

Abstract
The protease granzyme B (GrB) plays a key role in the cytocidal activity during cytotoxic T lymphocyte (CTL)-mediated programmed cell death. Multiple caspases have been identified as direct substrates for GrB, suggesting that the activation of caspases constitutes an important event during CTL-induced cell death. However, recent studies have provided evidence for caspase-independent pathway(s) during CTL-mediated apoptosis. In this study, we demonstrate caspase-independent and direct cleavage of the 45 kDa unit of DNA fragmentation factor (DFF45) by GrB both in vitro and in vivo. Using a novel and selective caspase-3 inhibitor, we show the ability of GrB to process DFF45 directly and mediate DNA fragmentation in the absence of caspase-3 activity. Furthermore, studies with DFF45 mutants reveal that both caspase-3 and GrB share a common cleavage site, which is necessary and sufficient to induce DNA fragmentation in target cells during apoptosis. Together, our data suggest that CTLs possess alternative mechanism(s) for inducing DNA fragmentation without the requirement for caspases.
Keywords: caspase/CTLs/DFF45/granzyme B
Introduction
Lymphocyte-mediated cytotoxicity is one of the most potent host defenses against intracellular pathogens, and oncogenic and virally infected cells through the induction of apoptosis of infected cells (Kagi and Hengartner, 1996; Shresta et al., 1998). Cytolytic effector cells induce two potent apoptotic pathways: the granule exocytosis and the Fas–Fas ligand (FasL) pathways. The granule exocytosis pathway is employed predominantly by CD8+, natural killer and lymphokine-activated killer cells. The direct interaction between effector and target cells causes realignment of the granules to contact points between effector and target cells. The major compounds of the granules are the pore-forming protein perforin and a subclass of neutral serine proteases known as granzymes, prototypically represented by granzyme B (GrB). GrB is an essential component of cytotoxic T lymphocyte (CTL)-mediated cytotoxicity, since GrB-deficient CTLs can not rapidly induce oligosomal DNA fragmentation in target cells (Heusel et al., 1994; Shresta et al., 1995). Despite its ability to enter target cells, GrB is unable to gain access to cellular substrates in the absence of perforin (Froelich et al., 1996; Shi et al., 1997; Pinkoski et al., 1998). However, GrB isolated from cytotoxic granules has been demonstrated to induce apoptosis when used in conjunction with either perforin, adenovirus (Ad2) exposure or detergent permeabilization in vitro (Shi et al., 1992; Chinnaiyan et al., 1996; Froelich et al., 1996).
The other apoptotic mechanism used by cytolytic effector cells involves the engagement of effector cell FasL to the cell surface receptor Fas found on target cells. The activation of this pathway triggers a well-characterized intracellular cascade involving cysteine proteases known as caspases, which ultimately leads to the typical biochemical and morphological hallmarks of apoptosis. Experiments using non-selective caspase inhibitors have implicated these proteases as common mediators of apoptotic cell death used by both granule exocytosis and FasL pathways (Sarin et al., 1996; Song et al., 1996).
Caspases exist in all living cells as inactive precursors that undergo proteolytic processing and activation when cells receive an apoptotic signal (Cohen, 1997; Nicholson and Thornberry, 1997; Villa et al., 1997; Budihardjo et al., 1999). Genetic analyses show clearly that caspase-3, -7, -8 and -9 are essential for apoptotic death (Hakem et al., 1998; Slee et al., 1999; Woo et al., 1999). A major biochemical hallmark of apoptosis is DNA fragmentation (Wyllie, 1980), which is mediated by the DNA fragmentation factor (DFF) complex. DFF is a heterodimeric protein composed of a 40 kDa nuclease and a 45 kDa inhibitory subunit known as DFF40/CPAN and DFF45, respectively (Liu et al., 1997, 1998; Halenbeck et al., 1998; Gu et al., 1999). Murine homologues of DFF40 and DFF45, namely caspase-activated deoxynuclease (CAD) and inhibitor of CAD (ICAD), respectively, have also been cloned and characterized (Enari et al., 1998). The DFF45/ICAD proteins possess inherent dual functions within the DFF complex: on the one hand, DFF45 serves as a specific molecular chaperone that facilitates the correct folding of DFF40, while on the other hand it inhibits DFF40 nuclease activity in growing cells (Enari et al., 1998; Halenbeck et al., 1998; Sakahira et al., 1998, 2000). Upon induction of apoptosis, DFF45 is cleaved by caspase-3 at amino acids 117 and 224, a step required for DFF40 to initiate DNA fragmentation in vivo and in vitro (Liu et al., 1997). The cleavage of murine ICAD at both caspase-3 sites is apparently required for activation of the CAD endonuclease (Sakahira et al., 1998). The cellular localization of the DFF complex in proliferating cells remains controversial. The original claim that the complex is located in the cytoplasm (Enari et al., 1998) has been challenged by others demonstrating a nuclear localization (Liu et al., 1998; Samejima and Earnshaw, 1998, 2000; Zhivotovsky et al., 1999; Lechardeur et al., 2000).
GrB displays an uncommon specificity for serine proteases that enables it to proteolytically cleave its substrates following aspartate residues (Odake et al., 1991; Poe et al., 1991; Caputo et al., 1999). GrB was found to process several members of the caspase family in vitro; however, only caspase-3 and caspase-8 have been shown to be direct substrates for GrB in vivo (Medema et al., 1997; Van de Craen et al., 1997; Atkinson et al., 1998), suggesting that GrB induces apoptosis by triggering the activation of caspases within target cells. Nevertheless, several lines of evidence suggest that GrB is also able to induce cell death by a caspase-independent intranuclear process. Cells overexpressing natural inhibitors of caspases such as Bcl-2, cytokine response modifier A (CrmA) and SPI-2, or treated with non-selective caspase inhibitors, are still sensitive to GrB-mediated apoptosis (Talanian et al., 1997; Atkinson et al., 1998). These studies, combined with recent reports showing that GrB may also directly process downstream targets of caspases such as nuclear mitotic apparatus protein (NuMa) and DNA-dependent protein kinase (catalytic subunit) (DNA-PKcs) in the absence of caspase activity (Andrade et al., 1998), suggest that GrB may not necessarily rely upon caspases and may bypass their involvement in eliciting target cell death.
Recently, Thomas et al. (2000) have shown that GrB can induce DNA fragmentation in the presence of broad range caspase inhibitors. In the present study, we have demonstrated caspase-independent and direct cleavage of DFF45 by GrB. We have also used a novel and selective non-peptidic caspase-3 and -7 inhibitor to show the ability of GrB to process DFF45 directly both in vitro and in vivo. Moreover, mutational analysis has revealed a common cleavage site of caspase-3 and GrB for DFF45 processing and the release of DFF40 endonuclease activity. This provision of a caspase-independent pathway for GrB-mediated cell death may guarantee the death of target cells in which the caspase cascade(s) is/are incomplete or strictly regulated by endogenous or exogenous caspase inhibitors.
Results
DFF45 is a nuclear protein and a substrate for granzyme B in vitro
The discovery that GrB is targeted to the nucleus was a key step in linking its proteolytic activity to nuclear changes associated with apoptosis, such as DNA fragmentation following DFF45 cleavage (Pinkoski et al., 1998). Attempts to determine the cellular localization of DFF45 in non-apoptotic cells have led to controversial results (Enari et al., 1998; Samejima and Earnshaw, 1998, 2000; Lechardeur et al., 2000). To determine whether the endo genous DFF45 and DFF40 are localized in the nucleus, Jurkat cells were labeled with DFF45 or DFF40 rabbit polyclonal antibodies (Abs) in combination with Ab to β-actin. Our results illustrated in Figure 1A confirm the exclusive localization of DFF45 and DFF40 to the nucleus of labeled cells. This is in sharp contrast to β-actin, which as expected was localized mostly in the cytosol (Figure 1A). Of note, high cytoplasmic background observed using anti-DFF45 or anti-DFF40 rabbit polyclonal Abs was also found using rabbit pre-immune serum or secondary Ab alone (Figure 1A). Our results thus demonstrate the exclusive nuclear localization of DFF45 and DFF40 in Jurkat cells.


Fig. 1. Nuclear compartmentalization and efficient in vitro processing of DFF45 protein by GrB. (A) Double labeling confocal immunofluorescence microscopy of Jurkat cells. Jurkat cells were stained by anti-β-actin monoclonal antibodies and FITC-coupled anti-mouse IgG. DFF45 and DFF40 were detected by using either anti-DFF45 or anti-DFF40 rabbit polyclonal antibodies, respectively, and TRITC-coupled anti-rabbit IgG as described in Materials and methods. Controls were performed by labeling cells with either the secondary TRITC-coupled anti-rabbit IgG alone, or with pre-immune rabbit serum. Each picture is representative of at least 10 fields of two independent experiments at a magnification of 1000×. (B) Time-dependent cleavage of DFF45 by GrB. rDFF45 (12 ng) was incubated for the times indicated with 1 U of GrB (lanes 1–9) or with 10 U of caspase-3 (lanes 10–18) at 37°C. Reactions were stopped by adding Laemmli sample buffer and analyzed by western blotting with anti-DFF45 Ab. Results are representative of three separate experiments. (C) Dose-dependent cleavage of DFF45 by GrB. Purified rDFF45 (12 ng) was incubated with different concentrations of GrB at 37°C for 20 min and subjected to western blot analysis using anti-DFF45 antiserum.
