5 mM H2O2. However, when cells were cultured with poor sulfur sources and then exposed to cystine, they transiently exhibited a greatly increased susceptibility to H2O2, with <1% surviving the challenge. Cell death was due to an unusually rapid rate of DNA damage, as indicated by their filamentation, a high rate of mutation among the survivors, and DNA lesions by a direct assay. Cell-permeable iron chelators eliminated sensitivity, indicating that intracellular free iron mediated the conversion of H2O2 into a hydroxyl radical, the direct effector of DNA damage. The cystine treatment caused a temporary loss of cysteine homeostasis, with intracellular pools increasing about eightfold. In vitro analysis demonstrated that cysteine reduces ferric iron with exceptional speed. This action permits free iron to redox cycle rapidly in the presence of H2O2, thereby augmenting the rate at which hydroxyl radicals are formed. During routine growth, cells maintain small cysteine pools, and cysteine is not a major contributor to DNA damage. Thus, the homeostatic control of cysteine levels is important in conferring resistance to oxidants. More generally, this study provides a new example of a situation in which the vulnerability of cells to oxidative DNA damage is strongly affected by their physiological state.
H2O2 is formed in bacteria when molecular oxygen oxidizes redox enzymes (32, 33). Endogenously produced H2O2 can slowly oxidize proteins, forming methionine sulfoxide and carbonyl adducts (14, 35). The more significant cellular damage, however, is likely to occur to DNA. Hydrogen peroxide itself cannot directly oxidize DNA, but it reacts very rapidly with transition metals to form a hydroxyl radical (·OH). Hydroxyl radicals attack both sugar and base moieties, leading to sugar fragmentation, strand scission, and base adducts (18). The death of Escherichia coli exposed to moderate doses of H2O2 is primarily due to DNA damage (17, 20).
Oxidative DNA damage occurs in three steps:
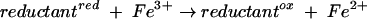 |
(1) |
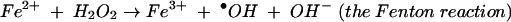 |
(2) |
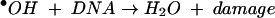 |
(3) |
where reductantred and reductantox are reduced and oxidized reductant, respectively. Both Cu+ and Fe2+ are capable of reacting with H2O2 to form ·OH in vitro. However, Fe2+ is evidently the coreactant in vivo, since cell-permeable iron chelators protect DNA from exogenous H2O2 (21). E. coli contains a small (ca. 20 μM) pool of “free” iron that is not integrated into proteins, and it is this iron that catalyzes the Fenton reaction. Indeed, mutants that have high levels of intracellular free iron are especially vulnerable to oxidative DNA damage (24, 29, 51).
What remains unclear is the identity of the iron reductant (equation 1). When cells are exposed to millimolar concentrations of H2O2, DNA damage occurs continuously for up to 30 min, even though any preexisting free Fe2+ should be oxidized within a few seconds (k = 76 M−1 s−1) (52). Thus, the rereduction of oxidized iron is an important part of the damage process. In fact, when reducing equivalents are depleted by carbon restriction, E. coli becomes resistant to H2O2, suggesting that in some situations the rate of iron reduction can determine the pace of oxidative DNA damage (20).
Superoxide sufficed as the iron reductant in in vitro model systems (10, 28). However, subsequent reports have shown that O2− is not an important reductant of free iron in vivo. First, given the rate constant for iron reduction by O2− (105 M−1 s−1 for Fe3+-ATP) (7) and the concentration of O2− in wild-type cells (10−10 M) (19), the half time for iron reduction would approach 20 h, far too long to support the rate of DNA damage that is observed. Second, even anaerobic cells, which are devoid of O2−, suffer rapid DNA damage when H2O2 is added (23).
These results prompted a search for other biomolecules that might reduce free iron in vivo. Both thiols (10−3 M) and NAD(P)H (10−3 M) (44, 45) can transfer electrons to free iron in vitro and are far more abundant in vivo than is O2−. Free reduced flavins are also efficient reductants of iron, and in fact, their accumulation in respiration-inhibited cells causes a large increase in vulnerability to DNA damage (53). However, the basal sensitivity to H2O2 persists in flavin reductase-deficient cells, indicating that under most conditions iron reduction occurs by some other pathway.
In this work we sought an explanation for the observation that cystine exposure can transiently potentiate the bactericidal effect of H2O2 (6, 9, 11, 12). We found that when cysteine homeostasis is disrupted, intracellular cysteine acts as an adventitious reductant of free iron and thereby promotes oxidative DNA damage.
MATERIALS AND METHODS
Chemicals, enzymes, and media.
Hydrogen peroxide (30% [wt/vol]), glutathione (GSH), d- and l-cystine, l-cysteine, homocystine, sodium sulfide, sodium sulfite, sodium sulfate, sodium thiosulfate, dithiothreitol (DTT), djenkolic acid, 5,5′-dithio-bis(2-nitrobenzoic acid) (DTNB), trimethoprim (TMP), thymine, ferric chloride, ferric sulfate, ferrous sulfate, dipyridyl, desferrioxamine mesylate, EDTA, 5-sulfosalicylic acid (5-SSA), N-ethylmorpholine, agarose, and ethidium bromide were purchased from Sigma. Trichloroacetic acid (TCA) was from EM Science. Beef liver catalase and GSSG (oxidized form of GSH) were from Roche. Tris, glucose, and glycerol were purchased from Fisher.
Luria broth (LB) contained 10 g of Bacto Tryptone, 5 g of yeast extract, and 5 g of sodium chloride (all per liter) (34). Minimal medium contained minimal A salts (34) (except that ammonium sulfate was replaced by ammonium chloride), 0.2% glucose, 1 mM MgCl2, a 0.5 mM concentration (total) of 18 amino acids (omitting cysteine and methionine), and 5 mg of thiamine per liter. Sulfur sources (0.5 mM) were added as indicated. Media were supplemented with the following antibiotics when selection was required: ampicillin (100 μg/ml), chloramphenicol (20 μg/ml), kanamycin (40 μg/ml), and tetracycline (10 μg/ml). Water was purified with a Labconco Water Pro PS system.
Strains and culture conditions.
All E. coli strains and plasmids used in this study are listed in Table 1. Mutations were introduced into strains by P1 transduction (34). Mutations in gshA were confirmed by measurements of intracellular GSH levels by the DTNB-GSSG reductase recycling assay (2). Mutations in cysA were detected by screening bacteria for the inability to grow on minimal medium containing sulfate. The optical density at 600 nm (OD600) was measured for each culture. Cultures were grown aerobically for at least six generations to an OD600 of 0.1 to 0.2.
TABLE 1.
