

Abstract
Newly synthesized proteins in the endoplasmic reticulum (ER) must fold and assemble correctly before being transported to their final cellular destination. While some misfolded or partially assembled proteins have been shown to exit the ER, they fail to escape the early secretory system entirely, because they are retrieved from post-ER compartments to the ER. We elucidate a mechanistic basis for this retrieval and characterize its contribution to ER quality control by studying the fate of the unassembled T-cell antigen receptor (TCR) α chain. While the steady-state distribution of TCRα is in the ER, inhibition of retrograde transport by COPI induces the accumulation of TCRα in post-ER compartments, suggesting that TCRα is cycling between the ER and post-ER compartments. TCRα associates with BiP, a KDEL protein. Disruption of the ligand-binding function of the KDEL receptor releases TCRα from the early secretory system to the cell surface, so that TCRα is no longer subject to ER degradation. Thus, our findings suggest that retrieval by the KDEL receptor contributes to mechanisms by which the ER monitors newly synthesized proteins for their proper disposal.
Keywords: COPI/KDEL receptor/protein degradation/quality control/TCRα
Introduction
Secretory proteins are synthesized by ribosomes and translocated co-translationally or post-translationally to the endoplasmic reticulum (ER) through the translocon, Sec61p complex (Corsi and Schekman, 1996; Rapoport et al., 1999). These newly synthesized proteins interact with ER molecular chaperones, such as immunoglobulin heavy chain-binding protein (BiP), calnexin, calreticulin and protein disulfide isomerase, to become properly folded and assembled into a mature protein complex for transport along the secretory pathway. Misfolded or partially unassembled proteins are retained in the ER with ER chaperones. Thus, ER chaperones also participate in a quality control mechanism to prevent transport-incompetent proteins from being exported out of the ER (Hammond and Helenius, 1995; Ellgaard et al., 1999). ER chaperones act on those retained proteins either for further maturation, or for degradation, which protects the ER from the destructive consequence of protein aggregation (Gething and Sambrook, 1992). Such degradation, termed ER-associated degradation (ERAD) (Bonifacino and Klausner, 1994), is accomplished by the translocation of proteins to the cytosol through the translocon, followed by ubiquitylation, and then degradation via the proteasome system (Bonifacino and Weissman, 1998; Brodsky and McCracken, 1999).
Upon escape from the ER to reach post-ER compartments, which include the ER–Golgi intermediate compartment (IC; also referred to as tubular vesicular structures) and the Golgi complex, imperfect proteins have been shown to be transported back to the ER. In a tumor cell line that could not assemble class I molecules correctly, the unassembled class I molecules were shown to exit the ER to reach the Golgi complex, where they were then retrieved to the ER (Hsu et al., 1991). Moreover, misfolded vesicular stomatitis virus (VSV) G proteins expressed in Chinese hamster ovary (CHO) cells, which have an ER distribution, were also demonstrated to be cycling between the ER and post-ER compartments (Hammond and Helenius, 1994). Thus, protein sorting in the post-ER compartments that involves recognition and retrograde transport to the ER has been suggested to contribute to ER quality control. However, the underlying mechanism of this retrieval, and its significance to overall mechanisms of ER quality control, remain unclear.
Intracellular transport of proteins is mediated by membrane carriers that include coated vesicles and tubular structures. From the ER, proteins are packaged into the COPII-coated vesicles to initiate their transport from the ER (Rothman and Wieland, 1996; Schekman and Orci, 1996). Subsequently, proteins are transferred to the IC, which then moves along microtubules to the Golgi apparatus (Bonfanti et al., 1998). Upon arrival at the Golgi complex, proteins are sorted to the peripheral compartments of the cell, such as endosomes and plasma membrane. Also, proteins can be retrieved to the ER by retrograde transport at either the IC or the Golgi complex (Allan and Balch, 1999) by COPI-coated vesicles (Letourneur et al., 1994).
Selective retrograde transport of proteins from post-ER compartments to the ER is achieved by multiple mechanisms (Teasdale and Jackson, 1996). Transmembrane proteins that have a C-terminal dilysine (KKXX) sequence, such as type I ER membrane proteins (Nilsson et al., 1989; Jackson et al., 1990), ERGIC53 (Itin et al., 1995) and p24 family proteins (Fiedler et al., 1996), are transported in COPI vesicles, because the COPI coat recognizes the dilysine motif directly (Cosson and Letourneur, 1994). However, soluble proteins that reside in the lumen of membrane compartments can also be retrieved to the ER. Ones such as BiP have a C-terminal Lys-Asp-Glu-Leu (KDEL) sequence (Munro and Pelham, 1987) that is recognized by the KDEL receptor in the post-ER compartments (Lewis and Pelham, 1992), and are then sorted into COPI vesicles for retrograde transport (Orci et al., 1997). Retrograde transport that is independent of COPI has also been suggested recently (Girod et al., 1999; White et al., 1999); however, its mechanistic details remain poorly characterized.
Besides its retrieval function, the KDEL receptor has been suggested to play a role in regulating COPI transport (Aoe et al., 1997, 1998). ADP-ribosylation factor 1 (ARF1), a Ras-like small GTPase, regulates the formation of COPI vesicles (Rothman and Wieland, 1996). Like all small GTPases, the activation of ARF1 requires a guanine nucleotide exchange factor, and its deactivation requires a GTPase-activating protein (GAP). GAP acts by promoting the hydrolysis of GTP on ARF1 to GDP (Cukierman et al., 1995). This action has been demonstrated to be required for the proper sorting of cargo proteins into COPI vesicles (Lanoix et al., 1999; Pepperkok et al., 2000). Significantly, ligand binding on the luminal side of the KDEL receptor also induces its interaction with ARF GAP1 on the cytoplasmic side of the receptor. As a result, ARF GAP1 is recruited from the cytosol to the membranes to activate GAP activity on ARF1 (Aoe et al., 1999). Thus, these findings suggest that the KDEL receptor does not merely function as a passive cargo protein of COPI transport, but is capable of modulating this transport pathway.