To investigate whether DFF45 is a GrB substrate in vitro, purified recombinant DFF45 (rDFF45) was incubated with GrB at 37°C for various time intervals (Figure 1B, lanes 1–9) and subjected to western blot analysis using DFF45 antiserum. Cleavage of DFF45 by caspase-3 was also studied under similar conditions (Figure 1B, lanes 10–18). As expected, caspase-3 cleaved DFF45 in a time-dependent manner, generating 28 and 31 kDa fragments corresponding to cleavage at residues D117 or D224, respectively. Treatment of DFF45 with GrB also resulted in a time-dependent cleavage. However, only the 28 kDa product was observed, suggesting that GrB and caspase-3 might share a common cleavage site on DFF45. In addition, a slight shift was observed in the 45 kDa precursor protein in the presence of GrB relative to the untreated protein, suggesting additional processing of DFF45 at one or more uncharacterized sites. Similar results were obtained in three separate experiments. In order to investigate GrB’s efficiency in cleaving DFF45 in vitro, a dose–response analysis was performed (Figure 1C). Purified rDFF45 was incubated with different concentrations of GrB, ranging over three logs, at 37°C for 20 min and subjected to western blot analysis using anti-DFF45 antiserum. The results revealed that concentrations of GrB as low as 1.56 nM were sufficient to cleave DFF45 in vitro. Catalytic efficiency, as defined by the catalytic constant (Kcat/Km) of the cleavage of DFF45 by GrB, was calculated using radiolabeled in vitro transcribed and translated proteins in time course experiments (30 min). Caspase-3 shows a 10-fold higher efficacy in cleaving DFF45 (Kcat/Km = 1.2 ± 0.2 × 105 M–1 s–1) when compared with GrB (Kcat/Km = 1.2 ± 0.5 × 104 M–1 s–1). Control experiments were performed with [35S]Cys-labeled caspase-3 as a substrate. As reported previously, GrB cleaves caspase-3 as efficiently as DFF45 (Kcat/Km = 2.4 ± 0.7 × 104 M–1 s–1) (Andrade et al., 1998).
Granzyme B shares a cleavage site with caspase-3 on DFF45 and exhibits additional specificity
To investigate further whether GrB and caspase-3 share the same cleavage site(s), we mutated the aspartic acid of the first (D117), second (D224) or both (D117 and D224) caspase-3 cleavage sites on DFF45 to a glutamic acid (E) residue, thereby generating three DFF45 mutants: DFF45-M1, DFF45-M2 and DFF45-M12, respectively (Figure 2A). DFF45-WT, -M1, -M2 and -M12 genes were then cloned into the pcDNA3.1His-tagged vector under the T7 promoter, and the proteins were expressed in an in vitro transcription and translation system generating a 48 kDa [35S]Met-labeled and His-tagged DFF45. These were incubated with GrB (Figure 2B, lanes 1–4) or caspase-3 (Figure 2B, lanes 5–8) for the times indicated and visualized using autoradiography. The concentrations of caspase-3 and GrB were chosen to give very similar cleavage on IETD-AMC at 25°C. As shown in Figure 2B, caspase-3 cleaved DFF45-WT at two sites, generating fragments of 28, 31 and 12 kDa, corresponding to cleavage at residues D117, D224, or both, respectively. On the other hand, processing of DFF45-WT by GrB generated two bands migrating at 45 and 28 kDa. Mutation of D117 in DFF45-M1 protein clearly abrogated the 28 kDa fragment of DFF45 in the presence of caspase-3 and GrB, suggesting that both enzymes share a common cleavage site at D117. The inability to detect the 12 kDa fragment corresponding to amino acids 224–331 is due to the absence of methionine in this fragment. Conversely, mutation of D224 in DFF45-M2 leads to the complete elimination of the 31 kDa band by caspase-3, but has no effect on DFF45 processing by GrB. In addition, examination of the double mutant DFF45-M12 results in a total loss of DFF45 processing by caspase-3, while affecting only the generation of the 28 kDa fragment during GrB treatment. Importantly, the generation of the 45 kDa product during GrB treatment of DFF45 was not affected by any of these mutations. These results are representative of three independent experiments.
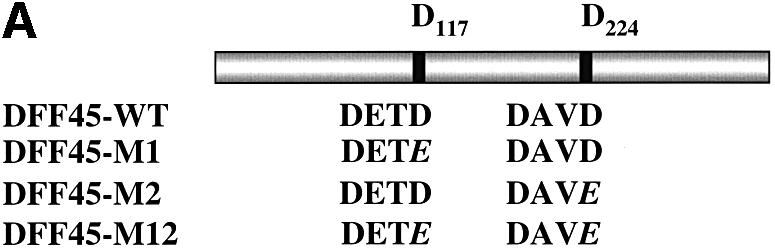


Fig. 2. GrB shares the DETD117↓ cleavage site with caspase-3 and exhibits an additional specificity at VTGD6↓ on DFF45. (A) Schematic diagram of wild-type DFF45 (DFF45-WT) and caspase-3 cleavage site mutants (DFF45-M1, -M2, -M12). Overlap PCR was used to mutate aspartic acid (D) residues of the first (D117), second (D224) or both caspase-3 cleavage sites to glutamic acid (E). Caspase-3 cleavage sites are indicated. (B) Radiolabeled His-tagged DFF45 proteins were incubated in the presence of 1 U of GrB (lanes 1–4) or 10 U of caspase-3 (lanes 5–8) for the indicated time intervals, and the reactions were analyzed by autoradiography. Incubation of DFF45-WT or its mutants with GrB or caspase-3 showed that GrB shares the D117 cleavage site with caspase-3 and exhibits additional proteolytic activity. These results are representative of three separate experiments. (C) Identification of the additional GrB cleavage site on DFF45. cDNAs representing DFF45 fragments (DFF45-F1, -F2 and -F3 corresponding to amino acids 1–117, 118–224 and 225–334, respectively) were cloned, and radiolabeled proteins were generated as described in Materials and methods. (D) Radiolabeled proteins were treated as in (B). Only DFF45-F1, corresponding to the N-terminus (1–117), was cleaved by GrB. These results are representative of two separate experiments.
To identify this novel cleavage site, cDNAs corresponding to the three fragments generated following caspase-3 cleavage, representing amino acids 1–117 (DFF45-F1), 118–224 (DFF45-F2) and 225–331 (DFF45- F3), were amplified and cloned separately into the pcDNA3.1-His-tagged vector (Figure 2C). Radiolabeled proteins were then expressed as described in Materials and methods. As expected, the experimental results illustrated in Figure 2D revealed the inability of caspase-3 to process these fragments further. However, GrB was capable of further processing the DFF45-F1 fragment, which contains the N-terminal moiety of DFF45 (amino acids 1–117). Disappearance of DFF45-F1 during GrB treatment suggests the removal of methionine residues at the start codon of this His-tagged fragment and indicates that this cleavage site is close to the N-terminus of DFF45. Analysis of the N-terminal sequence revealed the presence of an aspartic acid residue that might be recognized and cleaved by GrB (VTGD6). Mutation of D6 to E6 completely prevented GrB cleavage of DFF45 at this site (data not shown). Identical results were obtained from two separate experiments. The results gathered from these mutational analyses further confirmed the ability of both caspase-3 and GrB to cleave DFF45 following the D117 residue. In addition, GrB was found to cleave at an additional site near the N-terminus of DFF45.
DNA fragmentation induced by GrB is not abrogated by a caspase-3-specific inhibitor
Previous studies have reported that caspases play an intermediate role in GrB-induced DNA fragmentation (Medema et al., 1997; Van de Craen et al., 1997; Atkinson et al., 1998). Thus, to determine whether GrB can induce DNA fragmentation in a caspase-independent manner, we used a novel, non-peptidic, specific inhibitor of caspases -3 and -7: isatin sulfonamide (Figure 3A, compound 1) (Lee et al., 2000). First, we tested the ability of this compound to block Fas-mediated apoptosis. Fas-elicited apoptosis was triggered by cross-linkage of cell surface Fas using murine anti-human anti-Fas mAb (M3). The results show that compound 1 inhibits phosphatidylserine exposure and DNA fragmentation during Fas engagement (Figure 3B). This effect was dose dependent, with maximal inhibition occurring at 75 µM. A control, 600-fold less active analog of compound 1, compound 2 (Figure 3B), did not inhibit Fas-mediated apoptosis even at the higher concentrations used. Western blot analysis revealed that compound 1 inhibited further processing of p20 to the p17 and p15 subunits of caspase-3 (Figure 3B) and caspase-7 processing (data not shown). In addition, compound 1 inhibited the activity of both caspase-3 and caspase-7, since cleavage of DFF45 did not occur, as demonstrated by the lack of appearance of its proteolytic fragments (Figure 3B, lower panel). Together, these results indicate that compound 1 blocks Fas-induced apoptosis and DNA fragmentation through specific inhibition of caspase-3 and -7, the sole caspases that appear to cleave DFF45 (Liu et al., 1999). These results are representative of two separate experiments.




