

Abstract
The erythropoietin receptor (EpoR) is required for the proliferation and survival of committed erythroid lineage cells. Previous studies have utilized receptor mutations to show the requirement for the distal half of the cytoplasmic domain of the EpoR and receptor tyrosines for activation of signaling pathways potentially critical to Epo function. To extend these studies to in vivo erythropoiesis, we have created two mutant strains of mice. One strain (H) contains a truncation of the distal half of the cytoplasmic domain, while the second strain (HM) contains the same truncation as well as the mutation of the residual tyrosine (Y343) to a phenylalanine. Strikingly, both strains of mice are viable, with only slight alterations in constitutive erythropoiesis or in in vitro assays of red cell lineage function. Challenging H mutant mice with continuous injections of Epo results in an erythrocytosis that is not seen in HM mice. The results demonstrate that neither the distal region nor receptor tyrosines are essential for in vivo EpoR function, but contribute to receptor function in a subtle manner.
Keywords: cytokine signaling/erythropoiesis/erythropoietin/Jak2 kinase/receptor
Introduction
The production of erythrocytes is tightly regulated through the production of erythropoietin (Epo) and its ability to function through its receptor to support the survival and expansion of erythroid progenitors (Krantz, 1991). The Epo receptor (EpoR) is a member of the cytokine receptor family and functions to couple Epo binding to the activation of the Janus protein tyrosine kinase Jak2 (Witthuhn et al., 1993). Following the activation of Jak2, multiple sites on the receptor are tyrosine phosphorylated, as well as on substrates that are recruited to the receptor complex. One of the challenges of studying signal transduction by the EpoR, as with all members of the cytokine receptor superfamily, has been to define the physiological consequences of the activation of individual signaling pathways. Two general approaches have been used to date. One approach has utilized mutant stains of mice that lack components of various signaling pathways. However, this is limited by the potential involvement of the signaling pathways in other receptor systems. The other approach has utilized receptor mutations that have lost or retained the ability to activate signaling pathways when expressed in cell lines.
The first studies of EpoR mutants came to the unexpected conclusion that the distal half of the cytoplasmic domain of the EpoR was not essential for receptor function in cell lines and, indeed, negatively influenced receptor functions (D’Andrea et al., 1991). The concept of a negative role for the distal domain of the receptor gained support from the observation that there were mutations of the human EpoR that deleted 70–79 amino acids of the distal region of the receptor and were associated with a phenotype of erythrocytosis (De La Chapelle et al., 1993; Sokol et al., 1995; Arcasoy et al., 1997). Interestingly, all the cases reported to date are heterozygous for the mutation. The biochemical basis for the increased functionality of the distally truncated receptor is not known precisely. However, it has been shown that the distal region of the EpoR contains tyrosines that can be bound by the SH2 domains of the protein tyrosine specific phosphatase SHP-1 (Yi et al., 1995), a phosphatase that, when deleted in mice, results in the hyperproliferation of many cell lineages (Shultz et al., 1993). In one report, it was suggested that recruitment to the receptor complex is critical to bring SHP-1 in proximity to Jak2 (Klingmuller et al., 1995). However, subsequent studies have demonstrated that SHP-1 can interact directly with Jak2 and, consequently, the requirement for receptor tyrosine for recruitment is unclear (Jiao et al., 1996).
A variety of studies have been directed at the identification of phosphorylated tyrosine residues in the EpoR that are recognized by proteins that may be important in transduction of a proliferative or differentiative response. For example, the signal transducers and activators of transcription 5a and 5b (Stat5a/b) can bind to multiple sites of tyrosine phosphorylation within the receptor, including Y343. In the context of a truncated receptor that only contains Y343, mutation of Y343 to a phenylalanine eliminates the ability of the receptor to recruit and activate Stat5a/b (Gobert et al., 1996; Quelle et al., 1996). The consequences of this mutation have been variably reported to have no consequence (Quelle et al., 1996), to reduce the proliferative function of the receptor (Damen et al., 1995a; Gobert et al., 1996) and/or to promote differentiation as well as hemoglobinization (Iwatsuki et al., 1997; Gregory et al., 1998).
Other sites of tyrosine phosphorylation within the receptor have been shown to be critical for recruitment of the p85 regulatory subunit of phosphatidylinositol 3-kinase (PI-3K) (Damen et al., 1995b). Gab1 and Gab2 have been implicated in Epo signaling, and shown to require the distal region of the receptor, with Gab2 specifically requiring Y425 and/or Y367 for recruitment to the receptor complex (Wickrema et al., 1999). Epo-induced calcium channel activation has been shown to require Y460 in the distal region of the receptor (Miller et al., 1999). Activation of AP-1 transcription factors has been reported to require the presence of at least one of multiple tyrosines, including Y343 (Bergelson et al., 1998). Lyn has been reported to physically associate with the EpoR through Y464 and/or Y479 in the distal region (Chin et al., 1998), while Syp has been reported to bind to Y425, which corresponds to Y401 in the commonly used nomenclature. (Tauchi et al., 1996). Using retrovirally transduced mutants of a constitutive active form of the EpoR, recent studies concluded that efficient red blood cell development was dependent upon receptor cytoplasmic domain tyrosine residues. However, the functionality of a tyrosine-less receptor could be restored by multiple individual tyrosines, although Y343 and Y479 uniquely supported immature burst-forming progenitors (Longmore et al., 1998). Transfection of mutant receptors into EpoR-deficient fetal liver cells has identified a novel, Stat5a/b-independent pathway that requires Y479 and is necessary and sufficient for Epo responses (Klingmuller et al., 1997). However, the role of the recruitment of the above proteins is unclear since the sites required for their recruitment and activation are in the distal half of the cytoplasmic domain.
The above experiments with transfected EpoR mutants have several limitations. In many cases, the possibility exists that the cell lines used are transformed to the extent that a signaling pathway that is essential in normal hematopoiesis is obviated in the cell line by transforming events that allowed its establishment. The other difficulty is to obtain receptor expression levels that are comparable to that occurring during normal erythroid lineage differentiation. Because of these limitations, we have generated mutant strains of mice by modification of the endogenous receptor gene to contain the most commonly studied EpoR mutations. Quite strikingly, the deletion of the distal half of the cytoplasmic domain had very little consequence on in vivo erythropoiesis, although it provided some support to the concept that the distal region negatively influences receptor function to a small degree. Removal of the last tyrosine (Y343) in the truncated receptor similarly had only a very minor effect on receptor function and tended to eliminate the increase in receptor function seen with the truncation and returned the receptor function to a level comparable to that of the wild-type receptor.