In this study, we have examined how the KDEL receptor participates in ER quality control by studying the unassembled T-cell antigen receptor (TCR) α chain as a model system. TCR consists of at least six polypeptides (TCRα, β, CD3γ, δ, ε and ζ). Proper assembly is required for TCR to be expressed on the cell surface (Klausner et al., 1990). Unassembled TCRα has been demonstrated to be retained in the ER (Chen et al., 1988; Bonifacino et al., 1989) and degraded by ERAD in lymphocytes, as well as other cell types, when it is expressed as a single subunit (Huppa and Ploegh, 1997; Yu et al., 1997; Yang et al., 1998). Surprisingly, we find that a significant fraction of TCRα is exported from the ER and then retrieved to the ER from the post-ER compartments by retrograde transport. This retrieval is mediated by the KDEL receptor, and when disrupted, a significant fraction of TCRα can be detected to escape ERAD. Thus, our findings suggest an important role for the KDEL receptor in ER quality control.
Results
TCRα cycles in the early secretory system
A heterologous expression system has been used extensively to study the fate of unassembled TCRα chain by transfecting TCRα into COS cells that normally do not express the TCR. Without the other TCR subunits, the α subunit has been shown to be retained in the ER and targeted for degradation (Huppa and Ploegh, 1997; Yu et al., 1997; Yang et al., 1998). However, because an increasing number of proteins have been appreciated to maintain their distribution in the ER by being transported out and then cycling back (Pelham, 1996), we tested whether TCRα might also exhibit such behavior. Upon treatment with bafilomycin A1, a vacuolar H+-ATPase inhibitor that has been shown to impose a relative block on the retrograde arm of bidirectional transport between the ER and the Golgi complex (Palokangas et al., 1998), confocal microscopy revealed significantly increased co-localization of TCRα and ERGIC53 (Figure 1). The co-localization induced by bafilomycin also involved the redistribution of ERGIC53 to a pattern that was more compact and juxtanuclear (compare control with treatment condition in Figure 1). This effect is consistent with previous characterization of ERGIC53, which is known to cycle dynamically among the compartments of the early secretory system, which include the ER, IC and the Golgi complex (Itin et al., 1995). That is, when retrograde transport was blocked, ERGIC53 could be induced to accumulate at the Golgi complex (Palokangas et al., 1998).

Fig. 1. TCRα expressed in COS cells was retained in the ER. Perturbation of the retrograde transport from post-ER compartments to the ER enhanced the co-localization of TCRα and ERGIC53. COS cells were transiently transfected with TCRα. Forty hours later, the cells were fixed without treatment (control) or after incubation in the presence of bafilomycin A1 (10–6 M) at 37°C for 3 h (baf A1). The cells were evaluated by confocal laser scan microscopy with double labeling using a rabbit anti-murine TCRα antiserum and a mouse anti-ERGIC53 mAb. The pre-Golgi region was enlarged (#). Scale bar represents 10 µm.
Because of these known effects on the itinerary of proteins cycling in the early secretory compartments, we sought to examine the distribution of TCRα in more detail by subcellular fractionation. In the control condition, when no perturbation was added, most of TCRα was in the ER fractions as a p38 form, as judged by co-fractionation with calnexin. In this control setting, we could also detect a larger form (p43) in post-ER fractions, as judged by co-fractionation with the IC marker p58 (Bloom and Brashear, 1989) (Figure 2A). This is a surprising finding, as previous characterization of TCRα revealed that p38 is an immature glycoprotein that has not received Golgi-specific modification, while p43 represents a mature glycoprotein that has received Golgi-specific modification (Samelson, 1985; Yu et al., 1997). Consistent with the possibility that TCRα cycles out of the ER, when cells were treated with bafilomycin, TCRα accumulated markedly in post-ER fractions as the p43 form (Figure 2B). To ascertain that the p43 form of TCRα indeed represented those outside the ER, we assessed whether it had Golgi-specific glycosylation. Immunoblotting for TCRα from the different fractions that were either treated with endoglycosidase H (endo H) or subjected to mock treatment, we found that the p38 form was endo H sensitive, while the p43 form was endo H resistant (Figure 2C). These results, taken together with the above morphological findings, suggest that a significant fraction of TCRα was cycling between the ER and the Golgi complex.

Fig. 2. Subcellular fractionation on continuous sucrose gradients revealed the accumulation of Golgi-glycosylated TCRα in post-ER fractions. HeLa cells were transiently transfected with TCRα, and collected without treatment (A) or after incubation in the presence of bafilomycin A1 (10–6 M) at 37°C for 6 h (B). PNSs from the transfected cells were separated on continuous sucrose gradient (20–50%). Twelve fractions were obtained from each sample (fraction 1, top; fraction 12, bottom). An aliquot of each fraction was analyzed by SDS–PAGE. The distribution of TCRα, Golgi p58 and calnexin was determined by direct western blotting. While a 38 kDa core-glycosylated TCRα was localized in the ER fractions, a 43 kDa Golgi-glycosylated TCRα was found in the post-ER fractions. Aliquots of the ER [fraction 9 in (A)] and the post-ER [fraction 2 in (B)] fractions were treated with endo H and analyzed by SDS–PAGE. The membrane was immunoblotted using a rabbit anti-murine TCRα antiserum (C). A 43 kDa TCRα (p43) in the post-ER fraction acquired resistance to endo H digestion, while a 38 kDa TCRα (p38) in the ER fraction remained sensitive.
As a cycling itinerary involved active sorting in the post-ER compartments to retrieve TCRα back to the ER (Pelham, 1996), we next examined how this retrieval was accomplished. Retrograde transport from the post-ER compartments to the ER is mediated in many cases by the COPI transport vesicles (Letourneur et al., 1994; Orci et al., 1997). Thus, we examined the effect of acutely inactivating the function of COPI in vivo. A temperature-sensitive CHO cell line has been created that degrades ε-COP upon shift to a non-permissive temperature. Without ε-COP, the COPI complex could not be assembled to function (Guo et al., 1994). Thus, we used this cell line to examine the role of COPI in recycling TCRα from post-ER compartments. When transfected with TCRα and examined at the permissive temperature, TCRα in this mutant cell line had a similar distribution to that seen in COS cells, appearing in an ER distribution with little co-localization with ERGIC53 (Figure 3A). However, upon shift to the non-permissive temperature, a significant fraction of TCRα became co-localized with ERGIC53 in a compact juxtanuclear distribution. This co-localization was also confirmed by quantitative confocal microscopy. The intensities of TCRα and ERGIC53 within the cell are represented by fluorescent pixels that are quantifiable. Thus, quantitative scanning was performed to compare between areas suggested to have co-localization (as represented by yellow in the merge view of Figure 3A) with the rest of the cell (Figure 3B). The highest intensities of co-localization between TCRα and ERGIC53 correlated with the suggested area of co-localization in the merge view of Figure 3A. Thus, these findings suggested that the recycling of TCRα from post-ER compartments required the function of COPI.