Fig. 3. GrB induces DNA fragmentation in the absence of caspase-3 activation. (A) Structures of the caspase-3/-7-specific inhibitor (compound 1) and a 600-fold less active analog (compound 2). (B) Compound 1 blocks Fas-mediated apoptosis in a dose-dependent fashion. Jurkat cells were pre-treated with different concentrations of compound 1 (lanes 7–12) or compound 2 (lanes 1–6). Apoptosis was induced by cross-linking cell surface Fas receptors using mAb to Fas (M3, 20 µg/ml). Abrogation of Fas-induced apoptosis by compound 1 was monitored by DNA fragmentation, annexin V externalization, caspase-3 cleavage and DFF processing. No effect was noticed for compound 2 even at the highest concentration used (lanes 1–6). (C) DFF cleavage and DNA fragmentation occur in the absence of caspase-3 activation. Jurkat cells (1 × 106 cells per well) were pre-treated with 75 µM compound 1 or compound 2. Apoptosis by GrB/Ad2 was induced as described in Materials and methods. Cell death was assayed as in (B). Pre-treatment of the cells with the caspase-3-specific inhibitor compound 1 did not prevent DFF processing and DNA fragmentation induced by GrB (lane 6). Pre-treatment of cells with the caspase-3-specific inhibitor compound 1 (lane 6), but not its analog compound 2 (lane 5), or DMSO alone (lane 4) resulted in a significant decrease in annexin V externalization. Untreated Jurkat cells (lane 1) and Jurkat cells treated with GrB or Ad2 alone (lanes 2 and 3, respectively) showed no significant sign of apoptosis. These results are representative of two separate experiments. (D) Jurkat cells (1 × 105 cells per well) were pre-treated with 75 µM compound 1 or compound 2, and apoptosis was induced by recombinant GrB/perforin as described in Materials and methods. Cell death was assayed by 51Cr release. Pre-treatment with compound 1, but not compound 2, or DMSO blocked further processing of caspase-3 to the p17 active form (lanes 6, 4 and 5, respectively, upper panel) and processing of caspase-mediated cleavage of Rho-GDI substrate (lanes 6, 4 and 5, respectively, lower panel). Blockage of caspase-3 activity had no effect on DFF processing (lane 6, middle panel). Untreated Jurkat cells (lane 1) and those treated with perforin or GrB alone (lanes 2 and 3, respectively) showed no significant sign of apoptosis. These results are representative of three separate experiments.
Experiments were then performed to determine whether GrB could still induce DNA fragmentation even in the absence of caspase activity in vivo. For this purpose, GrB-induced apoptosis was triggered using a previously established method utilizing Ad2 infection (Pinkoski et al., 1998). Jurkat cells were induced to undergo apop tosis by GrB in the presence or absence of compound 1. Compound 2 and dimethylsulfoxide (DMSO) were used as controls. The results illustrated in Figure 3C reveal that pre-treatment of Jurkat cells with DMSO (lane 4) or compound 2 (lane 5) did not prevent apoptosis induced by GrB/Ad2 treatment, as demonstrated by phosphatidylserine externalization, caspase-3 activation, DFF processing and DNA fragmentation. In contrast, Jurkat cells triggered to undergo apoptosis by GrB/Ad2 in the presence of compound 1 showed a significant, 7-fold decrease in cell death, as measured by annexin V (n = 2); however, there was no blockage in DFF45 processing, as demonstrated by the degradation of the DFF45 precursor and the appearance of its proteolytic products (28 and 12 kDa fragments) or DNA fragmentation (lane 6). Under similar conditions, caspase-3 accumulated as a 20 kDa fragment and was not completely processed to the enzymatically active p17 and p15 subunits. Although GrB was able to process the DFF complex in the absence of caspase-3, the induction of DNA fragmentation was less than that seen in the presence of active caspase-3. The lower efficacy of DFF45 cleavage by GrB (Kcat/Km = 1.2 ± 0.5 × 104 M–1 s–1), which is one order of magnitude less than that of caspase-3-mediated cleavage of DFF45 (Kcat/Km = 1.2 ± 0.2 × 105 M–1 s–1), could explain the latter result. Similar results were obtained in two independent experiments.
When GrB was introduced into Jurkat target cells in the presence of sublytic concentrations of perforin, it induced cell death, as assayed by the percentage of specific 51Cr release, as described previously (Beresford et al., 1999). Jurkat cells were loaded with GrB and a sublytic concentration of perforin for 3 h at 37°C in the presence or absence of the caspase-3-specific inhibitor, as shown in Figure 3D. Pre-treatment of Jurkat cells with DMSO (lane 4) or compound 2 (lane 5) did not block apoptosis induced by GrB and perforin. In contrast, pre-treatment with compound 1 abrogated caspase-3 activity by preventing further processing of the p20 subunit to p17 and p15 subunits (lane 6, upper panel), but was unable to block processing of the DFF complex (lane 6, middle panel). To rule out the possibility of caspase-3 activity in the presence of compound 1, we monitored by western blotting the processing of Rho GDP-dissociation inhibitor (Rho-GDI), a caspase-3 substrate (Na et al., 1996). Rho-GDI processing from 28 to 23 kDa was not blocked in the presence of either compound 2 or DMSO when cells were treated with GrB and perforin. However, under similar conditions, the caspase-3-mediated cleavage of Rho-GDI was completely inhibited in cells pre-treated with compound 1, indicating complete inhibition of caspase-3 activity. When Jurkat cells were treated with either perforin (lane 2) or GrB (lane 3) alone, there was no evidence for caspase activation. Taken together, the results from GrB/Ad2 and GrB/perforin experiments clearly demonstrate that GrB can bypass effector caspases to induce DNA fragmentation through the direct cleavage of DFF45 in vivo. Results are representative of three separate experiments.
Cleavage of DFF45 at D117 is necessary and sufficient to induce DFF40 release and DNA fragmentation in vivo and in vitro
To determine the effect of the shared cleavage site on the induction of DNA fragmentation in vivo, Jurkat cells were stably transfected with DFF-WT, DFF-M1, DFF-M2 and DFF-M12 and cloned by limiting dilution (Figure 4A). Expression of transfected mutant and of wild-type DFF45 was monitored by the presence in transfected cells of a higher molecular weight form of DFF45 (48 kDa), due to the presence of an alternative initiation codon in the expression vector. RT–PCR analysis of the transfected cells using oligonucleotide probes specific for the two initiation sites further confirmed that these cells express comparable levels of exogenous wild-type and mutant forms of DFF45 (data not shown). Fas-elicited apoptosis was triggered using the anti-Fas mAb M3. GrB-induced cell death was accomplished by the GrB/Ad2 or GrB/perforin system, as described in Materials and methods. Analysis of DNA fragmentation during Fas- or GrB/Ad2-induced apoptosis revealed that the expression of DFF45-M1 completely inhibited DNA fragmentation (Figure 4B, lanes 4 and 10, respectively). On the other hand, expression of DFF45-WT (Figure 4B, lanes 3 and 9) or DFF45-M2 (Figure 4B, lanes 5 and 11) under similar conditions had no effect on the induction of DNA fragmentation. As expected, expression of DFF45-M12 inhibited DNA fragmentation induced by both stimuli (Figure 4B, lanes 6 and 12). Untreated (Figure 4B, lanes 1 and 7) or empty vector transfected (Figure 4B, lanes 2 and 8) Jurkat cells were used as controls. Similar results were obtained following exposure of these cells to GrB and perforin (data not shown). The results shown here were confirmed using nine different clones from each of the DFF45-WT and DFF45 mutants obtained from two separate, stable transfection experiments. Taken together, these results indicate that while cleavage of DFF45 at D224 has no effect on the induction of DNA fragmentation, cleavage at D117 is both necessary and sufficient to induce DFF40 release and DNA fragmentation by caspase-3 or GrB in vivo.



Fig. 4. Cleavage of DFF45 at residue D117 is necessary and sufficient to induce DNA fragmentation. (A) Jurkat cells were stably transfected with SRα Neo/DFF45-WT, -M1, -M2 or -M12 (lanes 2–5, respectively). The empty SRα Neo vector was transfected in Jurkat cells and used as a control (lane 1). Expression of the transfected (exogenous) and native (endogenous) DFF45 is indicated. (B) Stably transfected Jurkat cells were triggered to undergo apoptosis by either Fas (lanes 2–6) or GrB/Ad2 (lanes 8–12) for 5 h as described in Materials and methods. Following the reaction, fragmented DNA was extracted and analyzed on a 2% agarose gel. Mutation of residue D117 completely abolished DNA fragmentation following GrB- (lane 10) or Fas (lane 4)-induced apoptosis. The results are representative of three separate experiments. (C) Direct cleavage of DFF45 by GrB triggers CAD nuclease activity in a cell-free system. The human CAD was expressed in the in vitro transcription and translation system in the absence (lanes 3 and 7) or presence (lanes 4 and 8) of 160 ng of rDFF45 as described in Materials and methods. CAD nuclease activity using plasmid DNA as a substrate was determined in the presence of 0.12 µM GrB (lanes 5–8) or 0.2 µM caspase-3 (lanes 1–4). Samples containing plasmid alone (lanes 1 and 5) or rDFF45 alone (lanes 2 and 6) were subjected to a similar procedure in the presence of GrB (lanes 5 and 6) or caspase-3 (lanes 1 and 2). The results are representative of two separate experiments.