Results
The roles of the distal region of the EpoR and receptor tyrosines have been extensively studied in transfected cell lines. To address the role of the distal region under more physiological conditions, two mouse strains were created with the gene-targeting strategies illustrated in Figure 1A. The H strain of mice was obtained by using homologous recombination to delete the distal 108 amino acids of the EpoR in ES cells. Since the resulting receptor would contain one tyrosine (Y343), a second strain of mice (HM) was created in which the distal region was deleted and Y343 was mutated to phenylalanine. ES cells containing the appropriately modified receptor loci were injected into blastocysts to obtain chimeric mice, which were then bred to obtain germline transmission. Heterozygous mice for the modified loci were interbred to obtain mice that were homozygous for each locus. Mice homozygous for the H and HM mutations were obtained, and were grossly normal; they were subjected to a more detailed analysis, as described below.

Fig. 1. Generation of EpoR-H and EpoR-HM mice. (A) Targeting strategy. Solid boxes with numbers 1–8 indicate exons of the EpoR gene. Arrows labeled a and b indicate PCR primers used for geno typing. E, EcoRI. (H/HM) indicates H and HM mutation of exon 8. The H mutation was produced by truncation of the C-terminal 108 amino acids. The HM mutation was produced by the same truncation and a mutation of Y343 to F343 on exon 8. Homologous recombination replaced exon 8 with the H or HM mutant DNA, resulting in a truncated receptor with only one tyrosine residue on its cytoplasmic domain (EpoR-H) or a truncated receptor with Phe substitution for residue Y343 (EpoR-HM). (B) PCR analysis of tail clippings from wild-type and mutant mice: +/+, wild type; +/–, heterozygous for wild type and H allele; –/–, homozygous for H allele. Genomic DNA from tail clippings was used for PCR with primers a and b. The same PCR analysis was used to identify EpoR-HM mice (data not shown). (C) Southern blot analysis of DNA from offspring derived from heterozygous matings of wild-type and H mice. Genomic DNA was digested with EcoRI and probed with the 0.8 kb genomic DNA fragment indicated in (A). (D) Semi-quantitative RT–PCR analysis of EpoR, EpoR-H and EpoR-HM mRNA in adult bone marrow cells. The PCR products are 667 bp for EpoR wild-type allele, 349 bp for EpoR-H or EpoR-HM mutant allele and 900 bp for β-actin. The numbers indicate the number of PCR cycles performed. (E) Schematic diagram of wild-type, H and HM EpoR. The relative positions of tyrosine residues are marked.
To ensure that the H and HM strains contained the appropriately modified receptor loci, PCR and RT–PCR approaches were used. PCR analysis and Southern blot analysis of tail tissues revealed the segregation of the anticipated genomic DNA fragments (Figure 1B and C). To examine expressed sequences, RNA from bone marrow cells of control and mutant strains was used to prepare cDNA, which was used for PCR (Figure 1D). As indicated in Figure 1, the upstream primer was in coding sequences in exon 7, and therefore the cDNA derived from expressed sequences could be distinguished from genomic DNA by the absence of the intronic sequences between exons 7 and 8 (93 bp). As illustrated, bone marrow cells from the H or HM strains contained RNA that yielded products of the size expected for deletion of the distal region of the receptor. Based on the similarities of the yields of the PCR products with various numbers of cycles of amplification, the levels of expression of the mutated receptors are comparable to those of the wild-type receptor. Sequencing of the PCR products further substantiated the presence of the predicted receptor truncation, as well as the presence of the Y343F mutation in the HM strain of mice (data not shown).
Since the mice had no overt phenotype, we wanted to establish that the mutant receptors were functionally altered. Previous studies with cell lines have demonstrated that truncated receptors retaining Y343 mediate a stronger activation of Stat5a/b tyrosine phosphorylation and DNA binding activity than the wild-type receptor (Quelle et al., 1996). However, the mutation of Y343 to phenylalanine eliminates activation of Stat5a/b by the truncated receptor. Therefore, we stimulated bone marrow cells with Epo and examined the extent of activation of Stat5a/b. As illustrated (Figure 2), stimulation of bone marrow cells from wild-type mice resulted in the activation of Stat5a/b DNA binding activity, as assessed by electrophoretic mobility shift assay (EMSA). Epo stimulation of bone marrow cells from the H mutant strain resulted in a reproducibly stronger activation of Stat5a/b, consistent with studies of this receptor mutant in transfected cell lines. The activation of Stat5a/b by interleukin-3 (IL-3) is provided as an internal control, and is comparable in bone marrow preparations from wild-type and H mutant mice. Lastly, the activation of Stat5a/b was not detected following Epo stimulation of bone marrow cells from HM mutant mice, again consistent with studies with this receptor mutant transfected in cell lines, although IL-3-induced levels of Stat5a/b activation were comparable to wild-type cells. Together, the results demonstrate that the mutant mouse strains contain receptor mutations with properties predicted from the studies of these receptors in cell lines.
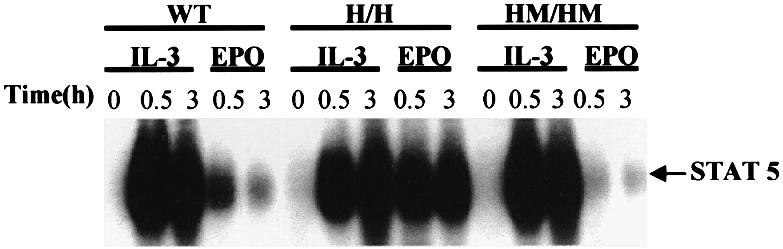
Fig. 2. Effect of EpoR-H or EpoR-HM on the activation of Stat5a/b in bone marrow cells. Bone marrow cells from femurs of adult (≥4 weeks) mice were isolated and suspended in α-MEM medium. After 4 h of growth arrest, cells were stimulated with either 10 ng/ml recombinant mIL-3 or 50 U/ml recombinant hEpo for 30 min or 3 h. Whole-cell extracts were prepared and analyzed by EMSA. γ-32P-labeled DNA from the Stat5a/b binding site in the β-casein promoter was used as binding sequence for the assay.
As indicated above, viable mice were obtained that contained either the H or the HM mutation, suggesting that these mutations did not grossly affect erythropoiesis, and consequently more detailed studies were carried out. To initially assess the consequences on embryonic erythropoiesis, the distributions of offspring from crosses of heterozygous mice were examined. As indicated in Table I, the distribution of H and HM genotypes was that predicted from Mendelian segregation, suggesting the absence of any embryonic lethality among the homozygous mutant mice. Since the HM mutation reduces or eliminates the ability to activate Stat5a/b, the distribution of genotypes from a cross of mice heterozygous for Stat5a/b deficiency is also presented. Similar to the H and HM strains, the distribution of genotypes was that predicted by Mendelian segregation, demonstrating the absence of any embryonic lethality associated with Stat5a/b deficiency.