E. coli strains and plasmids used in this study
| Strain or plasmid | Genotype | Source or reference |
|---|---|---|
| Strains | ||
| AB1157 | F−thr-1 leuB6 proA2 his-4 thi-1 argE2 lacY1 galK2 rpsL supE44 ara-14 xyl-15 mtl-1 tsx-33 | 4 |
| ALN2 | As AN387 plus fre::kan | 53 |
| ALN34 | As AN387 plus polA12(Ts) | Lab collection |
| AN387 | F−rpsL gal | 3 |
| BW25113 | lacIqrrnB ΔlacZ hsdR514 ΔaraBAD ΔrhaBAD | William Metcalf |
| BW9091 | As AB1157 plus xth-1 | Stuart Linn |
| CAG18468 | nupC510::Tn10 λ−rph-1 | E. coli Genetic Stock Center |
| CAG18475 | metC162::Tn10 λ−rph-1 | E. coli Genetic Stock Center |
| DHB4 | F′ lac-pro lacIqIΔ(ara-leu)7697 araD139 ΔlacX74 galE galK rpsL phoR ΔphoA PvuII | 43 |
| JF2201 | As AB1157 plus gshB::kan ΔlacZYA pro | James Fuch |
| JI367 | As MG1655 plus ΔkatG katE12::Tn10 | 46 |
| JM2314 | cysA97 araD139 Δ(argF-lac)169 flhB5301 Δ(his-gnd)296 relA1 fda-2(Ts) galP79 rpsL150 deoC1 λ− | E. coli Genetic Stock Center |
| JTG10 | As AB1157 plus gshA::kan | 16 |
| MC4100 | araD139 Δ(argF-lac)U169 rpsL150 relA1 flhB5301 deoC1 ptsF25 rbsR | Lab collection |
| MC4100Δ299 | As MC4100 plus ORF299::kan (=ydeD::kan) | Tobias Dassler |
| MG1655 | F wild type | E. coli Genetic Stock Center |
| OD503 | As AB1157 plus ΔsufS | Lab collection |
| PK4331 | As MG1655 plus iscS::kan | Patricia Kiley |
| RL165 | cysK511 thr-1 leuB6 fhuA2 lacY1 supE44 gal-6 λ−trp-1 hisG1(Fs) rpsL9 malT1(λr) xylA7 mtlA2 ΔargH1 thi-1 | E. coli Genetic Stock Center |
| SP31 | As AB1157 plus xth-1 gshA | P1(JTG10) × BW9091 |
| SP32 | As CAG18468 plus cysA97 | P1(CAG18468) × JM2314 |
| SP34 | As AB1157 plus cysA97. . .nupC::Tn10 gshA::kan xth-1 | P1(SP32) × SP31 |
| SP53 | As BW25113 plus cysB::cam | This study |
| SP55 | As AB1157 plus ORF299::kan (=ydeD::kan) | This study |
| WP840 | As DHB4 plus gor522. . .mini-Tn10TC | Jon Beckwith |
| Plasmids | ||
| pACYC184 | Cmr Tcr p15A origin | Abigail Salyers |
| pKD3 | Cmr AmroriRγ origin | William Metcalf |
| pKD46 | AmrrepA101(Ts) oriR101 origin | William Metcalf |
H2O2 sensitivity.
Cells were grown in minimal medium containing glucose and a 0.5 mM concentration of the indicated sulfur source (usually sulfate). When the culture reached an OD600 of 0.1 to 0.2, a second sulfur source (typically cystine) was added. After 3 min, the culture was challenged with 2.5 mM H2O2. At timed intervals, 20 μl of the culture was removed and diluted in LB containing catalase (1,300 U/ml of LB) to stop further killing. The diluted bacteria were mixed in LB soft agar and spread on LB plates. The surviving cells were counted after overnight incubation at 37°C.
Measurement of DNA damage by quantitative PCR (qPCR) assays.
Total genomic DNA was isolated from 5 ml of culture using a DNeasy Tissue kit (Qiagen). The extracted DNA was quantified using a PicoGreen dsDNA quantitation reagent (Molecular Probes) with lambda DNA as a standard. For primers, 10-kb fragments near fumC regions were used. Primer sequences were as follows: 5′-GGCGTGAACTCGCAAAATATTACGATTCAGCC (forward primer) and 5′-AGGGCAACGGAACACCCGCCCAGAGCATAACC (reverse primer). PCR was performed using an Expand Long Template PCR system (Roche). The 25-μl PCR mixture contained 0.05 to 0.5 ng of genomic DNA as a template, a 300 nM concentration (each) of the two primers, 350 μM each of the four deoxynucleoside triphosphates (Promega), 10× PCR buffer with 1.75 mM MgCl2, and 0.75 μl of DNA polymerase mix. Thermal cycling was performed with a PCR Express (Hybaid) cycler. The genomic DNA was initially denatured for 1 min at 94°C, and then the DNA was subjected to 25 cycles of PCR, with 1 cycle consisting of denaturation at 94°C for 15 s and annealing and extension at 68°C for 12 min. A final extension step at 72°C was performed for 10 min at the completion of the profile. PCR products were separated by 1% agarose gel electrophoresis, stained with ethidium bromide, scanned with a PhosphorImager (model 425; Molecular Dynamics), and quantified with ImageQuant software (Molecular Dynamics).
Mutagenesis rate.
To measure the rate of mutagenesis, Thy− cells were selected with TMP. TMP is a dihydrofolate reductase inhibitor that depresses the growth of Thy+ cells but not that of Thy− cells on plates supplemented with thymine (1). Two-hundred fifty microliters of culture was mixed with F-top agar (8 g of agar, 8 g of NaCl [both per liter]) supplemented with thymine (1 mg/ml) and TMP (0.1 mg/ml), and the culture was spread on LB plates. To determine the total number of viable cells, the culture was mixed with F-top agar supplemented with thymine only. The rate of mutagenesis was calculated by dividing the total number of Thy− cells by the number of viable cells.
Intracellular free iron measurement by EPR.
Cultures were grown in 1 liter of minimal medium containing sulfate. When the OD600 reached 0.1 to 0.2, cystine was added. After 3 min, cells were harvested, and the pellets were resuspended in 9 ml of the same medium. One milliliter of 0.2 M desferrioxamine mesylate was added, and the culture was incubated at 37°C for 15 min with shaking. The cells were then centrifuged and washed twice with 5 ml of cold 20 mM Tris-Cl (pH 7.4) buffer. The cells were finally resuspended in 400 μl of the cold Tris buffer containing 10% glycerol. Two hundred microliters of the cell suspension was loaded into a quartz electron paramagnetic resonance (EPR) tube, immediately frozen in dry ice, and stored at −80°C until analysis. Ferric sulfate standards were mixed with desferrioxamine mesylate and prepared in the same Tris buffer containing glycerol. The concentration of iron in the standard samples was determined from the value ɛmM at 420 nm of 2.865 cm−1. The EPR signals were measured with a Varian Century E-112 X-band spectrophotometer equipped with a Varian TE102 cavity and temperature controller. The spectrometer settings were as follows: field center, 1,570 G; receiver gain, 3,200 G; field sweep, 400 G; modulation amplitude, 12.5 G; temperature, −125°C; and power, 30 mW. The measured EPR signals were converted to approximate intracellular concentrations by normalization to the cell density by using the following relationship: 1 ml of a culture of E. coli on minimal medium containing glucose at an OD600 of 1 comprises 0.57 μl of intracellular volume (19).