To further confirm that cleavage of DFF45 by GrB directly induces functional DFF40 release and DNA fragmentation, a functional DFF complex was synthesized using rabbit reticulocyte lysate in a cell-free system, as described in Materials and methods. DFF40 nuclease activity was then determined in the presence of either caspase-3 or GrB using plasmid DNA as a substrate. Caspase-3 was able to cleave DFF45 with subsequent triggering of DFF40 nuclease activity (Figure 4C, lane 4). Likewise, GrB showed a similar ability to cleave the DFF45 complex and to induce DFF40 nuclease activity resulting in DNA degradation (Figure 4C, lane 8). Neither DFF45 nor DFF40 alone was able to induce DNA degradation in the presence of caspase-3 (lanes 2 and 3) or GrB (lanes 6 and 7). These results demonstrate directly that the cleavage of DFF45 by GrB is sufficient to induce DFF40 release and DNA degradation. These results are representative of two independent experiments. GrB used in this assay was purified from mammalian cytolytic granules that could be contaminated with small amounts of other proteases displaying similar physiochemical properties (i.e. caspase-3). To exclude the possibility that such contaminants could be responsible for the effect observed, GrB was added to radiolabeled rDFF45 in vitro. A cleavage pattern characteristic of GrB, but not of caspase-3, for DFF45 processing was observed (data not shown), confirming that GrB was indeed responsible for the observed cleavage and subsequent activation of DNase activity.
Discussion
The existence of caspase-independent proteolytic pathway(s) during CTL-mediated cell death has been suggested. Data reported here demonstrate the ability of GrB to cleave DFF45 efficiently and directly in the nucleus, leading to DNA fragmentation in the absence of caspase-3 activity. Our data are consistent with a recent report in which GrB was shown to process DFF45 directly and induce DNA fragmentation in the absence of caspase activity (Thomas et al., 2000). Using Trypan blue staining, the authors demonstrated that caspase-3 is not required during CTL-mediated killing for the induction of DNA fragmentation in target cells. We, on the other hand, demonstrate herein the ability of GrB to directly and efficiently cleave DFF45, both in vitro and in vivo, and to induce DNA fragmentation in a caspase-independent pathway during GrB-induced cell death. Furthermore, using DFF45 mutants, we have characterized the GrB cleavage sites on DFF45, thereby providing direct molecular evidence for the mechanism of caspase- independent DFF40 activation during CTL-mediated apoptosis. In addition, a mutant DFF45 molecule that can not be cleaved by GrB (DFF45-M1) was found to exert a dominant-negative activity by blocking DNA fragmentation during apoptosis.
Our in vitro analysis using radiolabeled DFF45 concurs with previous reports clearly demonstrating the ability of caspase-3 to cleave wild-type DFF45 at both the D117 and D224 sites. However, treatment of wild-type DFF45 or its mutants with GrB revealed that it could also cleave DFF45 at D117, indicating a shared specificity with caspase-3. In addition, GrB was found to cleave DFF45 at another site (VTGD6↓). The functional role of this cleavage, if any, remains to be explored. Interestingly, sequence comparisons between human DFF45 and murine ICAD revealed the absence of the D6 site in ICAD, which could explain the inability of GrB to cleave ICAD at this position (Wolf et al., 1999).
Non-selective peptide inhibitors of caspases block DNA fragmentation induced by GrB in vivo. However, well characterized natural inhibitors of caspases, such as CrmA and Bcl-2, are not able to protect target cells from CTL attack (Strasser et al., 1991; Vaux et al., 1992; Sutton et al., 1997). Furthermore, none of the caspase-deficient mice tested exhibits complete resistance to GrB-induced apoptosis (Woo et al., 1999). Since non-selective inhibitors inhibit several caspases in addition to caspase-3, we used a novel and selective non-peptidic protease inhibitor of the effector caspases -3 and -7: isatin sulfonamide (compound 1) (Lee et al., 2000). Processing of caspase-3 occurs via the generation of a p20 intermediate that is subsequently processed to a p17 fragment (Schlegel et al., 1996). Binding of peptide-based inhibitors to the p20 subunit blocks caspase-3 activity (Martin et al., 1996). Unlike the peptide-based caspase inhibitors, compound 1 exerts its selectivity for caspases -3 and -7 by interacting primarily with the S2 binding pocket and not with the primary aspartic acid pocket (S1). Our immunoblot analysis clearly showed that compound 1 blocks further processing of the p20 subunit of caspase-3 to p17 and p15 subunits in a dose-dependent fashion in Fas-mediated apoptosis. During GrB-mediated apoptosis, the processing of the p20 intermediate to the p17 fragment was also blocked by compound 1, but had no effect on DFF45 cleavage or DNA fragmentation. The absence of Rho-GDI processing by GrB in the presence of compound 1 clearly indicates that (i) the p20 subunit of caspase-3 has no enzymatic activity capable of substrate cleavage and (ii) there is no residual caspase-3 activity present in compound 1-treated cells, which excludes the involvement of caspase-3 in the induction of DFF45 cleavage and DNA fragmentation. These results clearly indicate the existence of an alternative, caspase-independent cascade for GrB-mediated DNA fragmentation and induction of apoptosis, as depicted in Figure 5. This further provides evidence that CTL-mediated apoptosis is initiated in the absence of caspase activity, and that activation of caspases serves to amplify the apoptotic process. The ability of the lymphocyte granule-induced cytotoxicity pathway to efficiently cleave downstream caspase substrates, such as the DNA-PKcs and NuMa (Andrade et al., 1998), in the absence of caspase activity, is consistent with this proposal.

Fig. 5. Mechanism of GrB-induced DNA fragmentation in the absence of caspase-3 activity. Following the induction of cellular cytotoxicity, GrB enters target cells and becomes activated in the presence of perforin. Rapid translocation of GrB from the cytoplasm to the nucleus leads to nuclear substrate cleavage. Direct cleavage of DFF45 by GrB induces DNA fragmentation via bypassing of caspase activation. GrB-induced caspase activity will serve to augment the apoptotic process.
Our data from in vivo mutational analysis using human DFF45 differ from that reported by Sakahira et al. (1998) on murine ICAD. These authors have shown that both D117 and D224 sites on murine ICAD must be cleaved by caspase-3 to induce DNA fragmentation. Surprisingly, and in contrast to other published data (Liu et al., 1998), the authors also demonstrated that overexpression of exogenous wild-type DFF45 is able to block DNA fragmentation following the induction of cell death. In contrast, we found that cleavage following only the D117 residue in human DFF45 is both necessary and sufficient for DFF40 activation and induction of DNA fragmentation. Mutation of D117, but not D224, completely abrogated DNA fragmentation. Blocking of DNA fragmentation in DFF45- M1 transfectants occurred without affecting other morphological and biochemical features accompanying cell death (E.Sharif-Askari, in preparation; Figure 4B). The discrepancy between our results and those published by Sakahira et al. (1998) might stem from higher overexpression of exogenous DFF45/ICAD in their system relative to ours. With a higher exogenous DFF45/ICAD substrate concentration, it is possible that the presence of excess, uncleaved DFF45/ICAD inhibited DFF40/CAD nuclease activity following its release and activation by caspase-3. In agreement with this hypothesis, Mitamura et al. (1998) have shown that following activation of DFF40 by caspase-3, addition of exogenous, uncleaved DFF45 completely inhibits DFF40 DNase activity. Thus, the presence of uncleaved DFF45 may function as a trap for released DFF40 endonuclease, thereby preventing DNA fragmentation.
Species-specific differences between human DFF45 and murine ICAD could also account for the discrepancy between our results and those of Sakahira et al. (1998). The presence of a putative leucine zipper motif at the N-terminal region of murine ICAD, but not of human DFF45, may enforce a stronger avidity between ICAD and CAD, thus requiring cleavage at both D117 and D224, especially since the interaction between DFF45/ICAD and DFF40/CAD occurs through the N-terminal fragment (Gu et al., 1999). Furthermore, species-specific differences as to the mechanisms of release and activation of DFF40/CAD have also been demonstrated with the Drosophila melanogaster dICAD/dCAD (Yokoyama et al., 2000). Indeed, activation of dCAD needs processing of both dICAD and dCAD by caspase-3, while activation of DFF40/CAD requires only the cleavage of DFF45/ICAD.
Although it is largely accepted that GrB induces cell death through the activation of caspases, it also initiates caspase-independent pathways that contribute to target cell death (Sarin et al., 1997; Talanian et al., 1997). It has been reported that in cells lacking caspase-3, DFF45 is still cleaved in response to apoptotic stimuli, suggesting that other protease(s) may also cleave DFF45 (Janicke et al., 1998; Woo et al., 1999). Several recent studies demonstrated that GrB can enter the cytoplasm of target cells in the absence of perforin, an event that is not sufficient, on its own, to trigger target cell death (Trapani, 1995; Froelich et al., 1996; Trapani et al., 1996; Shi et al., 1997). Perforin treatment or Ad2 infection leads to the induction of cell death accompanied by a rapid nuclear translocation and the accumulation of GrB (Jans et al., 1996; Trapani et al., 1996; Pinkoski et al., 1998). It is, therefore, of interest that the caspase targets described previously (Andrade et al., 1998), as well as lamin (Zhang et al., 2001) and the substrate described herein, all of which are cleaved directly by GrB in vitro and in vivo, are localized in the nucleus. Nevertheless, we can not exclude the remote possibility that GrB cleaves an unknown caspase or protease that has the ability to induce DNA fragmentation in the absence of caspase-3 activity.