Fig. 3. Blocking of the COPI transport enhanced the co-localization of TCRα and ERGIC53 in COPI mutant cells. LdlF CHO cells were transiently transfected with TCRα and myc-tagged ERGIC53 at 34°C. After incubation at a non-permissive temperature (39.5°C) for 6 h, the cells were fixed and evaluated by confocal laser scan microscopy with double labeling for TCRα with a rabbit anti-murine TCRα antiserum and for ERGIC53 with an anti-myc mAb (9E10) (A). Scale bar represents 10 µm. The pixel intensity (arbitrary units) in the fluorescence of TCRα (red) or ERGIC53 (green) was recorded by confocal laser scan microscopy along the line from the bottom to the top in the lower panel (B). The peak intensities of TCRα and ERGIC53 were co-localized well at a non-permissive temperature (39.5°C).
Retrieval of TCRα by the KDEL receptor
A dilysine (KKXX) motif on the cytoplasmic portion of transmembrane proteins mediates their retrograde transport to the ER, because COPI binds directly to this motif (Cosson and Letourneur, 1994). While TCRα is a transmembrane protein, it contains no such motif in its cytoplasmic domain. However, another possibility is suggested by studies on the ER chaperone BiP. BiP has been shown to leak out to the post-ER compartments, and then retrieved to the ER, because it contains a C-terminal KDEL motif that is recognized by the KDEL receptor (Munro and Pelham, 1987). Significantly, misfolded VSV-G protein that had previously been thought to be retained strictly in the ER has been shown to cycle in the early secretory system (Hammond and Helenius, 1994). During its cycling, VSV-G was detected in a complex with BiP. Thus, one possibility is that the KDEL receptor can retrieve TCRα, if TCRα were bound to a KDEL ligand, such as BiP. Consistent with this possibility, co-precipitation studies revealed an association between TCRα and BiP (Figure 4). This association was specific, because another ER chaperone, GRP94, did not co-precipitate with TCRα.
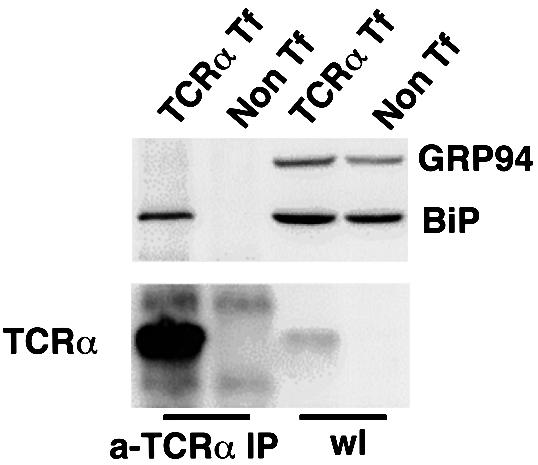
Fig. 4. Unassembled TCRα associated with a KDEL protein, BiP. HeLa cells were transiently transfected with TCRα, and then lysed and immunoprecipitated with an anti-murine TCRα mAb (A2B4) followed by SDS–PAGE. The membrane was immunoblotted using mouse anti-BiP mAb (SPA-827), which also recognizes GRP94, or a rabbit anti-murine TCRα antiserum. Immunoprecipitates from cells transfected with TCRα contained BiP, not GRP94 (a-TCRα IP). Direct immuno blotting of cell lysates (wl) revealed similar levels of BiP and GRP94 in either transfected (TCRα Tf) or non-transfected cells (Non Tf).
To provide further evidence that the KDEL receptor participates in the recycling of TCRα, we examined the effect of perturbing the ligand-binding function of the KDEL receptor. First, as the KDEL receptor binds to the KDEL ligands at the Golgi complex and then releases them at the ER, we examined whether increasing the expression of the KDEL receptor would shift the distribution of the TCRα away from the ER. Upon overexpression of the KDEL receptor, we detected by confocal microscopy more TCRα in a compact juxtanuclear distribution that had increased co-localization with the overexpressed KDEL receptor (Figure 5). To confirm that the effect of overexpressing the KDEL receptor on the distribution of TCRα was related to the ligand-binding function of the KDEL receptor, we also compared the effect of a point mutant that has been previously characterized to be defective in ligand binding (Townsley et al., 1993). Overexpression of this mutant receptor did not result in similar redistribution of TCRα (Figure 5).

Fig. 5. Unassembled TCRα co-localized with the KDEL receptor. COS cells were transiently transfected with TCRα and the wild-type KDEL receptor-myc, or with TCRα and a ligand-binding defective mutant KDEL receptor R169N-myc. Cells were fixed and evaluated by confocal laser scan microscopy with double labeling for TCRα with a rabbit anti-murine TCRα antiserum and for the KDEL receptors with an anti-myc mAb (9E10). The pre-Golgi region was enlarged (#). Scale bar represents 10 µm.
To rule out the possibility that overexpressing either the wild-type or the mutant KDEL receptor might be introducing an effect that was not related to the ligand-binding function of the KDEL receptor, we sought another approach to verify the above findings. We had shown previously that activation of the KDEL receptor through ligand binding on its luminal side induced the KDEL receptor to interact with the ARF GAP1 on the cytoplasmic side (Aoe et al., 1998). Thus, one possibility is that the complex of TCRα with BiP represented a ligand that would also activate the KDEL receptor to interact with GAP. Upon overexpression of TCRα, we detected an increased interaction between the KDEL receptor and ARF GAP1 (Figure 6). This level of increase was similar to a previously characterized enhanced association, when a chimeric KDEL ligand, which was created by appending the KDEL sequence to lysozyme (lysozyme–KDEL), was overexpressed (Aoe et al., 1998). Thus, this finding, when combined with the above observations on receptor overexpression, suggested that, most likely, TCRα was being retrieved by the KDEL receptor, because it was complexed with BiP, a known ligand for the receptor.