Construction of a cysB knockout mutant.
The cysB locus was disrupted by the method described by Datsenko and Wanner (13). Briefly, a chloramphenicol cassette region from pKD3 plasmid was amplified by PCR using the primers with cysB homology extensions. Primer sequences were as follows (the cysB extensions are underlined): 5′-CGCTATATTGTTGAGGTGGTCAATCATAACTGTAGGCTGGAGCTGCTTCG (forward primer) and 5′-AGAGCGCAATGCGACAGCCGCATCAACGACCATATGAATATCCTCCTTAG (reverse primer). The 1.1-kb PCR products were cleaned with a QIAquick PCR purification kit (Qiagen), and the E. coli BW25113 strain harboring pKD46 plasmid was transformed with the linear DNA by electroporation with a Gene Pulser apparatus (Bio-Rad Laboratories). Camr Amps transformants were selected at 37°C. The cysB disruption was verified by PCR and by the inability of mutants to grow in minimal medium containing sulfate.
Intracellular thiol measurement by HPLC.
GSH and other thiols react with monobromobimane (mBBr), a fluorescent dye. After the thiols were separated by high-pressure liquid chromatography (HPLC), the thiol-bimane derivatives can be quantified by fluorescence detection (38, 42). Samples were prepared under conditions similar to those described by Anderson (2). First, 25 ml of culture was harvested, and the cell pellets were resuspended in 250 μl of 5% 5-sulfosalicylic acid (5-SSA). The cell suspension was held on ice for 10 min. After the precipitates were removed by centrifugation, the supernatants were used for thiol derivatization. The derivatization reaction mixture contained 340 μl of water, 120 μl of the 5-SSA supernatants, 100 μl of 1 M N-ethylmorpholine, and 20 μl of 0.1 M mBBr (Calbiochem). After 20 min in the dark at room temperature, 20 μl of glacial acetic acid was added to decrease the pH. The samples were wrapped in foil to reduce the formation of mBBr degradation products and stored at −20°C until analysis.
HPLC separation was performed on a Beckman System Goldliquid chromatograph equipped with an Intelligent Fluorescence Detector (model FP-920; Jasco). The fluorescence detector was operated at an excitation wavelength of 400 nm and an emission wavelength of 475 nm. Twenty microliters of the derivatized sample was injected into a μBondapak C18 Cartridge column (4.6 by 250 mm) (particle size, 10 μm; Waters). Thiols were resolved by isocratic elution for 30 min at room temperature using 1 ml of solvent A (14.2% methanol-0.25% glacial acetic acid [pH 3.9]) per min. Between samples, the column was cleaned for 10 min with solvent B (90% methanol-0.25% glacial acetic acid [pH 3.9]) and reequilibrated with solvent A for 10 min. GSH and cysteine peaks were quantified by using a standard curve.
Determination of cellular acid-soluble thiols.
Total acid-soluble thiols were measured by the method of Lawley and Thatcher (27) with some modifications. Cystine-treated cultures were centrifuged and washed at room temperature with minimal medium containing sulfate and 1 mM EDTA. The cells were lysed by passage through a French pressure cell (SLM Aminco) and centrifuged. Proteins were precipitated from the supernatant fractions by the addition of cold TCA (final concentration of 5%). After 10 min on ice, the acidified samples were clarified by centrifugation, and 100 μl of the clear supernatants was added to 900 μl of a DTNB solution (200 μg/ml in 0.2 M sodium phosphate buffer [pH 7.6]). The absorption spectrum at 410 nm was measured immediately. GSH was used for the thiol standard, and the thiol content was normalized to the amount of total protein. The protein concentration was determined by a Coomassie protein assay reagent (Pierce) using egg albumin as a standard.
Iron reduction assays.
Iron reduction by thiols was performed in an anaerobic Coy chamber (85% N2, 10% H2, and 5% CO2). All reagents and stock solutions were prepared in anaerobic H2O. In a cuvette, 10 μM ferric chloride and 3 mM cysteine or GSH in 20 mM Tris-Cl buffer (pH 7.4) were mixed together. At each time point, aliquots were added to 300 μM Ferene to stop the reaction and to chelate the reduced iron. The cuvette was tightly capped and immediately taken to a spectrophotometer (model DU 640; Beckman) to measure the absorption spectrum at 562 nm.
DNA strand break assays.
DNA strand breaks were assayed by measuring the relaxation of supercoiled plasmid to an open circular form (36, 41). pACYC184 plasmid DNA was isolated from 9 ml of culture using a Qiaprep spin miniprep kit (Qiagen). The final DNA pellet was resolved in 200 μl of 50 mM TE (Tris-EDTA) buffer (pH 7.9), and the DNA concentration was determined by a PicoGreen dsDNA quantitation reagent (Molecular Probes). In an anaerobic Coy chamber, 33 ng of the pACYC184 plasmid in 3.5 mM NaHCO3 buffer (pH 7.2) with 10 μM FeCl3, 20 μM GSH or cysteine, and 50 μM H2O2 was added to the DNA. At each time interval, 2,600 U of catalase/20 μl of reaction mixture was added to stop the H2O2 reaction. A loading dye was mixed with the reaction mixture, and the sample was electrophoresed in a 1% agarose gel. The gel was stained with ethidium bromide, and the bands were scanned with a phosphorimager and quantified by Image Gauge software (Fuji).
RESULTS
Cystine treatment transiently sensitizes cells to H2O2.
E. coli AB1157 wild-type cells grown in minimal medium containing glucose with sulfate as the sole sulfur source were not particularly sensitive to H2O2, as >75% survived a 3-min challenge with 2.5 mM H2O2 (Fig. 1A). The same was true of cells grown with cystine as the sole sulfur source. However, when sulfate-grown cells were treated with cystine for 3 min, only about 0.4% survived the subsequent exposure to H2O2. Other E. coli K-12 strains, such as MG1655, AN387, and BW25113, demonstrated the same cystine-mediated H2O2 sensitivity (data not shown).
FIG. 1.

A cystine pulse transiently sensitizes cells to H2O2. (A) E. coli AB1157 (wild-type) cells were grown to log phase in minimal medium containing sulfate (circles) or cystine (squares). Cystine (0.5 mM) was added (solid symbols) or not added (open symbols) to the cells, and 3 min later, 2.5 mM H2O2 was added. At intervals, cells were diluted, and viability was determined by colony formation. (B) AB1157 cells were grown to log phase in medium containing sulfate. At time zero, cystine (0.5 mM) was added. At time points, aliquots were removed and challenged for 3 min with 2.5 mM H2O2.