Table I. Genotype distribution of heterozygote breeding.
| Strain | +/+ | +/– | –/– |
|---|---|---|---|
| EpoR-H | 73 (27%) | 127 (47%) | 70 (26%) |
| EpoR-HM | 37 (27%) | 69 (50%) | 31 (23%) |
| Stat5a/b | 103 (25%) | 219 (52%) | 97 (23%) |
For each strain, offspring from heterozygous matings of the mutant alleles on a background of C57Bl/6 × 129Sv were genotyped as described in Materials and methods. The expected Mendelian distribution of offspring is 25% +/+, 50% +/– and 25% –/–. The observed frequencies are not statistically different from those predicted based on the use of a χ2 goodness of fit test.
Analysis of adult mice also indicated the lack of any unexpected lethality or pathology. In particular, HM mice did not develop splenomegaly that is consistently seen in Stat5a/b-deficient mice. This splenomegaly is due to extramedullary hematopoiesis and includes a striking expansion of Ter119-positive erythroid lineage progenitors (Moriggl et al., 1999). Since splenomegaly was not observed in the HM mutant mice, the results suggest that the splenomegaly seen in Stat5a/b-deficient mice is not due to Stat5a/b deficiency in the erythroid lineage. Consistent with this conclusion, crossing the Stat5a/b deficiency onto a Rag2-deficient background to eliminate the T-cell alterations associated with Stat5a/b deficiency eliminates the splenomegaly (our unpublished results).
A previous study reported that Stat5a/b deficiency is associated with visually evident severe embryonic anemia (Socolovsky et al., 1999). Therefore, it would be anticipated that a comparable phenotype would be observed in HM embryos, since they lack the ability to activate Stat5a/b. Photographs of E12.5 embryos of the various genotypes are shown in Figure 3. As illustrated, H and HM homozygous embryos were visually indistinguishable from wild-type embryos. Stat5a/b-deficient embryos are also depicted in Figure 3 for comparison and are similarly indistinguishable from wild-type embryos. For comparison, Jak2- or EpoR-deficient embryos are presented to illustrate the readily observable anemia that is associated with these mutants.

Fig. 3. Morphology of EpoR-H and EpoR-HM embryos. Representative photographs of embryos at E12.5 from wild type (A), EpoR-H (B) and EpoR-HM (C). Photographs of Jak2–/– (D), EpoR–/– (E) and Stat5a/b–/– (F) at E12.5 are shown for comparison.
To assess the effects of the mutations on erythropoiesis, colony assays were performed with either fetal liver cells from E12.5 embryos or bone marrow cells of adult mice (Figure 4). As illustrated, the numbers of CFU-E per 105 fetal liver cells from H and HM mutant embryos were comparable to those of wild-type embryos, and also comparable to those obtained with fetal liver cells from Stat5a/b-deficient mice. However, the numbers of BFU-E were slightly reduced in H mutant fetal liver cell preparations relative to wild type, and further reduced in HM mutant fetal liver cell preparations. As a control, and for comparison, the colony-forming response to IL-3 is also shown and is comparable in all strains. In the bone marrow of adult mice, the numbers of CFU-E were increased ∼2-fold in H mutant mice, while the numbers in HM mutant mice were comparable to those in wild-type or Stat5a/b-deficient mice. Lastly, BFU-E from bone marrow from adult mice were only marginally increased in H mutant mice and marginally decreased in HM mutant mice to a level comparable to the BFU-E response of Stat5a/b mice. As a control, and for comparison, the colony-forming response to IL-3 is also shown and was comparable among the various strains of mice. The BFU-E and CFU-E responses to Epo were also compared with decreasing doses of Epo, and the mutant strains had comparable dose–response curves (data not shown).

Fig. 4. In vitro colony formation of hematopoietic progenitors from wild-type, EpoR-H, EpoR-HM and Stat5a/b–/– mice in response to various cytokines. The numbers of colonies/105 cells formed from E12.5 fetal liver cell cultures (A) or from bone marrow cell cultures (B) are plotted. The mean and standard deviation are shown from six independent assays. The two-tailed P values for comparison of the various mutant strains with wild-type mice or embryos are >0.01, with the exception of the BFU-E colonies for bone marrow from HM mice (P = 0.004) and of CFU-E colonies for fetal liver cells from H mice (P = 0.00006). Cells from E12.5 embryos or femurs of adult mice were prepared in α-MEM medium containing 2% FBS and counted in the presence of 3% acetic acid to lyse erythrocytes. Diluted cell suspensions and cytokines were mixed with Methocult 3230 to a final concentration of 0.9% methylcellulose. For CFU-E assay, cells were cultured in 0.2 U/ml recombinant hEpo. For BFU-E assay, cells were cultured in 3 U/ml recombinant hEpo and 10 ng/ml recombinant murine IL-3. IL-3 colony assays were performed in the presence of 10 ng/ml recombinant mIL-3. The plating conditions, cell concentration for each assay, culture conditions and colony scoring methods were exactly as described previously (Parganas et al., 1998; Teglund et al., 1998).
Embryonic and adult hematocrits provide another potential means of assessing the consequences of the receptor mutations on embryonic erythropoiesis. We therefore compared the embryonic hematocrits of H and HM mutant embryos with those of wild-type or Stat5a/b-deficient embryos as controls. The hematocrits of wild-type and Stat5a/b-deficient heterozygous and homozygous embryos or adults are also shown in Figure 5 for comparison. Also for comparison are shown the white and red cell blood counts (Figure 5A and B) for wild-type, H and HM mice. The hematocrit values for individual mice or embryos are illustrated in Figure 5C and D, respectively. The mean values for these and additional embryos and animals are shown in Figure 5E. As illustrated by the mean values (Figure 5E), Stat5a/b-deficient embryos had consistently lowered hematocrits from the onset of fetal liver erythropoiesis, throughout the last half of embryonic development and as adults. The values ranged from 70 to 90% of those of control embryos or adult mice. The values at E13.5 are comparable to recently reported hematocrits for Stat5a/b-deficient embryos at E13.5 (Socolovsky et al., 1999), with the exception that we rarely find individual embryos with severely decreased hematocrits. As illustrated (Figure 5D and E), H mutant embryos at E14.5, E15.5 and E18.5 consistently had hematocrits that were slightly higher than those of wild-type embryos. Moreover, the hematocrits of HM embryos at these ages were consistently lower than those of wild-type embryos. In adult mice, the hematocrits of H mutant mice were slightly higher than those of wild-type mice, while the hematocrits of HM mutant mice were consistently lower. For comparison and for an indication of the significance of the differences seen, the normal hematocrits of two strains of mice are also shown (Figure 5E; BL, C57Bl/6 and C3H/HeJ).