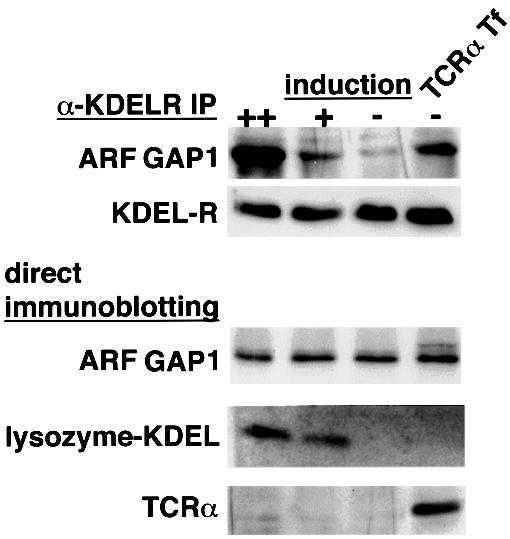
Fig. 6. Overexpression of unassembled TCRα induced the interaction of the KDEL receptor and ARF GAP1. A HeLa cell line that stably expresses the myc-tagged wild-type KDEL receptor and inducibly expresses lysozyme–KDEL by dexamethasone was induced for lysozyme–KDEL overexpression for 48 h (++) or 24 h (+), uninduced (–), or transiently transfected with TCRα without induction (TCRα Tf), and then lysed and immunoprecipitated with an anti-myc mAb, followed by immunoblotting with an anti-ARF GAP1 antiserum or an anti-myc mAb (α-KDELR IP). The whole-cell lysates from the above were evaluated for the expression of ARF GAP1, lysozyme–KDEL and TCRα by immunoblotting with an anti-ARF GAP1 antiserum, an anti-lysozyme antiserum or an anti-murine TCRα antiserum, respectively (direct immunoblotting).
A role for retrieval in ER quality control
Despite perturbing its recycling itinerary, not all TCRα appeared to cycle out of the ER, as judged by a residual ER distribution even in perturbation conditions that would result in a relative block in its retrieval (see Figures 1 and 3). Thus, to assess the physiological significance of TCRα cycling in the early secretory system, we examined how its recycling contributes to an important arm of ER quality control, ERAD. The natural ligands of the KDEL receptor are soluble ER chaperones, and only a minor fraction normally leaks from the ER because these proteins are mostly retained in the ER by a calcium-dependent mechanism (Booth and Koch, 1989). To saturate ligand binding by the KDEL receptor, the chimeric protein, lysozyme–KDEL, was used, because it was efficiently transported to the Golgi complex (Lewis and Pelham, 1992). Thus, to assess the role of TCRα recycling, we examined the effects of overexpressing the fusion protein, lysozyme–KDEL. First, to examine how TCRα recycling contributes to the total pool of TCRα being degraded in the ER, we examined in a metabolic pulse–chase experiment whether a fraction of TCRα would be protected from degradation when its recycling by the KDEL receptor was inhibited by the overexpression of lysozyme–KDEL. Expression of TCRα alone led mainly to a p38 form that was largely degraded by 3 h (Figure 7). However, in cells that co-overexpressed lysozyme–KDEL, TCRα acquired a p43 form by 3 h. These findings suggested that the p43 form was likely to represent the form of TCRα that escaped the recycling itinerary, as this form did not accumulate to any significant extent in the control setting.
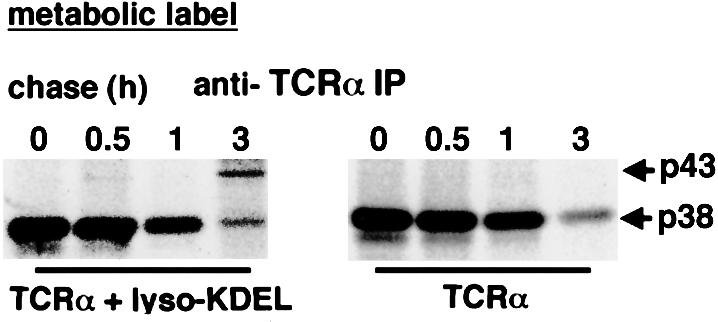
Fig. 7. Saturation of the KDEL receptor by lysozyme–KDEL released TCRα from the early secretory system. HeLa cells were transiently transfected with TCRα alone (right panel) or with TCRα and lysozyme KDEL (left panel), pulse-labeled, and then assessed for degradation at different chase times (0–3 h). TCRα was obtained by immuno precipitation with an anti-murine TCRα mAb (A2B4). Although a core-glycosylated 38 kDa TCRα was degraded, a Golgi-glycosylated 43 kDa TCRα remained significantly after 3 h in the presence of lysozyme–KDEL.
While only ∼10% of newly synthesized TCRα was transformed into a stable pool, as reflected by p43 (Figure 7), an important consideration was that this stable pool accumulated over time, while the p38 form (which represented the ER form) was constantly being degraded. Thus, to gain insight into this issue, we examined whether p43 could be detected to escape from the early secretory system by performing a surface-labeling experiment. Cells were co-transfected with TCRα and lysozyme–KDEL, and then subjected to surface biotinylation to label all surface proteins. Subsequently, cells were lysed and TCRα was immunoprecipitated. By this method, a significant pool of TCRα could be detected on the cell surface as the p43 form (Figure 8A). As a control, in cells that overexpressed the fusion protein lysozyme–AARL, which was not a ligand for the KDEL receptor, TCRα was not detected on the cell surface. Western blotting of total lysates derived from the overexpressed cells revealed that both fusion proteins were expressed to a similar extent (Figure 8B). Finally, to judge the extent that the p43 pool was increased upon disruption of retrieval by the KDEL receptor, we also compared the relative levels of p38 and p43 in whole-cell lysates. In cells that overexpressed lysozyme–KDEL, p43 represented ∼30% of total TCRα (represented by p38 and p43 combined) (Figure 8B). On the other hand, in cells that overexpressed lysozyme–AARL, p43 represented only ∼10% of total TCRα. Thus, this finding, together with the above observations that p43 was escaping ERAD by being transported to the cell surface upon disruption of retrieval by the KDEL receptor, suggested that this retrieval was likely to play a significant role in helping to maintain ER quality control.