The period of sensitivity after cystine addition was relatively brief. Over the subsequent 2 h, cells regained their typical resistance to H2O2 (Fig. 1B). In analyzing this phenomenon, we sought first to identify the mechanism of cell injury and then to explore its connection to the cystine treatment.
The cystine-mediated H2O2 killing effect results from DNA damage.
The lack of a shoulder (Fig. 1A) suggested that the killing may be due to DNA damage. Indeed, most of the “dead” cells filamented in the hours following the H2O2 exposure (data not shown). Filamentation commonly occurs in E. coli cells that have suffered extensive DNA damage but whose catabolic and biosynthetic pathways are functional (8).
qPCR assays confirmed that cystine-H2O2-treated cells suffered gross DNA damage. The qPCR method detects any lesions or strand breaks which block the progression of the PCR DNA polymerase (54, 55); when damaged DNA is used as a template, fewer PCR products are formed than when intact DNA is used. As shown in Fig. 2, cystine-H2O2 treatment reduced by 10-fold the amount of PCR product. Poisson analysis of the samples shown in Fig. 2 indicated approximately 2,900 blocking lesions per genome in the cystine-H2O2-treated cells compared to <290 in cells treated with H2O2 alone.
FIG. 2.

Cystine-H2O2 treatment generates abundant DNA damage. Log-phase E. coli AB1157 cells were treated with either 0.5 mM cystine, 2.5 mM H2O2, or cystine and H2O2, or left untreated as a control (con). Total genomic DNA was isolated, and qPCR was performed, using equivalent amounts of template DNA for each reaction mixture. PCR products were scanned, and the relative fluorescence was normalized to the untreated control. Values are the means and standard deviations (error bars) from three experiments.
The same result was obtained upon analysis when a polA(Ts) strain was challenged at the restrictive temperature. DNA polymerase I is required for most DNA repair pathways, so this result indicates that the effect of cystine is to quicken the process of DNA damage rather than to inhibit DNA repair.
Cystine-H2O2 treatment also caused a high rate of mutagenesis. The frequency of Thy+ to Thy− conversion increased approximately 100-fold (Fig. 3). All these data indicate that the toxic effect of the cystine-H2O2 treatment was due to an acceleration of DNA damage.
FIG. 3.

Cystine-H2O2 treatment accelerates mutagenesis. E. coli AB1157 cells were grown to log phase and exposed for 3 min to 0.5 mM cystine, 0.1 mM H2O2, or both cystine and H2O2 or left untreated as a control (con). After 3 min, catalase was added to scavenge H2O2, and cells were spread on LB plates containing thymine or thymine and TMP. Values are the means and standard deviations (error bars) from three experiments. Note that the scale is exponential.
The DNA damage occurs via the Fenton reaction.
The addition of cell-permeable iron chelators (dipyridyl or desferrioxamine) fully blocked the cell killing (Fig. 4), confirming that the Fenton reaction (equation 2) was involved in the cystine-mediated H2O2 killing.
FIG. 4.

Conditions that confer H2O2 sensitivity. Different E. coli strains and treatments were studied and are grouped in sets of bars. For bars 1 to 3, AB1157 cells were grown to the log phase and treated with iron chelators for 5 min (none [bar 1], 1 mM dipyridyl [bar 2], and 20 mM desferrioxamine [bar 3]) before the addition of 0.5 mM cystine and 2.5 mM H2O2. For bars 4 to 11, AB1157 cells were treated with a 0.5 mM concentration of an alternative sulfur species (cysteine [bar 4], homocystine [bar 5], sulfide [bar 6], thiosulfate [bar 7], sulfite [bar 8], GSH [bar 9], GSSG [bar 10], and DTT [bar 11],) instead of cystine. For bars 12 and 13, AB1157 cells were grown in minimal medium containing cystine until the cells reached early log phase, and then the cells were washed twice and suspended in minimal medium containing sulfate for 1 h. For bar 13, 20 μg of chloramphenicol per μl was present during the period of growth on sulfate. Cystine was then added, and the cells were challenged with H2O2 for 3 min. For bars 14 and 15, BW25113 and SP53 (cysB) cells were grown in minimal medium containing djenkolic acid and tested for cystine-mediated H2O2 sensitivity. JTG10 (gshA) cells (bar 16) and WP840 (gor) cells (bar 17) were grown in minimal medium containing sulfate and tested for cystine-mediated H2O2 sensitivity. Values are the means and standard deviations (error bars) from five samples.
Three substances are required for the Fenton reaction to occur: H2O2, iron, and an electron donor. We reasoned that the cystine treatment must increase the concentration of one of these substances to accelerate DNA damage. Under our experimental conditions, the intracellular concentration of H2O2 is approximately the same as the external concentration (47). Indeed, a katG katE catalase-deficient mutant was not more sensitive than its wild-type parent, and it also showed the sensitization after cystine exposure (data not shown). Thus, the cystine treatment did not affect damage by altering H2O2 levels.
The sensitization was also not mediated by an enlargement of the pool of free iron. The amount of intracellular free iron was determined by EPR. Cystine-treated E. coli AB1157 cells (34 μM free iron) showed a 1.4-fold-higher signal than untreated cells (24 μM). This moderate increase does not seem to be substantial enough to explain the much larger increase in the rates of DNA damage and cell death.
The third possibility was that Fenton chemistry might be accelerated by an increase in the reductant that recycles oxidized iron (equation 1). A similar phenomenon occurs when cellular respiration is inhibited by cyanide (53) or nitric oxide (A. N. Woodmansee and J. A. Imlay, unpublished data). The block causes electron flow to be diverted to free flavins, which then efficiently reduce free ferric iron. Flavin reductase is the enzyme responsible for the reduction of FAD; thus, to ascertain whether cystine promotes DNA damage in the same way, we looked at cystine-mediated H2O2 sensitivity in a flavin reductase-deficient strain (E. coli ALN2). This mutant also was still sensitive to cystine-H2O2 treatment (0.3% survival), indicating that the cystine effect occurred by a different pathway.
CysB-stimulated cystine transport may be required for H2O2 sensitivity.
In light of the above results, an alternative hypothesis was that cystine exposure might increase the amount of intracellular thiols. GSH, the predominant intracellular thiol, can reduce iron and drive the Fenton reaction in vitro (45). As a first step in testing this idea, the phenomenology of sensitization by cystine was more closely defined. A working hypothesis was that the growth on sulfate would activate the CysB protein (26), which stimulates the synthesis of high-affinity cystine transporters (5). Sudden exposure to cystine might then cause its overaccumulation inside the cell, until corrective processes, such as its rerepression and the synthesis of exporters, might reduce cysteine back to normal levels.
Indeed, pregrowth on other CysB-activating sulfur species (djenkolic acid, sulfite, and thiosulfate) also caused H2O2 sensitivity upon subsequent cystine addition (data not shown). One hour of growth on sulfate was sufficient to confer sensitivity to cystine-H2O2 treatment; however, the sensitivity was avoided if chloramphenicol were present to block protein synthesis during this period of sulfur limitation (Fig. 4). Finally, a null mutation in cysB prevented sensitivity (Fig. 4). Thus, the activation of CysB by sulfur limitation and its subsequent induction of a protein were essential for H2O2 sensitivity.