Fig. 5. Hematocrits and blood cell counts of embryos or adult mice with the indicated genotypes. For adult mice (≥4 weeks old), white blood cell numbers (A), red blood cell numbers (B) and hematocrits (C and E) were machine scored by a Hemavet 3700R counter. For embryos (D and E), blood samples were acquired using 2 µl glass micropipettes and then briefly centrifuged to measure hematocrits. N, total number of mice used for the experiment. H × Stat5a/b–/– indicates mice cross-breeding from Stat5a/b null mice with EpoR-H mice. (E) Numbers indicate days of gestation. The P values for all the comparisons support statistically significant differences in the hematocrits. To indicate the types of P values associated with the results, in (C) the two-tailed P values for the means of the various mutants relative to wild type are: H, 1.3 × 10–3; HM, 8.0 × 10–12; Stat5a/b, 6.4 × 10–4; H/Stat5a/b, 9.2 × 10–7.
The above results consistently demonstrated increased hematocrits associated with the H mutation, whereas the HM mutation is associated with reduced levels. Since Y343 has been shown to be important for the activation of several signaling pathways, as well as Stat5a/b, we wished to determine whether the primary difference between the H and HM mutations was associated with the absence of Stat5a/b activation. To assess this, we crossed the H mutation onto the Stat5a/b-deficient background, thus eliminating the contribution of Stat5a/b while preserving other functions requiring Y343. As illustrated (Figure 5C), this resulted in reduced hematocrits that were comparable to those seen in Stat5a/b-deficient mice containing a wild-type receptor. The results would, therefore, suggest that the change in the hematocrits that occurs when Y343 is mutated in the context of the truncated receptor is due to the loss of the ability to activate Stat5a/b.
The distal region of the EpoR and Y343 have been implicated in the regulation of pathways that affect apoptosis. We therefore examined fetal liver cells from mutant embryos of E13.5 for increased apoptosis or increased sensitivity to apoptosis in the absence of Epo. Since no significant apoptosis was seen in fetal liver cells of any of the mutants when examined directly from the embryos (data not shown), the cells were cultured for 18 h in the presence or absence of Epo. The cells were then examined by fluorescence-activated cell sorting (FACS) analysis for the expression of the erythroid lineage marker Ter119, and for apoptosis by TUNEL staining. As illustrated (Figure 6), fetal liver cells from H or HM mutants cultured in the presence of 10 U/ml Epo showed no consistent differences in the levels of apoptosis, comparable to the fetal liver cells from wild-type embyros. When cultured in the absence of Epo, significant fractions of apoptotic cells were evident in cultures of wild-type cells (7.1%), slightly fewer apoptotic cells in cell cultures from H mutant embryos (5.83%) and significantly increased numbers of apoptotic cells in cultures from HM mutant embryos (25.5%). For comparison, typical results obtained with cells from Stat5a/b-deficient embryos are also illustrated. As above, no differences in the numbers of apoptotic cells were evident when fetal liver cells were examined directly from E13.5 embryos. However, culture of the cells in the absence of Epo resulted in an increased degree of apoptosis in cultures of Stat5a/b-deficient fetal liver cells comparable to that seen in the HM mutant mice (Figure 6B).

Fig. 6. FACS analysis of fetal liver cells at E13.5 with combined TUNEL–Ter119 staining. Numbers in plots indicate the percentage of sorted cell population in each quadrant. The upper right quadrant in each plot represents Ter119-positive erythrocytes with TUNEL-positive apoptotic cells. (A) Fetal liver cells from EpoR wild-type, EpoR-H or EpoR-HM embryos were isolated at E13.5 and cultured in α-MEM with 2% FBS for 18 h in the presence or absence of 10 U/ml recombinant hEpo. Cells were then fixed with formalin and double stained with TUNEL reagent and Ter119 antibody. The cell suspension was analyzed by FACScan. (B) Fetal livers from Stat5a/b wild-type embryos or Stat5a/b null embryos were used for TUNEL– Ter119 double staining. Note that the EpoR mutants were performed independently of the Stat5a/b mutant studies and, consequently, should not be compared across the two experiments, but rather the mutants should be compared with their respective wild-type controls.
As an approach to establish additional in vivo biological consequences of the receptor mutations, H and HM mice were challenged with repeated doses of Epo, and the hematocrits assessed (Figure 7). Comparable experiments were not carried out with Stat5a/b-deficient mice because of the splenomegaly and extramedullary hematopoiesis associated with the altered T-cell functions in these mice. Repeated injections of Epo (three times weekly) into wild-type mice over 30 days resulted in an increasing hematocrit during the period of injections. As illustrated, this increase was enhanced in H mutant mice, consistent with the concept that the receptor functions slightly better than the wild-type receptor. In contrast, there was a lower rate of increase in hematocrits in HM mice. The results are generally consistent with the concept that the H truncation results in a moderately more efficient receptor, while the mutation of the last tyrosine in the truncated receptor reduces the efficiency of the receptor.

Fig. 7. Response of the EpoR mutant mice to continual injections of Epo. Mice were injected three times during a week, as described in Materials and methods, with recombinant hEpo. To prevent anomalous results from frequent bleeding, the mice were separated into groups, which were bled only every 14 days. The average hematocrits for two mice are indicated by the individual symbols. The deviations among the samples for each symbol were between 2 and 5%; consequently, error bars are not included. The line presents the linear regression of the data points for each of the mutant strains.
Discussion
The results demonstrate the striking finding that neither the distal region of the EpoR nor receptor tyrosines are essential for embryonic and adult erythropoiesis in vivo. Numerous studies have examined the properties of mutated EpoRs in cell lines, particularly receptors lacking tyrosine residues, which would have suggested that such mutations would have a dramatic phenotype in vivo. This was not the case and specifically neither the H nor HM mutations were associated with any evidence of embryonic or adult lethality. This contrasts with deficiencies of the receptor-associated kinase Jak2 (Neubauer et al., 1998; Parganas et al., 1998) or the EpoR-regulated genes Bcl-X (Motoyama et al., 1995) and N-myc (Moens et al., 1993; Sawai et al., 1993), all of which exhibit, like deficiency of the EpoR (Wu et al., 1995), embryonic lethal phenotypes associated with profound deficiencies in expansion of erythroid lineage progenitors with dramatically increased apoptosis in vivo. The lack of consistency with the data with cell lines may reflect the need for additional pathways, perhaps in a cell line-dependent manner, for the maintenance of the cell lines as opposed to the transient requirement for Epo to mediate the expansion of erythroid lineage cells.