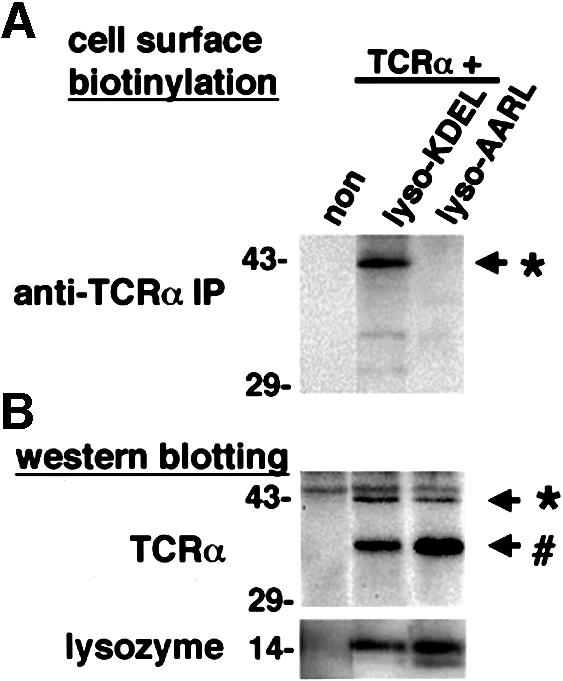
Fig. 8. Saturation of the KDEL receptor by a KDEL protein caused the cell surface expression of unassembled TCRα. (A) HeLa cells were transiently transfected with TCRα and lysozyme–KDEL, or TCRα and lysozyme–AARL. The left lane represents the mock transfection (non). Cells were surface biotinylated, and then lysed and immunoprecipitated with an anti-murine TCRα mAb (A2B4). The biotinylated proteins were analyzed by SDS–PAGE and assessed by chemiluminescence. (B) The whole-cell lysates from (A) were evaluated for the expression of TCRα and lysozymes by western blotting using a rabbit anti-murine TCRα antiserum or a rabbit anti-lysozyme antiserum, respectively. An asterisk represents the 43 kDa Golgi-glycosylated TCRα and a hash sign represents the 38 kDa ER-glycosylated TCRα.
Discussion
This study suggests an important role for the KDEL receptor in a post-ER retrieval mechanism that contributes to ER quality control. The TCRα chain that has been used as a model system to study ERAD is shown to cycle in the early secretory system. Retrieval of TCRα during this cycling is dependent on the KDEL receptor. Abrogating this retrieval results in a significant fraction of TCRα being released to the cell surface as a stable pool that is no longer subject to ER degradation.
Murine TCRα is a type I membrane protein containing four sites for N-glycosylation in its extracellular domain. If it is expressed as part of a complete TCR complex on the cell surface, it migrates as a 42–44 kDa band on SDS– PAGE (Samelson, 1985). Unassembled TCRα remains as a 38 kDa core-glycosylated precursor that is sensitive to digestion with endo H (Huppa and Ploegh, 1997; Yu et al., 1997; Yang et al., 1998). We have found by subcellular fractionation that TCRα could be detected in the post-ER fractions as a 43 kDa, endo H-resistant form (Figure 2). While this pool does not appear to be large in the control condition, when retrograde transport is not perturbed, an important consideration is that this distribution represents a steady-state distribution. Thus, to gain insight into the degree that TCRα cycles over time, perturbations in retrograde transport were used to show that a significant fraction of TCRα cycles out of the ER.
To elucidate how TCRα is retrieved during its cycling, we first showed that this retrieval is dependent on the function of COPI that is well characterized to mediate retrograde transport. However, the cytoplasmic domain of TCRα does not possess a dilysine motif that would provide a direct binding site for COPI, suggesting that an indirect mechanism is likely to be involved. In considering how such an indirect mechanism may occur, we were led by a previous observation that misfolded VSV-G proteins associate with BiP in post-ER compartments (Hammond and Helenius, 1994). While it had remained unclear whether this pool was recycled to the ER, we show in the current study that unassembled TCRα associates with BiP, and is retrieved to the ER in a fashion that depends on the ligand-binding function of the KDEL receptor. Taken together, these observations indicate that, most likely, retrieval of TCRα by the KDEL receptor is through a KDEL ligand, such as BiP, which complexes with TCRα. While we can not be certain that BiP solely mediates this retrieval, we do show that it is dependent on the ligand-binding function of the KDEL receptor. Thus, if not BiP, other KDEL protein(s) would necessarily be involved, as TCRα does not possess a KDEL motif in its luminal domain.
The finding that overexpression of lysozyme–KDEL disrupts retrieval of TCRα and leads to surface expression of TCRα suggested a way of assessing the relative contribution by the KDEL receptor to ER quality control. Previous studies have shown that TCRα is subject to ERAD, when expressed alone. Consistent with this, in a metabolic pulse–chase experiment, expression of TCRα alone led mainly to a p38 form that was degraded over time with kinetics characteristic of ER degradation. However, in cells that co-overexpress lysozyme–KDEL, TCRα acquires a p43 form at 3 h that remains stable (Figure 7). While this fraction represents only ∼10% of the initial total, an important consideration is that this fraction remains stable over a significantly longer period of time than its cohort, which is being constantly recycled to the ER for degradation. To gain insight into the importance of the retrieval function of the KDEL receptor, we examined whether TCRα could be detected on the cell surface upon disruption of ligand binding by the KDEL receptor. Whereas such a surface pool is undetectable when retrieval by the KDEL receptor is intact, a significant fraction is detected when retrieval is disrupted.