The exposure of CysB-activated cells to sulfur sources other than l-cystine (homocystine, sulfite, sulfide, thiosulfate, GSH, GSSG, and DTT) did not have any effect on H2O2 sensitivity (Fig. 4). d-Cystine also did not have any effect on H2O2 sensitivity (Fig. 5). l-Cysteine did sensitize cells.
FIG. 5.

Evidence that H2O2 sensitivity requires cystine uptake. E. coli AB1157 cells were grown in minimal medium containing sulfate until they reached log phase, and then they were exposed to different concentrations of d- or l-cystine for 3 min before 2.5 mM H2O2 was added. Viability was determined after 3 min of H2O2 exposure.
Not much is known about the cystine transport system in E. coli. However, in Salmonella enterica serovar Typhimurium, the CysB-inducible CTS-1 (cystine transport system 1) has a Km of about 2 μM. The dose of cystine necessary for the H2O2 sensitivity was also in the micromolar range and exhibited saturating kinetics, consistent with the saturation of a high-affinity transporter (Fig. 5). Unfortunately, the gene that encodes the high-affinity cystine transporter has not yet been identified, so its involvement could not be tested genetically.
Interestingly, ydeD mutants, which lack a cysteine exporter, remained sensitive to H2O2 slightly longer than did wild-type cells (e.g., after 15 min of cystine exposure, 0.07% versus 0.68% survived the H2O2 challenge).
The intracellular thiol pool is increased in cystine-treated cells.
The above data suggested that cystine import was responsible for sensitivity. The total acid-soluble thiol content of cystine-treated cells was only 1.8-fold higher than that of sulfate-grown cells, which does not seem commensurate with the acceleration of DNA damage. The predominant thiol is GSH, and a GSH-specific assay showed that its content also increased only about twofold. However, surprisingly, elimination of GSH by mutation (gshA, encoding γ-glutamylcysteinyl synthetase) blocked H2O2 sensitization (Fig. 4). A gshB mutant (GSH synthetase) also showed this phenotype (data not shown). Accordingly, DNA isolated from cells lacking GSH was found to be less damaged by cystine-H2O2 treatment (Fig. 6). These results gave us the initial impression that GSH may play the role as an iron reductant. However, given the fact that the degree of sensitization was far greater than the moderate increase in GSH levels, it was not clear whether GSH would function directly as a reductant.
FIG. 6.

GSH-deficient cells have less DNA damage than wild-type cells. E. coli JTG10 cells (gshA) were treated with both cystine and H2O2. Total genomic DNA was isolated, and qPCR was performed using four different amounts of template DNA. Data shown are for cells before (stippled bars) and after (gray bars) cystine-H2O2 treatment. Compare with the GSH+ parent (Fig. 2, cystine+H2O2 bar).
To study the thiol pool more closely, we resolved the soluble thiols by HPLC. In untreated cells, GSH was the sole abundant thiol (Fig. 7A); however, cysteine became abundant after the cystine treatment. Intracellular cysteine concentration rose about eightfold. Cysteine levels did not rise in a gshA mutant (Fig. 7B). This suggested that GSH is requisite for cystine uptake and/or reduction to cysteine. In fact, a GSH reductase mutant (gor), which cannot recycle GSSG, also did not exhibit an increase in cysteine content after cystine treatment (data not shown) and was resistant to H2O2 (Fig. 4).
FIG. 7.

Cystine treatment increases cysteine content. E. coli AB1157 (wild-type) (A) and JTG10 (gshA) (B) cells were grown in minimal medium containing sulfate until they reached log phase, and the cells were treated with 0.5 mM cystine for 3 min (thick line) or not treated (thin line). Acid-soluble thiols were isolated, labeled with mBBr fluorescent dye, and separated by HPLC. Cysteine and GSH peaks are indicated.
When cystine-treated cells were washed and suspended back into minimal medium containing sulfate, the intracellular cysteine level returned to its usual level by the first available time point (about 5 min). Those cells were also resistant to H2O2 (>50% survival). These data indicated a strong correlation between cysteine content and H2O2 sensitivity.
Cysteine is desulfurylated by a variety of enzymes, and it seemed possible that sulfide derived from cysteine might be the direct active agent. However, mutants lacking these enzymes, namely, cysteine desulfurase (IscS), selenocysteine lyase (SufS, or CsdB), β-cystathionase (MetC), and o-acetylserine sulfhydrylase A (CysK), all exhibited as much sensitivity to H2O2 after a cystine pulse as did wild-type cells (data not shown). If sulfide were the key species and desulfurylation were rate limiting, we would expect these mutations to have an impact.
Cysteine reduces iron and drives oxidative DNA damage in vitro.
The fact that sensitivity to H2O2 correlated better with cysteine than with total thiol levels prompted us to compare the abilities of different thiol species to reduce iron and drive Fenton chemistry. We also observed that GSH could reduce ferric iron in vitro; however, cysteine did so much more efficiently (Fig. 8). Rates of iron reduction were diminished by the presence of iron ligands such as ATP or citrate, yet cysteine was still more effective than GSH (data not shown).
FIG. 8.

Cysteine reduces ferric iron better than GSH does. Ferric chloride (10 μM) was mixed in anaerobic 20 mM Tris-HCl (pH 7.4) with 3 mM cysteine or GSH. At each time point, aliquots were removed and ferrous iron was assayed.
When Fe3+, cysteine or GSH, and H2O2 were mixed with pACYC184 plasmid, cysteine produced strand breaks in the DNA and shifted supercoiled-form bands to the relaxed-form bands (Fig. 9). When GSH was included as a reductant, however, no damage could be detected.
FIG. 9.

Cysteine efficiently drives Fenton-mediated DNA damage in vitro. In an anaerobic Coy chamber, 33 ng of pACYC184 plasmid was mixed in 3.5 mM NaHCO3 buffer (pH 7.2) with 10 μM FeCl3, 20 μM GSH or cysteine, and 50 μM H2O2. At each time point, the reaction was stopped by adding catalase. The reaction mixture was run in a 1% agarose gel. RF, relaxed form; SF, supercoiled form.
The reason for the superiority of cysteine over GSH is unclear but may derive from the ability of cysteine to form complexes with iron (31). In fact, the rate of iron reduction was not proportionate to cysteine concentration (data not shown), arguing against a simple rate-limiting electron transfer between cysteine and iron (see Discussion).
The cysteine content of cells under normal conditions is too low to promote DNA damage.