Viruses can express caspase inhibitors that allow infected cells to escape cell death; tumors also have built-in mechanisms that allow them to overcome caspase-mediated apoptosis (Beidler et al., 1995; Bump et al., 1995; Tewari et al., 1995; Xue and Horvitz, 1995; Macen et al., 1996; Deveraux et al., 1997; Irmler et al., 1997; Krajewska et al., 1997; Krajewski et al., 1997; Srinivasula et al., 1997; Thome et al., 1997; Uren and Vaux, 1997). The caspase-independent pathway of cell death described above might provide the host with apoptotic effector mechanisms that are insensitive to inhibitors of the execution components of the caspase cascade. In conclusion, the caspase-independent mechanism of DFF45 processing by GrB reported here offers additional/alternative cellular pathways during CTL-mediated cell death (Figure 5).
Materials and methods
Reagents and cell lines
Jurkat cells were grown in RPMI 1640 medium (Gibco BRL, Burlington, Canada) supplemented with 10% fetal calf serum. To generate stably transfected Jurkat cells expressing DFF45, cells were transfected by electroporation (240 V, 290 µF) in the presence of 10 µg of plasmid DNA, selected in 1 mg/ml G418 (Life Technologies, Inc.), and cloned by limiting dilution. GrB used for in vitro DFF45 cleavage experiments was purchased from Enzyme Systems Products (Dablin, CA). GrB used for the induction of apoptosis was prepared as described previously (Froelich et al., 1996; Beresford et al., 1999). Murine anti-human Fas monoclonal antibody (M3) was a generous gift from D.Lynch (Immunex) and was routinely used at 20 µg/ml. Rabbit anti-caspase-3 has been described (Alam et al., 1997). Rabbit anti-Rho-GDI was purchased from PharMingen. The caspase-3- and -7-specific inhibitor isatin sulfonamide (compound 1) and its 600-fold less active analog, compound 2, were a generous gift from SmithKline Beecham. Both compound 1 and compound 2 were dissolved in DMSO and stored at –20°C. Annexin V (Nexins Research B.V., Hoeven, The Netherlands) and propidium iodide (PI; Sigma) staining were performed as described previously (Alam et al., 1999).
Production of DFF fusion protein and generation of anti-DFF antibody
The cDNA encoding the entire open reading frame of DFF45 was reverse transcribed from total RNA of Jurkat cell lines and amplified by PCR using oligonucleotides (1) and (2) (see below). Amplified products were cloned into the BamHI and EcoRI sites of pBS-KS+ (Stratagene, La Jolla, CA) and sequenced. The DFF45 gene was then subcloned into the pGEX-2Tk vector (Pharmacia, Baie d’Urfé, Canada). After induction with isopropyl-β-d-thiogalactopyranoside (IPTG) (0.1 µM) for 3 h at 30°C, soluble glutathione S-transferase (GST) fusion protein was purified on glutathione–Sepharose 4B columns (Pharmacia), and the GST was removed by cleavage of the fusion protein with thrombin. Rabbits were immunized intramuscularly and subcutaneously by three injections of purified protein (250 µg per injection at 4 week intervals).
Mutagenesis and generation of DFF fragments
The oligonucleotide sequences used in this study for DFF45 cloning and mutagenesis were: (1) DFF-D: GCAGGATCCACCTTGTGGAGGATGGAGGTG; (2) DFF-F: CGCGAATTCTATGTGGGGTCCTGTCTGGCTCGCTTAGG; (3) DFF-M1D: GTAGATGAAACAGAGAGC GGGGCAG; (4) DFF-M1F: CTGCCCGCTCTCTGTTTCATCTAC; (5) DFF-M2D: GATGCAGTAGAGACGGGTATCAGCAG; (6) DFF-M2F: CTGCTGATACCCGTCTCTACTGCATC; (7) DFF-F1F: CGCGAATTCTAGTCTGTTTCATCTACATCAAAGG; (8) DFF-F2D: GGGATCCACCTTGTGGAGGATGAGCGGGGCAGGGTTGAGG; (9) DFF-F2F: CGCGAATTCTAGTCTACTGCATCCACCTCCTC; (10) DFF-F3D: GGGATCCACCTTGTGGAGGATGACGGGTATCAGCAGGAG.
The DFF45 mutants, DFF45-D117E (DFF45-M1), DFF45-D224E (DFF45-M2) and DFF45-D117E/D224E (DFF45-M12), were generated by a two-step overlapping PCR (Dillon and Rosen, 1990). In the first round of PCR, the following primer pairs were used: numbers 1–4 and 2–3 for DFF45-M1; 1–6 and 2–5 for DFF45-M2; and 1–4, 3–6 and 5–2 for DFF45-M12. In the second round of PCR, primers 1 and 2 were used for all mutants. Deletion mutants were generated by PCR using primers 1 and 7 for DFF45-F1 (amino acids 1–117), primers 8 and 9 for DFF45-F2 (amino acids 118–224) and primers 10 and 2 for DFF45-F3 (amino acids 225–331). PCR products were then cloned into pBS-KS+. The authenticity of all constructs was confirmed by dideoxy sequencing. DFF45-WT and mutants were then inserted into the BamHI and EcoRI sites of pcDNA3.1-His (Invitrogen) to produce N-terminal His-tagged proteins. In the transfection experiments, DFF45-WT, DFF45-M1, DFF45-M2 and DFF45-M12 were cloned into the SRα-neo vector.
Human DFF40 was amplified from Jurkat cells by RT–PCR using the upstream primer CCGCTCGAGAGATCTATGCTCCAGAAGCCCAAGAGCGTG and the downstream primer ATAAGAATGCGGCCGCTTCACTGGCGTTTCCGCACAGGC. The PCR product was then cloned into pGEM-T vector (Promega) and sequenced. The BglII–SalI fragment was subsequently cloned into the BamHI–XhoI of pBS-KS+ plasmid.
Assays for apoptosis
For anti-Fas-induced apoptosis, Jurkat cells (1 × 106 cells per well) were cultured at 37°C for various time intervals in 24-well culture plates in the presence of immobilized affinity-purified M3 mAb (20 µg/ml). GrB-induced target cell killing in the presence of Ad2 infection was carried out as described previously (Pinkoski et al., 1998). Briefly, Jurkat cells were pre-incubated with 1 µl of GrB (8.7 nM final concentration) in RPMI 1640 supplemented with 0.5% bovine serum albumin (BSA) for 1 h, followed by infection with type-2 Ad2 (10 plaque-forming units per cell) for an additional 4 h at 37°C. After Fas- or GrB/Ad2-induced apoptosis, the cells were collected, washed twice with phosphate-buffered saline (PBS) and analyzed for annexin V/PI double staining, caspase-3 and DFF45 cleavage, and DNA fragmentation. GrB loading with perforin was performed as described previously (Beresford et al., 1999). Briefly, cells (1 × 105) in 30–60 µl of Hanks’ balanced salt solution reaction buffer containing 1 mg/ml BSA, 1 mM CaCl2 and 1 mM MgCl2 were incubated with GrB (1 µM final concentration) and a sublytic concentration of perforin in the presence or absence of caspase-3 inhibitor for ∼3 h at 37°C. Cells were then treated for an additional 30 min with 1 mM phenylmethylsulfonyl fluoride before lysis in 2× SDS–PAGE sample buffer (0.125 M Tris–HCl pH 6.8, 4% SDS, 25% glycerol and 10% β-mercaptoethanol) to prevent further proteolytic digestion by GrB.
Assays for DNA fragmentation
Fragmented DNA was extracted using a phenol/chloroform extraction assay. Briefly, the cell pellet (5 × 105 cells) was resuspended in 25 µl of PBS, and an equal volume of phenol/chloroform/isoamylalcohol (l:1:0.1) was added. Following gentle agitation and centrifugation (10 000 g for 2 min), fragmented DNA was recovered, treated with RNase A for 1 h at 37°C and analyzed on 2% agarose gels containing ethidium bromide.