The first studies of mutant EpoRs indicated the existence of non-overlapping positive and negative regulatory domains (D’Andrea et al., 1991). In particular, the studies were the first to suggest that the distal half of the cytoplasmic domain negatively influenced EpoR signaling. Our results with modification of the receptor by gene targeting provide in vivo support to the concept that the distal region negatively affects receptor signaling. Consistent with the experiments in cell lines, the activation of Stat5a/b was increased and prolonged in bone marrow cells from H mutant mice stimulated with Epo. Unfortunately, due to the quantity of cells required and the heterogeneous nature of bone marrow cells, we have been unable to assess the Jak2 activation. Also consistent with a negative role of the distal region in regulating signaling, the hematocrits of H mutant mice were modestly increased and there was an increased response to repeated injections of Epo. However, the effects were not as striking as might have been predicted based on the mutant receptor studies or based on the mutations of the distal region of the EpoR in humans (De La Chapelle et al., 1993; Sokol et al., 1995; Arcasoy et al., 1997). In particular, individuals who are heterozygous for a truncation of 70 amino acids were reported to have an erythrocytosis in which hemoglobin levels were 1.33-fold higher than controls. In contrast, mice homozygous for a truncation of 108 amino acids have hemoglobin levels that are only 1.08-fold those of controls (data not shown). Lastly, it should be noted that whereas the erythrocytosis associated with the human receptor mutations has been observed with individuals heterozygous for the receptor mutations, mice heterozygous for the H mutation display no increase in hematocrits (data not shown). One possible basis for the difference is the extent of the truncations. In particular, the human truncations retain an additional tyrosine and this may provide an additional positive signal that contributes to the overall larger positive response.
In a recent study (Divoky et al., 2001), mutant mouse strains were created in which the endogenous murine EpoR gene was replaced with either the wild-type human EpoR gene or a carboxyl-truncated human EpoR gene as a model for the human primary familial congenital polycythemia associated with receptor truncations. Consistent with our studies with the H strain of mice, no loss of viability was observed. Also consistent with our results was the lack of striking changes in BFU-E or CFU-E. However, the extent of erythrocytosis, as assessed with hematocrits, was considerably greater than that seen in our H strain of mice. Moreover, erythrocytosis was evident in heterozygous animals for the truncation in combination with either the wild-type murine endogenous gene or the wild-type human gene. This is in contrast to our results in which any differences observed with the H mutation were only evident in the homozygous state. The basis for the differences is not known, but may well reside in the differences that exist between the human and murine receptors, and the extent of the truncations, as noted above. The other possibility might be mouse strain differences, although we have not seen any significant differences in H or HM mutant mice on a C57Bl/6 or Balb/c background.
Numerous studies have used receptor mutants to identify the domains and/or tyrosine in the EpoR that are required for the activation of signal transduction pathways. The potential role for many of these pathways was inferred from the activity of transfected mutated receptors or from the use of dominant-negative proteins, inhibitors or antisense reduction of protein levels. Because of this extensive body of research, it is particularly striking that a distally truncated receptor containing no tyrosines was able to sustain erythropoiesis at the levels observed. It must be concluded that either the activation of the pathways studied is not essential for receptor function or that there is some type of selection for compensating functions that is occurring in vivo and that is sufficiently efficient to completely obviate the role of the receptor tyrosines. Unfortunately, in most cases, it is not possible to determine whether signal transduction pathways are still activated in cells from HM mutant mice because of the requirement for large quantities of homogeneous cells. We are currently breeding the HM mutation on a background that is susceptible to Friend virus disease, as a way of obtaining large quantities of erythroid lineage cells for further biochemical studies (Koury et al., 1984). Using these mice, we will also be able to evaluate the cell surface expression patterns of the receptor mutants, which has not been possible with bone marrow cells. However, we would not expect that the expression of the mutants would be different since the gene modifications are quite distant from the promoter of the gene. Moreover, extensive studies with these mutants in transduced cell lines have consistently shown that, when expressed comparably, they are processed and expressed at the cell surface in comparable patterns.
In contrast to most of the signaling pathways, the sensitivity of EMSA allowed us to assess the ability of H and HM mice to recruit and activate Stat5a/b, for which Y343 is known to be necessary and sufficient for its activation. The results demonstrate that the strong activation of Stat5a/b that is seen in H mutant mice is completely eliminated in HM mutant mice. Thus, HM mice have not activated a compensating pathway that either allows Epo to activate Stat5a/b or results in a constitutive level of Stat5a/b activation comparable to that seen in Epo-stimulated cells. The HM mutation thus renders the mice specifically deficient in Stat5a/b function within the erythroid lineage, and allows a potentially more precise analysis of Stat5a/b function in erythroid cells than complete deletion of the gene because of the critical role that Stat5a/b play in other lineages of cells and particularly in T cells (Teglund et al., 1998; Moriggl et al., 1999).
Based on the published properties of erythropoiesis in Stat5a/b-deficient embryos from the strain of mice we produced (Socolovsky et al., 1999), we had anticipated a dramatic phenotype of severe fetal anemia in HM mice. However, many of the phenotypes that were reported were not evident in our studies of Stat5a/b-deficient embryos. In particular, HM mutant embryos, like Stat5a/b-deficient embryos, were morphologically indistinguishable from wild-type embryos. Moreover, no embryonic lethality was evident in either HM mutant or Stat5a/b-deficient embryos. The frequencies of BFU-E and CFU-E were comparable among wild-type, HM mutant and Stat5a/b-deficient embryos. Lastly, there was no apoptosis in fetal livers of HM mutant or Stat5a/b-deficient embryos, consistent with the absence of apoptosis in fetal livers of wild-type embryos. The basis for the differences in the results is yet to be resolved.
Consistent with the previous studies, however (Socolovsky et al., 1999), Stat5a/b-deficient embryos had hematocrits that were reduced relative to those of wild-type mice throughout embryonic development and as adults. The differences are comparable to that seen between different strains of mice. Importantly, a comparable reduction in hematocrits was seen in HM mutant embryos and adults. The possibility that the reduction seen in HM mutants is due to the loss of Stat5a/b function is supported by the observation that the increased hematocrits seen in H mutant embryos are reduced to that of Stat5a/b-deficient or HM mutant embryos when crossed onto the Stat5a/b-deficient background. The Stat5a/b functions that mediate the 10–30% reduction in hematocrits are unclear from studies of erythroid progenitors in vivo. However, fetal liver cells from both HM and Stat5a/b-deficient embryos are more susceptible to apoptosis when cultured in the absence of Epo than are wild-type or H mutant fetal liver cells. It has been proposed that Stat5a/b is required for the Epo-induced expression of Bcl-X (Socolovsky et al., 1999); however, when examined, the levels of Bcl-X protein in fetal liver erythroid cells are not detectably altered in Stat5a/b-deficient embryos (Marine et al., 1999).