The elevated cysteine concentration in cells after cystine treatment was about 1.5 mM. A larger issue was whether the 200 μM cysteine concentration found under normal conditions was responsible for the residual sensitivity of these cells to H2O2. To answer this question, we attempted to deplete cysteine pools by starving cells for all sulfur sources. E. coli SP34 (cysA gshA xth-1) cannot import sulfate and has no residual pool of GSH. This strain grew well in medium containing cystine; after it was washed and resuspended in medium containing sulfate, its growth stopped. Nevertheless, the resistance of this repair-defective strain to H2O2 was not significantly increased (3.7% of cell survival in medium containing cystine versus 4.8% in medium containing sulfate).
Unfortunately, the cysteine levels of the mutant were nearly at detection limits even prior to starvation, so we could not directly confirm that cysteine pools were subsequently depleted. However, that is the obvious implication of the cessation of growth. Therefore, these data suggest that at its usual intracellular concentration, cysteine is not the primary reductant that drives Fenton chemistry.
DISCUSSION
Homeostatic mechanisms limit cysteine concentration and minimize sensitivity to oxidants.
Our results show that supranormal levels of intracellular cysteine cause sensitivity to oxidative DNA damage. In this study, those levels were achieved by sulfur restriction, leading to the synthesis of high-affinity sulfur transporters, followed by exposure to cystine (Fig. 10). Apparently, the subsequent rapid import of cystine causes the cell to overshoot the usual set point. Homeostasis is gradually restored over the next generation, perhaps by the inactivation of transporters.
FIG. 10.

Mechanism of cystine-mediated H2O2 sensitivity. The CysB protein, which is activated during growth on relatively poor sulfur sources, stimulates the concerted import and reduction of cystine. Reduced GSH is necessary for this process. When saturating cystine is provided to erstwhile sulfur-poor cells, intracellular cysteine pools transiently rise to supranormal levels. Free iron catalyzes electron transfer from excess cysteine to H2O2, generating hydroxyl radicals.
The normal level of cysteine in growing E. coli, 0.1 to 0.2 mM, is evidently insufficient to contribute significantly to DNA damage, and this may have been a factor in the determination of the set point. This may also speak to the reason that E. coli employs GSH, rather than free cysteine, as a thiol buffer (48). GSH and cysteine have similar reduction potentials (cysteine/cystine, −250 mV; GSH/GSSG, −264 mV at pH 7.4 [22]) and pKa values (cysteine, 8.3; GSH, 8.6); they differ in that GSH has a strikingly diminished ability to reduce free iron. Thus, the cell can maintain a pool of 2 to 4 mM GSH without an accompanying vulnerability to Fenton chemistry. GSH has even been proposed to serve as a stored form of cysteine (30), a concept that is rationalized by its lower reactivity.
Our observation that cysteine reduces iron more avidly than does GSH is in agreement with the observations of studies of transferrin reduction and reactive oxygen species production by other workers (25, 37, 56). The reason for the difference remains obscure. It has been proposed that ferric iron might coordinate with the sulfhydryl and carboxylate groups of cysteine (50). McAuliffe and Murray suggested that the interaction of sulfur, iron, and oxygen atoms in a three-center π system would promote facile electron transfer (31). The additional carboxylate group of GSH might disturb this geometry. Consistent with the suggestion of the formation of complexes, we observed that high rates of iron reduction were obtained in vitro with 20 μM cysteine and that further increases had only moderate additional effects upon ferrous iron formation. The implication is that low levels of cysteine were sufficient to complex the iron, so that collision frequency no longer controlled overall electron transfer rates. In vivo, of course, the presence of competing iron ligands would shift this saturation point to much higher concentrations of cysteine.
The rate of oxidative DNA damage depends upon the reduction of ferric iron.
A recent study from our lab found conditions under which the pool of free, reduced flavins (reduced flavin adenine dinucleotide [FADH2]) becomes enlarged and drives oxidative DNA damage. That phenomenon broadly resembles our observations with cysteine: both cysteine and FADH2 are unusually effective reductants of ferric iron in vitro, and it is evidently this activity that promotes oxidative DNA damage in vivo. The results of both experimental lines indicate that the rate of DNA damage is typically limited by whether reductants are available to recycle iron after it reacts with H2O2.
We were somewhat disappointed to realize that neither the cysteine nor flavin reduction pathways seem to be important for the DNA damage that occurs in the absence of a cystine pulse or respiratory block. What is the Fenton-driving reductant in that case? Experiments with gshA mutants indicate that it is not GSH (16), experiments with zwf pnt mutants indicate that it is not NADPH (53), and experiments with superoxide dismutase-overproducing cells indicate that it is not superoxide (23). Since GSH still reduces iron better than NADH, it must not be the latter, either (53). The cell presumably has a pool of free FADH2 even in the absence of flavin reductase, so this species could be responsible. The question is intriguing, because the level of the unknown reductant must determine to a significant extent the vulnerability of bacteria to oxidative damage. By point of contrast, the free intracellular iron in Saccharomyces cerevisiae remains in an oxidized form, a difference which may help protect that organism from H2O2 (49).
Why is GSH necessary for rapid cysteine accumulation?
We noted that cystine pulses failed to force cysteine accumulation in mutants that could not synthesize GSH or reduce GSSG. These mutants still grow on minimal medium containing sulfate, indicating that the CysB regulon remains functional. We tested whether cystine accumulated intracellularly in these mutants in the oxidized form and found that it did not. Thus, the transport process itself might require GSH as a cofactor. Although the mechanism of this linkage remains to be established, a rationale can be suggested: high intracellular levels of cystine would likely trigger the formation of mixed disulfides and intraprotein disulfide bonds. Indeed, Streptomyces coelicolor engages a response against “disulfide stress” that is separate from its response to reactive oxygen species (40). This threat would be obviated by linking cystine import to its reduction.
A variety of metabolic perturbations can create vulnerability to oxidative DNA damage.
Since the enzymes that adventitiously form H2O2 inside cells (33) are transcriptionally regulated, it seems certain that the rate at which DNA is oxidized will be indirectly affected by the metabolic strategy of the cell. However, it has gradually become clear that less-obvious metabolic perturbations can also have large impacts on the rate of damage. In this study, we found that vulnerability results from a transient loss of cysteine homeostasis. Similarly, superoxide stress threatens DNA by elevating the level of free iron (24), while nitric oxide does so by forcing the accumulation of free FADH2 (39; Woodmansee and Imlay, unpublished), an iron reductant like cysteine. A short period of hypersensitivity to H2O2 corresponds with entry into stationary phase (15), although the mechanism is not yet known. Thus, while the chemistry of oxidative DNA damage is largely understood, the physiological connections are just beginning to be revealed.
Acknowledgments
We thank Alex Smirnov for assistance with the EPR experiments conducted at the Illinois EPR research center. We also thank the colleagues cited in Table 1 who generously provided strains.
This study was supported in part by grant GM59030 from the National Institutes of Health.