Confocal immunofluorescence microscopy
Cells were centrifuged onto polylysine-coated glass microscope slides (Baxter Diagnostics Inc., Deerfield, IL), washed briefly with PBS, fixed in 3.7% formaldehyde (Sigma) for 15 min at room temperature and followed by a brief rinse with PBS. Cells were then permeabilized for 10 min in PBS ± 0.2% Triton X-100, washed three times with PBS and then incubated with PBS containing 3% BSA for 1 h at room temperature. After a brief wash with PBS, cells were incubated with either anti-DFF45 or anti-DFF40 polyclonal antibodies (Affinity BioReagents Inc., Golden, CO) diluted 1:200 in PBS ± 0.1% Tween-20 ± 0.2% BSA for 1 h at room temperature. Control cells were incubated with pre-immune serum (1:200). Cells were washed three times in PBS ± 0.1% Tween-20 (PBST) for 10 min, blocked for 1 h at room temperature and then incubated for 1 h at room temperature with mAbs to anti-β-actin (Sigma) at a 1:100 dilution, followed by 1:500-diluted tetramethylrhodamine isothiocyanate (TRITC)-coupled anti-rabbit IgG (Sigma) and 1:250-diluted fluorescein isothiocyanate (FITC)-coupled anti-mouse IgG (Sigma). Control cells were only incubated with the secondary TRITC-coupled anti-rabbit IgG (1:500). Between incubations, cells were washed with PBST and blocked for 1 h in PBS ± 3% BSA at room temperature. After three final washes with PBST, cells were washed once in PBS for 10 min and mounted in DABCO mounting medium (Sigma). Slides were analyzed with a confocal laser microscope (LSM 510-axioplan2; Zeiss, Jena, Germany) and photographs were taken with a CCD camera. Each photograph is representative of at least 10 fields of two independent experiments at 1000× magnification.
In vitro transcription and translation
35S-labeled DFF45 and DFF40 proteins were generated using an in vitro rabbit reticulocyte TNT kit (Promega) according to the manufacturer’s instructions. In brief, pcDNA3.1His-DFF45 or pBS-DFF40 (1 µg) was incubated with the TNT reaction mixture containing 25 µl of rabbit reticulocyte lysate, 2 µl of TNT reaction buffer, 1 µl of T7 RNA polymerase, 1 mM amino acid mixture minus cysteine, 2 µl of [35S]cysteine or 2 µl of [35S]methionine (10 mCi/ml) and 40 U of RNase inhibitor (Amersham Pharmacia Biotech, Inc.) in a total volume of 50 µl for 90 min at 30°C.
In vitro cleavage of DFF45 by GrB and caspase-3
For in vitro cleavage, recombinant DFF45 (12 ng) or 35S-labeled DFF45 (2 µl) was incubated in the presence of 10 U of caspase-3 (1 U corresponds to the amount of enzyme necessary to release 1 pmol of AMC per minute from DEVD-AMC cleavage at 25°C) in the caspase-3 reaction buffer (20 mM HEPES pH 7.0, 5 mM EDTA, 2 mM dithiothreitol, 10% sucrose) or 1 U of GrB (1 U corresponds to the amount of enzyme necessary to release 1 pmol of AMC per minute from IETD-AMC cleavage at 25°C) in 150 mM NaCl. The reactions were performed at 30°C in a final volume of 10 µl for various time intervals. The reactions were then stopped by the addition of an equal volume of 2× Laemmli buffer. Cleavage products were then separated by 15% SDS–PAGE and detected either by immunoblotting (see below) or phosphorimaging of the dried gels (Molecular Dynamics, Sunnyvale, CA).
Purification of active caspase-3
Purified caspase-3 was obtained after overexpression of His6-tagged caspase-3 in Escherichia coli. Briefly, full-length caspase-3 cDNA (Rheaume et al., 1997) was subcloned into the pRSETB vector (Invitrogen), allowing in-frame C-terminal histidine tagging. Plasmid pRSET-caspase-3 was introduced into E.coli DH5, and the protein was isolated from the soluble fraction, following induction with 0.2 mM IPTG at 30°C for 3 h, by affinity purification using Talon metal affinity resin (Clontech Laboratories, Inc.) and then concentrated in caspase-3 buffer (see above) using a Centricon-10 filter (Amicon). A second purification step was performed using FPLC to maximize the purity of the enzyme. The purity of the protein and assessment of its autoactivation were achieved by Coomassie Blue and silver staining of the protein. The protein concentration was determined using the Bradford assay. Caspase-3 activity was measured by spectrofluorometry using DEVD-AMC (Peptide Institute, Inc.). One unit is defined as the amount of enzyme that releases 1 pmol of AMC per minute at 25°C.
Calculation of catalytic constant
Catalytic constant (Kcat/Km) values were determined using purified caspase-3 (Calbiochem) or GrB (Enzyme Systems Products) on subsaturating substrate concentrations as described (Casciola-Rosen et al., 1996). 35S-labeled substrate and cleavage product bands were quantified on a PhosphorImager (Molecular Dynamics) and Kcat/Km values were calculated using the first-order rate equation Kcat/Km = –[ln (fraction of uncleaved substrate)/([enzyme] × time)] using at least three different time points and different enzyme concentrations.
Immunoblotting analysis
Following the reactions, cells destined for immunoblotting were washed with PBS, lysed in Laemmli buffer and boiled for 5 min. The samples were then resolved using 15% SDS–PAGE and transferred to nitrocellulose membrane (Hybond C Super; Amersham, Oakville, Canada). Blots were blocked overnight at 4°C in 5% non-fat dried milk in PBST (PBS + 0.5% Tween-20). Membranes were then incubated for 1 h with rabbit anti-human caspase-3 (1/2500), anti-DFF45 (1/2500) or anti-Rho-GDI (1/5000) at 25°C. The blots were then washed three times in PBST and re-incubated in the presence of horseradish peroxidase-conjugated goat anti-rabbit antibody (1:2500; Jackson Laboratories) for 1 h at 25°C. Following three washes with PBST, the proteins were detected with the enhanced chemiluminescence (ECL) kit (Amersham, Oakville, Canada).
Assay for DFF40 nuclease activity
For DFF40 nuclease activity, pBS-KS+/DFF40 (1 µg) was expressed in an in vitro transcription and translation system in the presence of 160–200 ng of recombinant DFF45 as described previously (Mukae et al., 1998). Three microliters of TNT reaction mixture containing DFF complex were then incubated with either caspase-3 (0.2 µM) or GrB (0.12 µM) for 60 min in DFF40 buffer (10 mM HEPES pH 7.4, 5 mM MgCl2, 50 mM NaCl, 1 mg/ml BSA and 5 mM EGTA) at 30°C in the presence of 1 µg of pcDNA-3.1His plasmid as the substrate. The DNase activity of DFF40 was then analyzed by electrophoresis on 1% agarose gels.
Acknowledgments
Acknowledgements
We are grateful to Drs C.Bleackley, M.Barry and J.Gauldie for kindly providing us with granule-purified GrB and adenovirus. We are also grateful to Dr C.Lazure for protein sequencing. We thank Dr M.Vaillancourt, L.M.Fawaz and L.Sabbagh for critical reading of the manuscript, and Dr M.Bourbonniere for valuable discussions. This work was supported by grant number MOP 38105 from the Canadian Institutes of Health Research (CIHR) to R.-P.S. E.S.-A. is the recipient of a CIHR doctoral research award. J.L. is supported by NIH grant AI 45587. R.-P.S. holds a CIHR scientist award.