In summary, while it might have been anticipated that the creation of mouse strains containing a severely truncated EpoR, which lacks any tyrosine residues, might have resulted in striking consequences to erythropoiesis, this was not the case. The results support the concept that many of the signal transducing events that have been well characterized with the use of mutant receptors in cell lines may have limited significance in defining the critical events in EpoR signaling. The availability of these mutant strains of mice will be particularly useful in allowing studies to focus on a more restricted set of signal transducing pathways, retained by the mutant receptors, in more physiologically relevant in vivo studies. The availability of these mutant strains will also allow examination of the possibility that the distal region of the receptor is required in very specific situations to modify or amplify a ‘core’ signal.
Materials and methods
Construction of targeting vector
The EpoR gene was isolated from the E14 (129/Ola mouse strain) genomic library in EMBL3 using EpoR full-length cDNA as a probe. Positive clones were restriction mapped and sequenced. Briefly, a XhoI fragment including intron 2–exon 8 of the EpoR gene was subcloned into pBluescript. For truncation of the receptor, a HindIII site was introduced just upstream of the stop codon by site-directed mutagenesis (5′-TATGTGGCCTGCTCCTAG-3′ to 5′-TATGAAGCTTGCTCCTAG-3′), and HindIII digestion utilizing a HindIII site on exon 8 and a newly created HindIII site was followed. For Y343F mutation, a 0.8 kb HindIII fragment utilizing HindIII site on intron 6 and exon 8 was subcloned into pBluescript. Site-directed mutagenesis was performed as described in the manufacturer’s protocol (Qiagen) and the plasmid containing the correct mutation was used for the construction of HM targeting vector. For negative selection, a diphtheria toxin A gene (DTA) driven by the polyoma virus enhancer cassette (Adachi et al., 1995) was inserted in the 5′ end of the targeting construct. A thymidine kinase promoter-driven neo cassette was inserted after the stop codon of exon 8 to confer positive selection.
Transfection of ES cells and generation of EpoR-H or EpoR-HM mice
W9.5 ES cells and E14 (129/Ola mouse strain) ES cells were cultured as previously described (Parganas et al., 1998). W9.5 ES cells were used for the generation of EpoR-H mice and E14 cells were used for the generation of EpoR-HM mice. Twenty-five micrograms of SalI-linearized EpoR-H or EpoR-HM plasmid construct were electroporated into the W9.5 or E14 ES cells. Twenty-four hours after electroporation, selection was performed in 350 µg/ml geneticin (G418; Gibco-BRL). Conditions for blastocyst injection of correctly targeted and karyotypically normal ES clones, and breeding to generate mice homozygous for the mutated EpoR gene, were performed essentially as described (van Deursen et al., 1993). ES clones were injected into C57Bl/6 blastocysts, and five clones from EpoR-H targeting and three clones from EpoR-HM targeting gave germline transmission. For PCR analysis, mice tails were clipped and digested overnight in lysis buffer (500 mM KCl, 100 mM Tris pH 8.3, 0.1 mg/ml gelatin, 1% NP-40, 1% Tween-20, 500 µg/ml proteinase K). PCR was carried out with primer a (5′-GAGTTTGAGGGTCTCTTCACC-3′) in exon 7 and primer b (5′-TAGGCTGGAGTCCTAGGAGC-3′) in exon 8 of the EpoR gene. Since primer b corresponds to a DNA sequence at the beginning of the stop codon in exon 8 and extends into the following non-coding 3′ sequences that are retained in the truncation, the 442 bp fragment is characteristically derived from the mutant containing the truncation, while the wild-type allele gives rise to a 760 bp fragment. For EpoR-HM mice, the PCR product was sequenced to confirm HM mutation. For Southern blot analysis, EcoRI–XhoI digestion was performed on isolated EpoR genomic DNA, and the 0.8 kbp fragment comprising exon 2 and an intron between exon 1 and exon 2 was used as a probe. Genomic DNA from mouse tail was digested by EcoRI and, after probing, the mutant allele shows a 4.5 kb fragment and the wild-type allele shows an ∼12 kb fragment. For RT–PCR analysis, total cellular RNA was extracted from bone marrow cells of adult mice (≥4 weeks) using RNAzol B (Tel-Test). First-strand cDNA synthesis using oligo(dT) primer was carried out as described in the manufacturer’s protocol (Gibco-BRL Superscript reverse transcriptase). PCR was performed using PCR primers a and b for the EpoR gene, and β-actin primers (5′-GTGACGAGGCCCAGAGCAAGAG-3′, 5′-AGGGGCCGGACT CATCGTACTC-3′) for the β-actin gene.
EMSA
Bone marrow cells (8.0 × 107) were collected from femurs of wild-type, EpoR-H and EpoR-HM mice. Cells were suspended in α-MEM medium with 2% fetal calf serum, 1% l-glutamine and appropriate amounts of antibiotics. Cells were then starved for 4 h and stimulated with 50 U/ml recombinant human Epo (hEpo; Amgen) or 10 ng/ml recombinant murine IL-3 (mIL-3; R & D systems) for 30 min or 3 h. Whole-cell extract was prepared by incubation in extraction buffer [50 mM Tris pH 8.0, 50 mM NaCl, 0.1 mM EDTA, 0.5 mM Na3VO4, 0.5% NP-40, 50 mM NaF, 10% glycerol, 20 mM β-mercaptoethanol, 1 mM dithiothreitol (DTT), 0.4 mM phenylmethylsulfonyl fluoride (PMSF), protease inhibitor cocktail] for 30 min at 4°C. For Stat5 DNA binding sequence, sense and antisense oligonucleotides representing the Stat5a/b site in β-casein promoter (5′-AGATTTCTAGGAATTCAATCC-3′) were synthesized, annealed to make double-stranded DNA and labeled with [γ-32P]ATP. Ten micrograms of cell extract were incubated in binding buffer [10 mM Tris, 50 mM NaCl, 0.1 mM EDTA, 0.2 mM PMSF, 1 mM DTT, 0.1% NP-40, 5% glycerol, 2 µg poly(dI–dC), 30 µg/ml bovine serum albumin] for 30 min at room temperature, and the reaction mixtures were separated on 4% native TBE gel.
Colony assay
Fetal liver cells were prepared from livers of E12.5 embryos, and bone marrow cells from femurs of wild-type, EpoR-H, EpoR-HM and Stat5a/b–/– mice. The cells were plated and the colonies scored exactly as described previously (Parganas et al., 1998; Teglund et al., 1998).