TCRα is likely to be only one of many newly synthesized proteins that complex with BiP during their maturation through folding and assembly in the ER. Thus, our finding that TCRα has a significant fraction on the cell surface suggests that other newly synthesized proteins would also be leaked out to the cell surface in significant levels if the retrieval function of the KDEL receptor were compromised. However, current evidence also suggests the possibility that a compromise of this retrieval may have a physiological role under specialized circumstances. In immature CD4–CD8– thymocytes, several KDEL proteins, such as BiP, calreticulin and GRP94, have been found on the cell surface (Wiest et al., 1997). Surface expression of KDEL proteins has been reported in a neural culture cell (Xiao et al., 1999b) and in activated T lymphocytes (Arosa et al., 1999). These KDEL proteins seem to associate with other proteins that are derived from the ER. Although the biological significance of their escape from the retrieval system is not well understood, certain KDEL proteins, such as calreticulin, have been demonstrated to have distinct functions outside the ER. Calreticulin is a KDEL protein that contributes to the folding of glycoproteins in the ER. However, when it is expressed on the cell surface (Xiao et al., 1999a), calreticulin may mediate cell adhesion or a mitogenic signal as a receptor for fibrinogen (Gray et al., 1995). Thus, modulating retrieval by the KDEL receptor may provide novel ways of regulating the function of certain proteins under specialized circumstances, and this issue will be explored in future studies.
Finally, our finding that overexpression of TCRα induces an interaction between the KDEL receptor and ARF GAP1 reveals an intriguing regulatory feedback mechanism. In previous studies, we have shown that activation of the KDEL receptor by its ligand induces the receptor to associate with ARF GAP1 (Aoe et al., 1998), and this interaction is critical for GAP to act on the GTP-bound form of ARF1 (Aoe et al., 1999). Other recent studies suggest that ARF1, COPI and ARF GAP1 form a complex (Goldberg, 1999), which has been referred to as a priming complex (Springer et al., 1999), to initiate formation of COPI vesicles. This complex is needed to ensure the proper incorporation of cargo proteins into COPI vesicles (Lanoix et al., 1999). Taken together, these observations suggest that ligand binding by the KDEL receptor would regulate the degree of COPI-mediated retrograde transport. This scenario also suggests a mechanism by which retrieval through the KDEL receptor is responsive to the level of KDEL ligands that leak out of the ER to reach the Golgi complex. During stress that increases the level of misfolded proteins in the ER, one possibility is that more of these misfolded proteins would leak from the ER. However, because of the mechanistic link between ligand binding by the KDEL receptor and its interaction with ARF GAP1, an important function of the KDEL receptor appears to be in integrating increased leakage that reaches the Golgi complex to increasing COPI-mediated retrograde transport from this organelle. In this manner, any increased leakage out of the ER would not result in increased leakage out of the early secretory system.
Materials and methods
Cells and reagents
COS-7 and HeLa cells were grown in a complete medium that consisted of Dulbecco’s modified Eagle’s medium (DMEM; Sigma Chemical Co., Irvine, UK) with 10% fetal calf serum (FCS), 2 mM glutamine, 50 µg/ml streptomycin and 50 U/ml penicillin G at 37°C in a 5% CO2 incubator. A HeLa cell that stably expresses the myc-tagged KDEL receptor and inducibly expresses lysozyme–KDEL was described (Aoe et al., 1998). Inducible lysozyme–KDEL was expressed by adding 100 nM dexamethasone for 24–48 h. Wild-type CHO cells and temperature-sensitive mutant ldlF cells (Guo et al., 1994) were grown in Ham’s F12 medium (Gibco-BRL, Rockville, MD) with 10% FCS, 2 mM glutamine, 50 µg/ml streptomycin and 50 U/ml penicillin G at 34°C in a 5% CO2 incubator.
The following antibodies were used: mouse monoclonal antibody (mAb) 9E10 against the myc epitope (ATCC, Rockville, MD), mouse mAb against Golgi p58 (Sigma Chemical Co., Irvine, UK), mouse mAb SPA-827 against BiP (Stressgen, Victoria, Canada), mouse mAb against ERGIC53 (kindly provided by H.-P.Hauri, Basel, Switzerland), mouse mAb AF8 against calnexin (kindly provided by M.M.Brenner, Boston, MA), mouse mAb A2B4 against murine TCRα 2B4 (Saito et al., 1987), rabbit polyclonal antiserum against lysozyme (Chemicon, Temecula, CA), rabbit polyclonal antiserum against murine TCR (kindly provided by M.Taniguchi, Chiba, Japan) and rabbit polyclonal antiserum against ARF GAP1 (kindly provided by D.Cassel, Haifa, Israel). Cyanine-conjugated donkey antibody against mouse IgG, cyanine-conjugated donkey antibody against rabbit IgG, indocarbocyanine-conjugated donkey antibody against mouse IgG and indocarbocyanine-conjugated donkey antibody against rabbit IgG were obtained from Jackson ImmunoResearch Laboratories, Inc. (West Grove, PA). Bafilomycin A1 was purchased from Sigma Chemical Co. (Irvine, UK).
Plasmids and transfection
The following cDNAs were used. The myc-tagged human KDEL receptor, and C-terminal KDEL- or AARL-tagged lysozyme have been described previously (Hsu et al., 1992; Aoe et al., 1998). The myc-tagged mutant KDEL receptor R169N (Townsley et al., 1993) was a gift from H.R.B.Pelham (Medical Research Council, Cambridge, UK). Myc-tagged ERGIC53 was kindly provided by H.-P.Hauri (Basel, Switzerland). The cDNA encoding murine TCRα 2B4 (Saito et al., 1987) was subcloned into a modified mammalian expression vector pCDLSRα, pXS (Hsu et al., 1992). Transfection with the calcium phosphate method was performed as described previously (Hsu et al., 1992).
Confocal laser scan microscopy
Cells on coverslips were fixed in 2% formaldehyde in phosphate-buffered saline (PBS) for 10 min at room temperature, and then processed as described previously (Hsu et al., 1992). The coverslips were mounted with PermaFluor (Immunon, Pittsburgh, PA). The labeled cells were examined using a confocal laser scan microscope (LSM510; Carl Zeiss Co., Ltd, Jena, Germany) fitted with krypton and argon lasers.