References
- Alam A., Braun,M.Y., Hartgers,F., Lesage,S., Cohen,L., Hugo,P., Denis,F. and Sekaly,R.P. (1997) Specific activation of the cysteine protease CPP32 during the negative selection of T cells in the thymus. J. Exp. Med., 186, 1503–1512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alam A., Cohen,L.Y., Aouad,S. and Sekaly,R.P. (1999) Early activation of caspases during T lymphocyte stimulation results in selective substrate cleavage in nonapoptotic cells. J. Exp. Med., 190, 1879–1890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrade F., Roy,S., Nicholson,D., Thornberry,N., Rosen,A. and Casciola-Rosen,L. (1998) Granzyme B directly and efficiently cleaves several downstream caspase substrates: implications for CTL-induced apoptosis. Immunity, 8, 451–460. [DOI] [PubMed] [Google Scholar]
- Atkinson E.A., Barry,M., Darmon,A.J., Shostak,I., Turner,P.C., Moyer,R.W. and Bleackley,R.C. (1998) Cytotoxic T lymphocyte-assisted suicide. Caspase 3 activation is primarily the result of the direct action of granzyme B. J. Biol. Chem., 273, 21261–21266. [DOI] [PubMed] [Google Scholar]
- Beidler D.R., Tewari,M., Friesen,P.D., Poirier,G. and Dixit,V.M. (1995) The baculovirus p35 protein inhibits Fas- and tumor necrosis factor-induced apoptosis. J. Biol. Chem., 270, 16526–16528. [DOI] [PubMed] [Google Scholar]
- Beresford P.J., Xia,Z., Greenberg,A.H. and Lieberman,J. (1999) Granzyme A loading induces rapid cytolysis and a novel form of DNA damage independently of caspase activation. Immunity, 10, 585–594. [DOI] [PubMed] [Google Scholar]
- Budihardjo I., Oliver,H., Lutter,M., Luo,X. and Wang,X. (1999) Biochemical pathways of caspase activation during apoptosis. Annu. Rev. Cell Dev. Biol., 15, 269–290. [DOI] [PubMed] [Google Scholar]
- Bump N.J. et al. (1995) Inhibition of ICE family proteases by baculovirus antiapoptotic protein p35. Science, 269, 1885–1888. [DOI] [PubMed] [Google Scholar]
- Caputo A., Parrish,J.C., James,M.N., Powers,J.C. and Bleackley,R.C. (1999) Electrostatic reversal of serine proteinase substrate specificity. Proteins, 35, 415–424. [PubMed] [Google Scholar]
- Casciola-Rosen L., Nicholson,D.W., Chong,T., Rowan,K.R., Thornberry,N.A., Miller,D.K. and Rosen,A. (1996) Apopain/CPP32 cleaves proteins that are essential for cellular repair: a fundamental principle of apoptotic death. J. Exp. Med., 183, 1957–1964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chinnaiyan A.M., Hanna,W.L., Orth,K., Duan,H., Poirier,G.G., Froelich,C.J. and Dixit,V.M. (1996) Cytotoxic T-cell-derived granzyme B activates the apoptotic protease ICE-LAP3. Curr. Biol., 6, 897–899. [DOI] [PubMed] [Google Scholar]
- Cohen G.M. (1997) Caspases: the executioners of apoptosis. Biochem. J., 326, 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deveraux Q.L., Takahashi,R., Salvesen,G.S. and Reed,J.C. (1997) X-linked IAP is a direct inhibitor of cell-death proteases. Nature, 388, 300–304. [DOI] [PubMed] [Google Scholar]
- Dillon P.J. and Rosen,C.A. (1990) A rapid method for the construction of synthetic genes using the polymerase chain reaction. Biotechniques, 9, 298–300. [PubMed] [Google Scholar]
- Enari M., Sakahira,H., Yokoyama,H., Okawa,K., Iwamatsu,A. and Nagata,S. (1998) A caspase-activated DNase that degrades DNA during apoptosis, and its inhibitor ICAD. Nature, 391, 43–50. [DOI] [PubMed] [Google Scholar]
- Froelich C.J. et al. (1996) New paradigm for lymphocyte granule-mediated cytotoxicity. Target cells bind and internalize granzyme B, but an endosomolytic agent is necessary for cytosolic delivery and subsequent apoptosis. J. Biol. Chem., 271, 29073–29079. [DOI] [PubMed] [Google Scholar]
- Gu J., Dong,R.P., Zhang,C., McLaughlin,D.F., Wu,M.X. and Schlossman,S.F. (1999) Functional interaction of DFF35 and DFF45 with caspase-activated DNA fragmentation nuclease DFF40. J. Biol. Chem., 274, 20759–20762. [DOI] [PubMed] [Google Scholar]
- Hakem R. et al. (1998) Differential requirement for caspase 9 in apoptotic pathways in vivo. Cell, 94, 339–352. [DOI] [PubMed] [Google Scholar]
- Halenbeck R., MacDonald,H., Roulston,A., Chen,T.T., Conroy,L. and Williams,L.T. (1998) CPAN, a human nuclease regulated by the caspase-sensitive inhibitor DFF45. Curr. Biol., 8, 537–540. [DOI] [PubMed] [Google Scholar]
- Heusel J.W., Wesselschmidt,R.L., Shresta,S., Russell,J.H. and Ley,T.J. (1994) Cytotoxic lymphocytes require granzyme B for the rapid induction of DNA fragmentation and apoptosis in allogeneic target cells. Cell, 76, 977–987. [DOI] [PubMed] [Google Scholar]
- Irmler M. et al. (1997) Inhibition of death receptor signals by cellular FLIP. Nature, 388, 190–195. [DOI] [PubMed] [Google Scholar]
- Janicke R.U., Ng,P., Sprengart,M.L. and Porter,A.G. (1998) Caspase-3 is required for α-fodrin cleavage but dispensable for cleavage of other death in apoptosis substrates in apoptosis. J. Biol. Chem., 273, 15540–15545. [DOI] [PubMed] [Google Scholar]
- Jans D.A., Jans,P., Briggs,L.J., Sutton,V. and Trapani,J.A. (1996) Nuclear transport of granzyme B (fragmentin-2). Dependence of perforin in vivo and cytosolic factors in vitro. J. Biol. Chem., 271, 30781–30789. [DOI] [PubMed] [Google Scholar]
- Kagi D. and Hengartner,H. (1996) Different roles for cytotoxic T cells in the control of infections with cytopathic versus noncytopathic viruses. Curr. Opin. Immunol., 8, 472–477. [DOI] [PubMed] [Google Scholar]
- Krajewska M., Wang,H.G., Krajewski,S., Zapata,J.M., Shabaik,A., Gascoyne,R. and Reed,J.C. (1997) Immunohistochemical analysis of in vivo patterns of expression of CPP32 (caspase-3), a cell death protease. Cancer Res., 57, 1605–1613. [PubMed] [Google Scholar]
- Krajewski S. et al. (1997) Immunolocalization of the ICE/Ced-3-family protease, CPP32 (Caspase-3), in non-Hodgkin’s lymphomas, chronic lymphocytic leukemias, and reactive lymph nodes. Blood, 89, 3817–3825. [PubMed] [Google Scholar]
- Lechardeur D. et al. (2000) Determinants of the nuclear localization of the heterodimeric DNA fragmentation factor (ICAD/CAD). J. Cell Biol., 150, 321–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee D. et al. (2000) Potent and selective nonpeptide inhibitors of caspases 3 and 7 inhibit apoptosis and maintain cell functionality. J. Biol. Chem., 275, 16007–16014. [DOI] [PubMed] [Google Scholar]
- Liu X., Zou,H., Slaughter,C. and Wang,X. (1997) DFF, a heterodimeric protein that functions downstream of caspase-3 to trigger DNA fragmentation during apoptosis. Cell, 89, 175–184. [DOI] [PubMed] [Google Scholar]
- Liu X., Li,P., Widlak,P., Zou,H., Luo,X., Garrard,W.T. and Wang,X. (1998) The 40-kDa subunit of DNA fragmentation factor induces DNA fragmentation and chromatin condensation during apoptosis. Proc. Natl Acad. Sci. USA, 95, 8461–8466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X., Zou,H., Widlak,P., Garrard,W. and Wang,X. (1999) Activation of the apoptotic endonuclease DFF40 (caspase-activated DNase or nuclease). Oligomerization and direct interaction with histone H1. J. Biol. Chem., 274, 13836–13840. [DOI] [PubMed] [Google Scholar]
- Macen J.L., Graham,K.A., Lee,S.F., Schreiber,M., Boshkov,L.K. and McFadden,G. (1996) Expression of the myxoma virus tumor necrosis factor receptor homologue and M11L genes is required to prevent virus-induced apoptosis in infected rabbit T lymphocytes. Virology, 218, 232–237. [DOI] [PubMed] [Google Scholar]
- Martin S.J. et al. (1996) The cytotoxic cell protease granzyme B initiates apoptosis in a cell-free system by proteolytic processing and activation of the ICE/CED-3 family protease, CPP32, via a novel two-step mechanism. EMBO J., 15, 2407–2416. [PMC free article] [PubMed] [Google Scholar]
- Medema J.P., Toes,R.E., Scaffidi,C., Zheng,T.S., Flavell,R.A., Melief,C.J., Peter,M.E., Offringa,R. and Krammer,P.H. (1997) Cleavage of FLICE (caspase-8) by granzyme B during cytotoxic T lymphocyte-induced apoptosis. Eur. J. Immunol., 27, 3492–3498. [DOI] [PubMed] [Google Scholar]
- Mitamura S., Ikawa,H., Mizuno,N., Kaziro,Y. and Itoh,H. (1998) Cytosolic nuclease activated by caspase-3 and inhibited by DFF-45. Biochem. Biophys. Res. Commun., 243, 480–484. [DOI] [PubMed] [Google Scholar]
- Mukae N., Enari,M., Sakahira,H., Fukuda,Y., Inazawa,J., Toh,H. and Nagata,S. (1998) Molecular cloning and characterization of human caspase-activated DNase. Proc. Natl Acad. Sci. USA, 95, 9123–9128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Na S., Chuang,T.H., Cunningham,A., Turi,T.G., Hanke,J.H., Bokoch,G.M. and Danley,D.E. (1996) D4-GDI, a substrate of CPP32, is proteolyzed during Fas-induced apoptosis. J. Biol. Chem., 271, 11209–11213. [DOI] [PubMed] [Google Scholar]