Blood cell counts and hematocrits
For adult mice, blood samples were taken from orbital sinus using a microcapillary tube. Twenty microliters of sample blood were used for analysis in a MASCOT Hemavet 3700R counter (CDC Technologies) and scored for white blood cells and red blood cells. Hematocrits were calculated by Hemavet counter based on red blood cell counts and mean corpuscular volume. For embryos, blood samples were taken from the carotid artery using heparinized 2 µl microcapillary tubes. Tubes were sealed at one end and centrifuged for 5 min at 3000 r.p.m. to measure hematocrits.
Flow cytometry and TUNEL staining
E13.5 fetal livers were isolated from embryos, and made into single-cell suspension by passing them through a G25 needle. Cells were cultured in α-MEM medium with 2% fetal bovine serum (FBS) for 18 h. For Epo stimulation, recombinant hEpo was added to a final concentration of 10 U/ml in culture medium. After culture, cells were centrifuged and washed with phosphate-buffered saline (PBS) containing 1% FBS. Cells were then centrifuged for 5 min at 1000 g and PBS decanted, followed by fixation with formalin for 20 min at room temperature and subsequent treatment with 0.1% Triton X-100 and 0.1% sodium citrate. Cells were placed on ice for 2 min and washed with PBS. TUNEL reaction was performed as described in the manufacturer’s protocol (In situ cell death detection kit, Fluorescein; Boehringer Manheim) and, after washing with PBS, Ter119 antibody was added, followed by incubation on ice for an hour and several washing with PBS. The cell suspension was then analyzed in a Becton-Dickinson FACScan using CellQuest software.
Epo challenge analysis
A total of 30 mice were selected for the experiment. The selection was gender balanced and mice of various ages were included. Epo (500 U/kg body weight) was injected into all 30 mice three times a week. Blood samples were drawn between injections and complete blood counts were performed. Hematocrits were monitored for 4 weeks. To prevent any anomaly in the experimental results due to frequent bleeding, mice were separated into five groups and scheduled to take turns for bleeding. A group of six mice consisted of two mice from each genotype: wild type, EpoR-H and EpoR-HM.
Acknowledgments
Acknowledgements
We would like to thank Linda Snyder, Kristen Rothammer and Melanie Loyd for technical support; Drs John Cleveland, Gerry Zambetti and Nick Carpino for helpful discussion; and Richard Morrigl for helping with HCT analysis of Stat5a/b embryos. This work was supported by the Cancer Center CORE grant CA21765, by grants RO1 DK42932 and PO1 HL53749, and by the American Lebanese Syrian Associated Charities (ALSAC).
References
- Adachi M., Suematsu,S., Kondo,T., Ogasawara,J., Tanaka,T., Yoshida,N. and Nagata,S. (1995) Targeted mutation in the Fas gene causes hyperplasia in peripheral lymphoid organs and liver. Nature Genet., 11, 294–300. [DOI] [PubMed] [Google Scholar]
- Arcasoy M.O., Degar,B.A., Harris,K.W. and Forget,B.G. (1997) Familial erythrocytosis associated with a short deletion in the erythropoietin receptor gene. Blood, 89, 4628–4635. [PubMed] [Google Scholar]
- Bergelson S., Klingmuller,U., Socolovsky,M., Hsiao,J.G. and Lodish,H.F. (1998) Tyrosine residues within the intracellular domain of the erythropoietin receptor mediate activation of AP-1 transcription factors. J. Biol. Chem., 273, 2396–2401. [DOI] [PubMed] [Google Scholar]
- Chin H., Arai,A., Wakao,H., Kamiyama,R., Miyasaka,N. and Miura,O. (1998) Lyn physically associates with the erythropoietin receptor and may play a role in activation of the Stat5 pathway. Blood, 91, 3734–3745. [PubMed] [Google Scholar]
- Damen J.E., Wakao,H., Miyajima,A., Krosl,J., Humphries,R.K., Cutler,R.L. and Krystal,G. (1995a) Tyrosine 343 in the erythropoietin receptor positively regulates erythropoietin-induced cell proliferation and Stat5 activation. EMBO J., 14, 5557–5568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damen J.E., Cutler,R.L., Jiao,H., Yi,T. and Krystal,G. (1995b) Phosphorylation of Y503 in the erythropoietin receptor (EpR) is essential for binding the p85 subunit of phosphatidylinositol (PI) 3-kinase and for EpR associated PI 3-kinase activity. J. Biol. Chem., 270, 23402–23408. [DOI] [PubMed] [Google Scholar]
- D’Andrea A.D., Yoshimura,A., Youssoufian,H., Zon,L.I., Koo,J.W. and Lodish,H.F. (1991) The cytoplasmic region of the erythropoietin receptor contains nonoverlapping positive and negative growth-regulatory domains. Mol. Cell. Biol., 11, 1980–1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De La Chapelle A., Traskelin,A.-L. and Juvonen,E. (1993) Truncated erythropoietin receptor causes dominantly inherited benign human erythrocytosis. Proc. Natl Acad. Sci. USA, 90, 4495–4499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Divoky V., Liu,Z., Ryan,T.M., Prchal,J.F., Townes,T.M. and Prchal,J.T. (2001) Mouse model of congenital polycythemia: homologous replacement of murine gene by mutant human erythropoietin receptor gene. Proc. Natl Acad. Sci. USA, 98, 986–991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gobert S. et al. (1996) Identification of tyrosine residues within the intracellular domain of the erythropoietin receptor crucial for STAT5 activation. EMBO J., 15, 2434–2441. [PMC free article] [PubMed] [Google Scholar]
- Gregory R.C., Jiang,N., Todokoro,K., Crouse,J., Pacifici,R.E. and Wojchowski,D.M. (1998) Erythropoietin receptor and STAT5-specific pathways promote SKT6 cell hemoglobinization. Blood, 92, 1104–1118. [PubMed] [Google Scholar]
- Iwatsuki K., Endo,T., Misawa,H., Yokouchi,M., Matsumoto,A., Ohtsubo,M., Mori,K.J. and Yoshimura,A. (1997) STAT5 activation correlates with erythropoietin receptor-mediated erythroid differentiation of an erythroleukemia cell line. J. Biol. Chem., 272, 8149–8152. [DOI] [PubMed] [Google Scholar]