Immunoprecipitation and immunoblotting
Transfected cells were removed from dishes, pelleted by centrifugation, and lysed for 30 min at 4°C in a buffer containing 1% Triton X-100, 50 mM Tris–HCl pH 7.4, 300 mM NaCl, 10 µg/ml aprotinin, 10 µg/ml leupeptin, 30 µg/ml N-acetyl-l-leucinal-l-leucinal-l-norleucinal (ALLN; Sigma Chemical Co., St Louis, MO) and 2 mg/ml iodoacetamide. The lysates were pre-cleared with protein A–Sepharose beads, and incubated with antibody-coupled protein A–Sepharose for 1 h at 4°C. Immuno precipitates were washed, boiled in a sample buffer, and separated by SDS–PAGE under reducing conditions. For immunoblotting, gels were transferred onto polyvinylidene fluoride membranes (Immobilon-P; Millipore Corp., Bedford, MA), blocked, incubated with primary antibodies followed by peroxidase-conjugated donkey anti-mouse or anti-rabbit IgG antibody, and developed by chemiluminescence (ECL; Amersham Pharmacia Biotech, Buckinghamshire, UK). Imaging was by LAS1000 and Image Gauge software (Fuji Photo Film Co. Ltd, Tokyo, Japan).
Metabolic labeling and chase experiment
Cells were transiently transfected with TCRα construct alone and/or with other constructs as described in the figure legends. Forty hours later, cells were pre-incubated in a labeling medium (DMEM without methionine supplemented with 2% FCS, 2 mM glutamine, 50 µg/ml streptomycin and 50 U/ml penicillin G) for 20 min at 37°C, labeled with [35S]methionine (Amersham Pharmacia Biotech) at 250 µCi/ml for 10 min, washed, and then chased in a complete medium for 0–3 h. The cells were collected, lysed, and subjected to immunoprecipitation with anti-murine TCRα mAb (A2B4) as above. The immunoprecipitates were washed, boiled in a sample buffer, and separated by SDS–PAGE under reducing conditions. The gels were then analyzed by BAS 2500 and Image Gauge software (Fuji Photo Film Co. Ltd).
Cell surface biotinylation
The transfected cells were washed three times with PBS, and incubated with 100 µg/ml Sulfo-NHS-LC-biotin (Pierce, Rockford, IL) in a buffer containing 0.1 M HEPES pH 8.0 for 1 h at 4°C. The cells were washed three times with DMEM and removed from the dishes, pelleted by centrifugation, lysed, and then subjected to immunoprecipitation with anti-murine TCRα mAb (A2B4) as above. The immunoprecipitates were washed, boiled in a sample buffer, and separated by SDS–PAGE under reducing conditions. The gels were transferred onto polyvinylidene fluoride membranes, blocked, incubated with the Vecstatin ABC kit (Vector Laboratories, Inc., Burlingame, CA), and developed by chemiluminescence. Imaging was by LAS1000 and Image Gauge software (Fuji Photo Film Co. Ltd).
Sucrose gradient
Cells were washed twice with ice-cold PBS, and once with homogenization buffer (10 mM triethanolamine, 1 mM EDTA, 250 mM sucrose), then removed from dishes, and pelleted by centrifugation at 800 g for 10 min. The pellet was resuspended in 1 ml of homogenization buffer containing 10 µg/ml aprotinin, 10 µg/ml leupeptin, 30 µg/ml ALLN, and homogenized by passing four times through a ball-bearing homogenizer (EMBL, Heidelberg, Germany). A post-nuclear supernatant (PNS) was obtained by centrifugation at 800 g for 10 min at 4°C. The PNS was loaded on continuous sucrose gradient (20–50%) prepared by Gradient Master (Biocomp Instruments, Inc., Fredericton, Canada). The PNS on the gradient was centrifuged at 40 000 r.p.m. for 2 h using a Beckman SW41Ti rotor. Twelve fractions were obtained from each sample. One-twentieth of each fraction was separated by SDS–PAGE under reducing conditions, then transferred onto polyvinylidene fluoride membranes. The distribution of TCRα, calnexin and Golgi p58 was determined by western blotting. Imaging was by LAS1000 and Image Gauge software (Fuji Photo Film Co. Ltd).
Acknowledgments
Acknowledgements
We thank Masaru Taniguchi and Takashi Nishino for support, and Hiroshi Ohno and Masahiko Sugita for helpful suggestions. This work was supported by Grants-in-Aids for Science Research from the Ministry of Education to T.A. and H.K., and a grant from Chiba University School of Medicine to T.A.
References
- Allan B.B. and Balch,W.E. (1999) Protein sorting by directed maturation of Golgi compartments. Science, 285, 63–66. [DOI] [PubMed] [Google Scholar]
- Aoe T., Cukierman,E., Lee,A., Cassel,D., Peters,P.J. and Hsu,V.W. (1997) The KDEL receptor, ERD2, regulates intracellular traffic by recruiting a GTPase-activating protein for ARF1. EMBO J., 16, 7305–7316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aoe T., Lee,A.J., van Donselaar,E., Peters,P.J. and Hsu,V.W. (1998) Modulation of intracellular transport by transported proteins: insight from regulation of COPI-mediated transport. Proc. Natl Acad. Sci. USA, 95, 1624–1629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aoe T., Huber,I., Vasudevan,C., Watkins,S.C., Romero,G., Cassel,D. and Hsu,V.W. (1999) The KDEL receptor regulates a GTPase-activating protein for ADP-ribosylation factor 1 by interacting with its non-catalytic domain. J. Biol. Chem., 274, 20545–20549. [DOI] [PubMed] [Google Scholar]
- Arosa F.A., de Jesus,O., Porto,G., Carmo,A.M. and de Sousa,M. (1999) Calreticulin is expressed on the cell surface of activated human peripheral blood T lymphocytes in association with major histocompatibility complex class I molecules. J. Biol. Chem., 274, 16917–16922. [DOI] [PubMed] [Google Scholar]
- Bloom G.S. and Brashear,T.A. (1989) A novel 58-kDa protein associates with the Golgi apparatus and microtubules. J. Biol. Chem., 264, 16083–16092. [PubMed] [Google Scholar]
- Bonfanti L. et al. (1998) Procollagen traverses the Golgi stack without leaving the lumen of cisternae: evidence for cisternal maturation. Cell, 95, 993–1003. [DOI] [PubMed] [Google Scholar]
- Bonifacino J.S. and Klausner,R.D. (1994) Degradation of proteins retained in the endoplasmic reticulum. In Ciechanover,A. and Schwartz,A.L. (eds), Cellular Proteolytic Systems. Wiley-Liss, New York, NY, pp. 137–160.