- Nicholson D.W. and Thornberry,N.A. (1997) Caspases: killer proteases. Trends Biochem. Sci., 22, 299–306. [DOI] [PubMed] [Google Scholar]
- Odake S., Kam,C.M., Narasimhan,L., Poe,M., Blake,J.T., Krahenbuhl,O., Tschopp,J. and Powers,J.C. (1991) Human and murine cytotoxic T lymphocyte serine proteases: subsite mapping with peptide thioester substrates and inhibition of enzyme activity and cytolysis by isocoumarins. Biochemistry, 30, 2217–2227. [DOI] [PubMed] [Google Scholar]
- Pinkoski M.J., Hobman,M., Heibein,J.A., Tomaselli,K., Li,F., Seth,P., Froelich,C.J. and Bleackley,R.C. (1998) Entry and trafficking of granzyme B in target cells during granzyme B–perforin-mediated apoptosis. Blood, 92, 1044–1054. [PubMed] [Google Scholar]
- Poe M., Blake,J.T., Boulton,D.A., Gammon,M., Sigal,N.H., Wu,J.K. and Zweerink,H.J. (1991) Human cytotoxic lymphocyte granzyme B. Its purification from granules and the characterization of substrate and inhibitor specificity. J. Biol. Chem., 266, 98–103. [PubMed] [Google Scholar]
- Rheaume E., Cohen,L.Y., Uhlmann,F., Lazure,C., Alam,A., Hurwitz,J., Sekaly,R.P. and Denis,F. (1997) The large subunit of replication factor C is a substrate for caspase-3 in vitro and is cleaved by a caspase-3-like protease during Fas-mediated apoptosis. EMBO J., 16, 6346–6354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakahira H., Enari,M. and Nagata,S. (1998) Cleavage of CAD inhibitor in CAD activation and DNA degradation during apoptosis. Nature, 391, 96–99. [DOI] [PubMed] [Google Scholar]
- Sakahira H., Iwamatsu,A. and Nagata,S. (2000) Specific chaperone-like activity of inhibitor of caspase-activated DNase for caspase-activated DNase. J. Biol. Chem., 275, 8091–8096. [DOI] [PubMed] [Google Scholar]
- Samejima K. and Earnshaw,W.C. (1998) ICAD/DFF regulator of apoptotic nuclease is nuclear. Exp. Cell Res., 243, 453–459. [DOI] [PubMed] [Google Scholar]
- Samejima K. and Earnshaw,W.C. (2000) Differential localization of ICAD-L and ICAD-S in cells due to removal of a C-terminal NLS from ICAD-L by alternative splicing. Exp. Cell Res., 255, 314–320. [DOI] [PubMed] [Google Scholar]
- Sarin A., Wu,M.L. and Henkart,P.A. (1996) Different interleukin-1β converting enzyme (ICE) family protease requirements for the apoptotic death of T lymphocytes triggered by diverse stimuli. J. Exp. Med., 184, 2445–2450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarin A., Williams,M.S., Alexander-Miller,M.A., Berzofsky,J.A., Zacharchuk,C.M. and Henkart,P.A. (1997) Target cell lysis by CTL granule exocytosis is independent of ICE/Ced-3 family proteases. Immunity, 6, 209–215. [DOI] [PubMed] [Google Scholar]
- Schlegel J., Peters,I., Orrenius,S., Miller,D.K., Thornberry,N.A., Yamin,T.T. and Nicholson,D.W. (1996) CPP32/apopain is a key interleukin 1β converting enzyme-like protease involved in Fas-mediated apoptosis. J. Biol. Chem., 271, 1841–1844. [DOI] [PubMed] [Google Scholar]
- Shi L., Kraut,R.P., Aebersold,R. and Greenberg,A.H. (1992) A natural killer cell granule protein that induces DNA fragmentation and apoptosis. J. Exp. Med., 175, 553–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi L., Mai,S., Israels,S., Browne,K., Trapani,J.A. and Greenberg,A.H. (1997) Granzyme B (GraB) autonomously crosses the cell membrane and perforin initiates apoptosis and GraB nuclear localization. J. Exp. Med., 185, 855–866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shresta S., Heusel,J.W., Macivor,D.M., Wesselschmidt,R.L., Russell,J.H. and Ley,T.J. (1995) Granzyme B plays a critical role in cytotoxic lymphocyte-induced apoptosis. Immunol. Rev., 146, 211–221. [DOI] [PubMed] [Google Scholar]
- Shresta S., Pham,C.T., Thomas,D.A., Graubert,T.A. and Ley,T.J. (1998) How do cytotoxic lymphocytes kill their targets? Curr. Opin. Immunol., 10, 581–587. [DOI] [PubMed] [Google Scholar]
- Slee E.A. et al. (1999) Ordering the cytochrome c-initiated caspase cascade: hierarchical activation of caspases-2, -3, -6, -7, -8, and -10 in a caspase-9-dependent manner. J. Cell Biol., 144, 281–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song Q. et al. (1996) Interleukin-1β-converting enzyme-like protease cleaves DNA-dependent protein kinase in cytotoxic T cell killing. J. Exp. Med., 184, 619–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srinivasula S.M. et al. (1997) FLAME-1, a novel FADD-like anti-apoptotic molecule that regulates Fas/TNFR1-induced apoptosis. J. Biol. Chem., 272, 18542–18545. [DOI] [PubMed] [Google Scholar]
- Strasser A., Harris,A.W. and Cory,S. (1991) bcl-2 transgene inhibits T cell death and perturbs thymic self-censorship. Cell, 67, 889–899. [DOI] [PubMed] [Google Scholar]
- Sutton V.R., Vaux,D.L. and Trapani,J.A. (1997) Bcl-2 prevents apoptosis induced by perforin and granzyme B, but not that mediated by whole cytotoxic lymphocytes. J. Immunol., 158, 5783–5790. [PubMed] [Google Scholar]
- Talanian R.V., Yang,X., Turbov,J., Seth,P., Ghayur,T., Casiano,C.A., Orth,K. and Froelich,C.J. (1997) Granule-mediated killing: pathways for granzyme B-initiated apoptosis. J. Exp. Med., 186, 1323–1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tewari M., Telford,W.G., Miller,R.A. and Dixit,V.M. (1995) CrmA, a poxvirus-encoded serpin, inhibits cytotoxic T-lymphocyte-mediated apoptosis. J. Biol. Chem., 270, 22705–22708. [DOI] [PubMed] [Google Scholar]
- Thomas D.A., Du,C., Xu,M., Wang,X. and Ley,T.J. (2000) DFF45/ICAD can be directly processed by granzyme B during the induction of apoptosis. Immunity, 12, 621–632. [DOI] [PubMed] [Google Scholar]
- Thome M. et al. (1997) Viral FLICE-inhibitory proteins (FLIPs) prevent apoptosis induced by death receptors. Nature, 386, 517–521. [DOI] [PubMed] [Google Scholar]
- Trapani J.A. (1995) Target cell apoptosis induced by cytotoxic T cells and natural killer cells involves synergy between the pore-forming protein, perforin, and the serine protease, granzyme B. Aust. N. Z. J. Med., 25, 793–799. [DOI] [PubMed] [Google Scholar]
- Trapani J.A., Browne,K.A., Smyth,M.J. and Jans,D.A. (1996) Localization of granzyme B in the nucleus. A putative role in the mechanism of cytotoxic lymphocyte-mediated apoptosis. J. Biol. Chem., 271, 4127–4133. [DOI] [PubMed] [Google Scholar]
- Uren A.G. and Vaux,D.L. (1997) Viral inhibitors of apoptosis. Vitam. Horm., 53, 175–193. [DOI] [PubMed] [Google Scholar]
- Van de Craen M., Van den Brande,I., Declercq,W., Irmler,M., Beyaert,R., Tschopp,J., Fiers,W. and Vandenabeele,P. (1997) Cleavage of caspase family members by granzyme B: a comparative study in vitro. Eur. J. Immunol., 27, 1296–1299. [DOI] [PubMed] [Google Scholar]
- Vaux D.L., Aguila,H.L. and Weissman,I.L. (1992) Bcl-2 prevents death of factor-deprived cells but fails to prevent apoptosis in targets of cell mediated killing. Int. Immunol., 4, 821–824. [DOI] [PubMed] [Google Scholar]
- Villa P., Kaufmann,S.H. and Earnshaw,W.C. (1997) Caspases and caspase inhibitors. Trends Biochem. Sci., 22, 388–393. [DOI] [PubMed] [Google Scholar]
- Wolf B.B., Schuler,M., Echeverri,F. and Green,D.R. (1999) Caspase-3 is the primary activator of apoptotic DNA fragmentation via DNA fragmentation factor-45/inhibitor of caspase-activated DNase inactivation. J. Biol. Chem., 274, 30651–30656. [DOI] [PubMed] [Google Scholar]
- Woo M., Hakem,A., Elia,A.J., Hakem,R., Duncan,G.S., Patterson,B.J. and Mak,T.W. (1999) In vivo evidence that caspase-3 is required for Fas-mediated apoptosis of hepatocytes. J. Immunol., 163, 4909–4916. [PubMed] [Google Scholar]
- Wyllie A.H. (1980) Glucocorticoid-induced thymocyte apoptosis is associated with endogenous endonuclease activation. Nature, 284, 555–556. [DOI] [PubMed] [Google Scholar]
- Xue D. and Horvitz,H.R. (1995) Inhibition of the Caenorhabditis elegans cell-death protease CED-3 by a CED-3 cleavage site in baculovirus p35 protein. Nature, 377, 248–251. [DOI] [PubMed] [Google Scholar]
- Yokoyama H., Mukae,N., Sakahira,H., Okawa,K., Iwamatsu,A. and Nagata,S. (2000) A novel activation mechanism of caspase-activated DNase from Drosophila melanogaster. J. Biol. Chem., 275, 12978–12986. [DOI] [PubMed] [Google Scholar]
- Zhang D., Beresford,P.J., Greenberg,A.H. and Lieberman,J. (2001) Granzyme A and B directly cleave lamins and disrupt the nuclear lamina during granule-mediated cytolysis. Proc. Natl Acad. Sci. USA, 98, 5746–5751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhivotovsky B., Samali,A., Gahm,A. and Orrenius,S. (1999) Caspases: their intracellular localization and translocation during apoptosis. Cell Death Differ., 6, 644–651. [DOI] [PubMed] [Google Scholar]