REFERENCES
- 1.Alikhanian, S. I., T. S. Iljina, E. S. Kaliaeva, S. V. Kameneva, and V. V. Sukodolec. 1965. Mutants of Escherichia coli K12 lacking thymine. Nature 206:848-849. [DOI] [PubMed] [Google Scholar]
- 2.Anderson, M. E. 1985. Determination of glutathione and glutathione disulfide in biological samples. Methods Enzymol. 113:548-555. [DOI] [PubMed] [Google Scholar]
- 3.Au, D. C.-T., G. N. Green, and R. B. Gennis. 1984. Role of quinones in the branch of the Escherichia coli respiratory chain that terminates in cytochrome o. J. Bacteriol. 157:122-125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bachmann, B. J. 1987. Derivations and genotypes of some mutant derivatives of Escherichia coli K-12, p. 1190-1219. In F. C. Neidhardt, J. L. Ingraham, K. B. Low, B. Magasanik, M. Schaechter, and H. E. Umbarger (ed.), Escherichia coli and Salmonella typhimurium: cellular and molecular biology. American Society for Microbiology, Washington, D.C.
- 5.Baptist, E. W., and N. M. Kredich. 1977. Regulation of l-cystine transport in Salmonella typhimurium. J. Bacteriol. 131:111-118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Berglin, E. H., M. K. Edmund, G. K. Nyberg, and J. Carlsson. 1982. Potentiation by l-cystine of the bactericidal effect of hydrogen peroxide in Escherichia coli. J. Bacteriol. 152:81-88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bielski, B. H. J., D. E. Cabelli, and R. L. Arudi. 1985. Reactivity of HO2/O2− radicals in aqueous solution. J. Phys. Chem. Ref. Data 14:1041-1062. [Google Scholar]
- 8.Brandi, G., M. Fiorani, C. Pierotti, A. Albano, F. Cattabeni, and O. Cantoni. 1989. Morphological changes in Escherichia coli cells exposed to low or high concentrations of hydrogen peroxide. Microbiol. Immunol. 33:991-1000. [DOI] [PubMed] [Google Scholar]
- 9.Brandi, G., L. Luzzi, P. Giacomoni, A. Albano, F. Cattabeni, and O. Cantoni. 1992. Differential effect of the amino acid cystine in cultured mammalian and bacterial cells exposed to oxidative stress. Mutat. Res. 281:157-161. [DOI] [PubMed] [Google Scholar]
- 10.Brawn, K., and I. Fridovich. 1981. DNA strand scission by enzymically generated oxygen radicals. Arch. Biochem. Biophys. 206:414-419. [DOI] [PubMed] [Google Scholar]
- 11.Cantoni, O., G. Brandi, A. Albano, and F. Cattabeni. 1995. Action of cystine in the cytotoxic responses of Escherichia coli cells exposed to hydrogen peroxide. Free Rad. Res. Commun. 22:275-283. [DOI] [PubMed] [Google Scholar]
- 12.Carlsson, J., G. P. D. Granberg, G. K. Nyberg, and M.-B. K. Edlund. 1979. Bactericidal effect of cysteine exposed to atmospheric oxygen. Appl. Environ. Microbiol. 37:383-390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Datsenko, K. A., and B. L. Wanner. 2000. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. USA 97:6640-6645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dukan, S., and T. Nystrom. 1999. Oxidative stress defense and deterioration of growth-arrested Escherichia coli cells. J. Biol. Chem. 274:26027-26032. [DOI] [PubMed] [Google Scholar]
- 15.Gort, A. S., D. M. Ferber, and J. A. Imlay. 1999. The regulation and role of the periplasmic copper, zinc superoxide dismutase of Escherichia coli. Mol. Microbiol. 32:179-191. [DOI] [PubMed] [Google Scholar]
- 16.Greenberg, J. T., and B. Demple. 1986. Glutathione in Escherichia coli is dispensable for resistance to H2O2 and gamma radiation. J. Bacteriol. 168:1026-1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hagensee, M. E., and R. E. Moses. 1989. Multiple pathways for repair of hydrogen peroxide-induced DNA damage in Escherichia coli. J. Bacteriol. 171:991-995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hutchinson, F. 1985. Chemical changes induced in DNA by ionizing radiation. Prog. Nucleic Acid Res. 32:116-154. [DOI] [PubMed] [Google Scholar]
- 19.Imlay, J. A., and I. Fridovich. 1991. Assay of metabolic superoxide production in Escherichia coli. J. Biol. Chem. 266:6957-6965. [PubMed] [Google Scholar]
- 20.Imlay, J. A., and S. Linn. 1986. Bimodal pattern of killing of DNA-repair-defective or anoxically grown Escherichia coli by hydrogen peroxide. J. Bacteriol. 166:519-527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Imlay, J. A., and S. Linn. 1988. Toxic DNA damage by hydrogen peroxide through the Fenton reaction in vivo and in vitro. Science 240:640-642. [DOI] [PubMed] [Google Scholar]
- 22.Jones, D. P., J. L. Carlsson, V. C. Mody, Jr., J. Cai, M. J. Lynn, and P. Sternberg, Jr. 2000. Redox state of glutathione in human plasma. Free Rad. Biol. Med. 28:625-635. [DOI] [PubMed] [Google Scholar]
- 23.Keyer, K., A. S. Gort, and J. A. Imlay. 1995. Superoxide and the production of oxidative DNA damage. J. Bacteriol. 177:6782-6790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Keyer, K., and J. A. Imlay. 1996. Superoxide accelerates DNA damage by elevating free-iron levels. Proc. Natl. Acad. Sci. USA 93:13635-13640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kojima, N., and G. W. Bates. 1979. The reduction and release of iron from Fe3+/transferrin/CO32−. J. Biol. Chem. 254:8847-8854. [PubMed] [Google Scholar]
- 26.Kredich, N. M. 1996. Biosynthesis of cysteine, p. 514-527. In F. C. Neidhardt, R. Curtiss III, J. L. Ingraham, E. C. C. Lin, K. B. Low, B. Magasanik, W. S. Reznikoff, M. Riley, M. Schaechter, and H. E. Umbarger (ed.), Escherichia coli and Salmonella: cellular and molecular biology, 2nd ed. ASM Press, Washington, D.C.