- Jiao H., Berrada,K., Yang,W., Tabrizi,M., Platanias,L.C. and Yi,T. (1996) Direct association and dephosphorylation of Jak2 kinase by SH2 domain-containing protein tyrosine phosphatase SHP-1. Mol. Cell. Biol., 16, 6985–6992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klingmuller U., Lorenz,U., Cantley,L.C., Neel,B.G. and Lodish,H.F. (1995) Specific recruitment of the hematopoietic protein tyrosine phosphatase SH-PTP1 to the erythropoietin receptor causes inactivation of JAK2 and termination of proliferative signals. Cell, 80, 729–738. [DOI] [PubMed] [Google Scholar]
- Klingmuller U., Wu,H., Hsiao,J.G., Toker,A., Duckworth,B.C., Cantley,L.C. and Lodish,H.F. (1997) Identification of a novel pathway important for proliferation and differentiation of primary erythroid progenitors. Proc. Natl Acad. Sci. USA, 94, 3016–3021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koury M.J., Sawyer,S.T. and Bondurant,M.C. (1984) Splenic erythroblasts in anemia-inducing Friend disease: a source of cells for studies of erythropoietin-mediated differentiation. J. Cell Physiol., 121, 526–532. [DOI] [PubMed] [Google Scholar]
- Krantz S.B. (1991) Erythropoietin. Blood, 77, 419–434. [PubMed] [Google Scholar]
- Longmore G.D., You,Y., Molden,J., Liu,K.D., Mikami,A., Lai,S.Y., Pharr,P. and Goldsmith,M.A. (1998) Redundant and selective roles for erythropoietin receptor tyrosines in erythropoiesis in vivo. Blood, 91, 870–878. [PubMed] [Google Scholar]
- Marine J.C. et al. (1999) SOCS3 is essential in the regulation of fetal liver erythropoiesis. Cell, 98, 617–627. [DOI] [PubMed] [Google Scholar]
- Miller B.A., Barber,D.L., Bell,L.L., Beattie,B.K., Zhang,M.Y., Neel,B.G., Yoakim,M., Rothblum,L.I. and Cheung,J.Y. (1999) Identification of the erythropoietin receptor domain required for calcium channel activation. J. Biol. Chem., 274, 20465–20472. [DOI] [PubMed] [Google Scholar]
- Moens C.B., Stanton,B.R., Parada,L.F. and Rossant,J. (1993) Defects in heart and lung development in compound heterozygotes for two different targeted mutations at the N-myc locus. Development, 119, 485–499. [DOI] [PubMed] [Google Scholar]
- Moriggl R. et al. (1999) Stat5 is required for IL-2 induced cell cycle progression of peripheral T cells. Immunity, 10, 249–259. [DOI] [PubMed] [Google Scholar]
- Motoyama N. et al. (1995) Massive cell death of immature hematopoietic cells and neurons in Bcl-x-deficient mice. Science, 267, 1506–1510. [DOI] [PubMed] [Google Scholar]
- Neubauer H., Cumano,A., Muller,M., Wu,H., Huffstadt,U. and Pfeffer,K. (1998) Jak2 deficiency defines an essential developmental checkpoint in definitive hematopoiesis. Cell, 93, 397–409. [DOI] [PubMed] [Google Scholar]
- Parganas E. et al. (1998) Jak2 is essential for signaling through a variety of cytokine receptors. Cell, 93, 385–395. [DOI] [PubMed] [Google Scholar]
- Quelle F.W., Wang,D., Nosaka,T., Thierfelder,W.E., Stravopodis,D., Weinstein,Y. and Ihle,J.N. (1996) Erythropoietin induces activation of Stat5 through association with specific tyrosines on the receptor that are not required for a mitogenic response. Mol. Cell. Biol., 16, 1622–1631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawai S., Shimono,A., Wakamatsu,Y., Palmes,C., Hanaoka,K. and Kondoh,H. (1993) Defects of embryonic organogenesis resulting from targeted disruption of the N-myc gene in the mouse. Development, 117, 1445–1455. [DOI] [PubMed] [Google Scholar]
- Shultz L.D., Schweitzer,P.A., Rajan,T.V., Yi,T., Ihle,J.N., Matthews,R.J., Thomas,M.L. and Beier,D.R. (1993) Mutations at the murine motheaten locus are within the hematopoietic cell protein tyrosine phosphatase (Hcph) gene. Cell, 73, 1445–1454. [DOI] [PubMed] [Google Scholar]
- Socolovsky M., Fallon,A.E., Wang,S., Brugnara,C. and Lodish,H.F. (1999) Fetal anemia and apoptosis of red cell progenitors in Stat5a–/–5b–/– mice: a direct role for Stat5 in Bcl-X(L) induction. Cell, 98, 181–191. [DOI] [PubMed] [Google Scholar]
- Sokol L., Luhovy,M., Guan,Y., Prchal,J.F., Semenza,G.L. and Prchal,J.T. (1995) Primary familial polycythemia: a frameshift mutation in the erythropoietin receptor gene and increased sensitivity of erythroid progenitors to erythropoietin. Blood, 86, 15–22. [PubMed] [Google Scholar]
- Tauchi T., Damen,J.E., Toyama,K., Feng,G.S., Broxmeyer,H.E. and Krystal,G. (1996) Tyrosine 425 within the activated erythropoietin receptor binds Syp, reduces the erythropoietin required for Syp tyrosine phosphorylation, and promotes mitogenesis. Blood, 87, 4495–4501. [PubMed] [Google Scholar]
- Teglund S. et al. (1998) Stat5a and Stat5b proteins have essential and non-essential, or redundant, roles in cytokine responses. Cell, 93, 841–850. [DOI] [PubMed] [Google Scholar]
- van Deursen J., Heerschap,A., Oerlemans,F., Ruitenbeek,W., Jap,P., ter Laak,H. and Wieringa,B. (1993) Skeletal muscles of mice deficient in muscle creatine kinase lack burst activity. Cell, 74, 621–631. [DOI] [PubMed] [Google Scholar]
- Wickrema A. et al. (1999) Engagement of Gab1 and Gab2 in erythropoietin signaling. J. Biol. Chem., 274, 24469–24474. [DOI] [PubMed] [Google Scholar]
- Witthuhn B., Quelle,F.W., Silvennoinen,O., Yi,T., Tang,B., Miura,O. and Ihle,J.N. (1993) JAK2 associates with the erythropoietin receptor and is tyrosine phosphorylated and activated following EPO stimulation. Cell, 74, 227–236. [DOI] [PubMed] [Google Scholar]
- Wu H., Liu,X., Jaenisch,R. and Lodish,H.F. (1995) Generation of committed erythroid BFU-E and CFU-E progenitors does not require erythropoietin or the erythropoietin receptor. Cell, 83, 59–67. [DOI] [PubMed] [Google Scholar]
- Yi T., Zhang,J., Miura,O. and Ihle,J.N. (1995) Hematopoietic cell phosphatase (HCP) associates with the erythropoietin receptor following Epo induced receptor tyrosine phosphorylation: identification of potential binding sites. Blood, 85, 87–95. [PubMed] [Google Scholar]