- 27.Lawley, P. D., and C. J. Thatcher. 1970. Methylation of deoxyribonucleic acid in cultured mammalian cells by N-methyl-N′-nitro-N-nitrosoguanidine. The influence of cellular thiol concentrations. Biochem. J. 116:693-707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lesko, S. A., R. J. Lorentzen, and P. O. P. Ts'o. 1980. Role of superoxide in deoxyribonucleic acid strand scission. Biochemistry 19:3023-3028. [DOI] [PubMed] [Google Scholar]
- 29.Liochev, S. I., and I. Fridovich. 1994. The role of O2·− in the production of HO·: in vitro and in vivo. Free Rad. Biol. Med. 16:29-33. [DOI] [PubMed] [Google Scholar]
- 30.Lu, S. C. 2000. Regulation of glutathione synthesis. Curr. Top. Cell. Regul. 36:95-116. [DOI] [PubMed] [Google Scholar]
- 31.McAuliffe, C. A., and S. G. Murray. 1972. Metal complexes of sulphur-containing amino acids. Inorg. Chim. Acta Rev. 6:103-121. [Google Scholar]
- 32.Messner, K. R., and J. A. Imlay. 2002. Mechanism of superoxide and hydrogen peroxide formation by fumarate reductase, succinate dehydrogenase, and aspartate oxidase. J. Biol. Chem. 277:42563-42571. [DOI] [PubMed] [Google Scholar]
- 33.Messner, K. R., and J. A. Imlay. 1999. The identification of primary sites of superoxide and hydrogen peroxide formation in the aerobic respiratory chain and sulfite reductase complex of Escherichia coli. J. Biol. Chem. 274:10119-10128. [DOI] [PubMed] [Google Scholar]
- 34.Miller, J. H. 1972. Experiments in molecular genetics. Cold Spring Harbor Laboratory, Cold Spring Harbor, N.Y.
- 35.Moskovitz, J., M. A. Rahman, J. Strassman, S. O. Yancey, S. R. Kushner, N. Brot, and H. Weissbach. 1995. Escherichia coli peptide methionine sulfoxide reductase gene: regulation of expression and role in protecting against oxidative damage. J. Bacteriol. 177:502-507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Muiras, M. L., P. U. Giacomoni, and P. Tachon. 1993. Modulation of DNA breakage induced via the Fenton reaction. Mutat. Res. 295:47-54. [DOI] [PubMed] [Google Scholar]
- 37.Nappi, A. J., and E. Vass. 1997. Comparative studies of enhanced iron-mediated production of hydroxyl radical by glutathione, cysteine, ascorbic acid, and selected catechols. Biochim. Biophys. Acta 1336:295-301. [DOI] [PubMed] [Google Scholar]
- 38.Newton, G. L., R. Dorian, and R. C. Fahey. 1981. Analysis of biological thiols: derivatization with monobromobimane and separation by reverse-phase high-performance liquid chromatography. Anal. Biochem. 114:383-387. [DOI] [PubMed] [Google Scholar]
- 39.Pacelli, R., D. A. Wink, J. A. Cook, M. C. Krishna, W. DeGraff, N. Friedman, M. Tsokos, A. Samuni, and J. B. Mitchell. 1995. Nitric oxide potentiates hydrogen peroxide-induced killing of Escherichia coli. J. Exp. Med. 182:1469-1479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Paget, M. S., V. Molle, G. Cohen, Y. Aharonowitz, and M. J. Buttner. 2001. Defining the disulphide stress response in Streptomyces coelicolor A3(2): identification of the sigmaR regulon. Mol. Microbiol. 42:1007-1020. [DOI] [PubMed] [Google Scholar]
- 41.Park, J., and R. A. Floyd. 1994. Generation of strand breaks and formation of 8-hydroxy-2′-deoxyguanosine in DNA by a thiol/Fe3+/O2− catalyzed oxidation system. Arch. Biochem. Biophys. 312:285-291. [DOI] [PubMed] [Google Scholar]
- 42.Parmentier, C., P. Leroy, M. Wellman, and A. Nicolas. 1998. Determination of cellular thiols and glutathione-related enzyme activities: versatility of high-performance liquid chromatography-spectrophotometric detection. J. Chromatogr. B 719:37-46. [DOI] [PubMed] [Google Scholar]
- 43.Prinz, W. A., F. Aslund, A. Holmgren, and J. Beckwith. 1997. The role of the thioredoxin and glutaredoxin pathways in reducing protein disulfide bonds in the Escherichia coli cytoplasm. J. Biol. Chem. 272:15661-15667. [DOI] [PubMed] [Google Scholar]
- 44.Rowley, D. A., and B. Halliwell. 1982. Superoxide-dependent formation of hydroxyl radicals from NADH and NADPH in the presence of iron salts. FEBS Lett. 142:39-41. [DOI] [PubMed] [Google Scholar]
- 45.Rowley, D. A., and B. Halliwell. 1982. Superoxide-dependent formation of hydroxyl radicals in the presence of thiol compounds. FEBS Lett. 138:33-36. [DOI] [PubMed] [Google Scholar]
- 46.Seaver, L. C., and J. A. Imlay. 2001. Alkyl hydroperoxide reductase is the primary scavenger of endogenous hydrogen peroxide in Escherichia coli. J. Bacteriol. 183:7173-7181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Seaver, L. C., and J. A. Imlay. 2001. Hydrogen peroxide fluxes and compartmentalization inside growing Escherichia coli. J. Bacteriol. 183:7182-7189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sies, H. 1999. Glutathione and its role in cellular functions. Free Rad. Biol. Med. 27:916-921. [DOI] [PubMed] [Google Scholar]
- 49.Srinivasan, C., A. Liba, J. A. Imlay, J. S. Valentine, and E. B. Gralla. 2000. Yeast lacking superoxide dismutase(s) show elevated levels of “free iron” as measured by whole cell electron paramagnetic resonance. J. Biol. Chem. 275:29187-29192. [DOI] [PubMed] [Google Scholar]
- 50.Taylor, J. E., J. F. Yan, and J. Wang. 1966. The iron(III)-catalyzed oxidation of cysteine by molecular oxygen in the aqueous phase: an example of a two-thirds-order reaction. J. Am. Chem. Soc. 88:1663-1667. [DOI] [PubMed] [Google Scholar]
- 51.Touati, D., M. Jacques, B. Tardat, L. Bouchard, and S. Despied. 1995. Lethal oxidative damage and mutagenesis are generated by iron in Δfur mutants of Escherichia coli: protective role of superoxide dismutase. J. Bacteriol. 177:2305-2314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Walling, C. 1975. Fenton's reagent revisited. Acc. Chem. Res. 8:125-131. [Google Scholar]
- 53.Woodmansee, A. N., and J. A. Imlay. 2002. Reduced flavins promote oxidative DNA damage in non-respiring E. coli by delivering electrons to intracellular free iron. J. Biol. Chem. 277:34055-34066. [DOI] [PubMed] [Google Scholar]
- 54.Yakes, F. M., Y. Chen, and B. Van Houten. 1996. PCR-based assays for the detection and quantitation of DNA damage and repair, p. 169-182. In G. P. Pfeifer (ed.), Technologies for detection of DNA damage and mutations. Plenum, New York, N.Y.
- 55.Yakes, F. M., and B. Van Houten. 1997. Mitochondrial DNA damage is more extensive and persists longer than nuclear DNA damage in human cells following oxidative stress. Proc. Natl. Acad. Sci. USA 94:514-519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yang, E. Y., A. Campbell, and S. C. Bondy. 2000. Configuration of thiols dictates their ability to promote iron-induced reactive oxygen species generation. Redox Rep. 5:371-375. [DOI] [PubMed] [Google Scholar]