- Bonifacino J.S. and Weissman,A.M. (1998) Ubiquitin and the control of protein fate in the secretory and endocytic pathways. Annu. Rev. Cell Dev. Biol., 14, 19–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonifacino J.S., Suzuki,C.K., Lippincott-Schwartz,J., Weissman,A.M. and Klausner,R.D. (1989) Pre-Golgi degradation of newly synthesized T-cell antigen receptor chains: intrinsic sensitivity and the role of subunit assembly. J. Cell Biol., 109, 73–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Booth C. and Koch,G.L. (1989) Perturbation of cellular calcium induces secretion of luminal ER proteins. Cell, 59, 729–737. [DOI] [PubMed] [Google Scholar]
- Brodsky J.L. and McCracken,A.A. (1999) ER protein quality control and proteasome-mediated protein degradation. Semin. Cell Dev. Biol., 10, 507–513. [DOI] [PubMed] [Google Scholar]
- Chen C., Bonifacino,J.S., Yuan,L.C. and Klausner,R.D. (1988) Selective degradation of T cell antigen receptor chains retained in a pre-Golgi compartment. J. Cell Biol., 107, 2149–2161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corsi A.K. and Schekman,R. (1996) Mechanism of polypeptide translocation into the endoplasmic reticulum. J. Biol. Chem., 271, 30299–30302. [DOI] [PubMed] [Google Scholar]
- Cosson P. and Letourneur,F. (1994) Coatomer interaction with di-lysine endoplasmic reticulum retention motifs. Science, 263, 1629–1631. [DOI] [PubMed] [Google Scholar]
- Cukierman E., Huber,I., Rotman,M. and Cassel,D. (1995) The ARF1 GTPase-activating protein: zinc finger motif and Golgi complex localization. Science, 270, 1999–2002. [DOI] [PubMed] [Google Scholar]
- Ellgaard L., Molinari,M. and Helenius,A. (1999) Setting the standards: quality control in the secretory pathway. Science, 286, 1882–1888. [DOI] [PubMed] [Google Scholar]
- Fiedler K., Veit,M., Stamnes,M.A. and Rothman,J.E. (1996) Bimodal interaction of coatomer with the p24 family of putative cargo receptors. Science, 273, 1396–1399. [DOI] [PubMed] [Google Scholar]
- Gething M.J. and Sambrook,J. (1992) Protein folding in the cell. Nature, 355, 33–45. [DOI] [PubMed] [Google Scholar]
- Girod A., Storrie,B., Simpson,J.C., Johannes,L., Goud,B., Roberts,L.M., Lord,J.M., Nilsson,T. and Pepperkok,R. (1999) Evidence for a COP-I-independent transport route from the Golgi complex to the endoplasmic reticulum. Nature Cell Biol., 1, 423–430. [DOI] [PubMed] [Google Scholar]
- Goldberg J. (1999) Structural and functional analysis of the ARF1– ARFGAP complex reveals a role for coatomer in GTP hydrolysis. Cell, 96, 893–902. [DOI] [PubMed] [Google Scholar]
- Gray A.J., Park,P.W., Broekelmann,T.J., Laurent,G.J., Reeves,J.T., Stenmark,K.R. and Mecham,R.P. (1995) The mitogenic effects of the B β chain of fibrinogen are mediated through cell surface calreticulin. J. Biol. Chem., 270, 26602–26606. [DOI] [PubMed] [Google Scholar]
- Guo Q., Vasile,E. and Krieger,M. (1994) Disruptions in Golgi structure and membrane traffic in a conditional lethal mammalian cell mutant are corrected by ε-COP. J. Cell Biol., 125, 1213–1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammond C. and Helenius,A. (1994) Quality control in the secretory pathway: retention of a misfolded viral membrane glycoprotein involves cycling between the ER, intermediate compartment, and Golgi apparatus. J. Cell Biol., 126, 41–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammond C. and Helenius,A. (1995) Quality control in the secretory pathway. Curr. Opin. Cell Biol., 7, 523–529. [DOI] [PubMed] [Google Scholar]
- Hsu V.W., Yuan,L.C., Nuchtern,J.G., Lippincott-Schwartz,J., Hammerling,G.J. and Klausner,R.D. (1991) A recycling pathway between the endoplasmic reticulum and the Golgi apparatus for retention of unassembled MHC class I molecules. Nature, 352, 441–444. [DOI] [PubMed] [Google Scholar]
- Hsu V.W., Shah,N. and Klausner,R.D. (1992) A brefeldin A-like phenotype is induced by the overexpression of a human ERD-2-like protein, ELP-1. Cell, 69, 625–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huppa J.B. and Ploegh,H.L. (1997) The α chain of the T cell antigen receptor is degraded in the cytosol. Immunity, 7, 113–122. [DOI] [PubMed] [Google Scholar]
- Itin C., Schindler,R. and Hauri,H.P. (1995) Targeting of protein ERGIC-53 to the ER/ERGIC/cis-Golgi recycling pathway. J. Cell Biol., 131, 57–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson M.R., Nilsson,T. and Peterson,P.A. (1990) Identification of a consensus motif for retention of transmembrane proteins in the endoplasmic reticulum. EMBO J., 9, 3153–3162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klausner R.D., Lippincott-Schwartz,J. and Bonifacino,J.S. (1990) The T cell antigen receptor: insights into organelle biology. Annu. Rev. Cell Biol., 6, 403–431. [DOI] [PubMed] [Google Scholar]
- Lanoix J., Ouwendijk,J., Lin,C.C., Stark,A., Love,H.D., Ostermann,J. and Nilsson,T. (1999) GTP hydrolysis by arf-1 mediates sorting and concentration of Golgi resident enzymes into functional COPI vesicles. EMBO J., 18, 4935–4948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letourneur F., Gaynor,E.C., Hennecke,S., Demolliere,C., Duden,R., Emr,S.D., Riezman,H. and Cosson,P. (1994) Coatomer is essential for retrieval of dilysine-tagged proteins to the endoplasmic reticulum. Cell, 79, 1199–1207. [DOI] [PubMed] [Google Scholar]
- Lewis M.J. and Pelham,H.R. (1992) Ligand-induced redistribution of a human KDEL receptor from the Golgi complex to the endoplasmic reticulum. Cell, 68, 353–364. [DOI] [PubMed] [Google Scholar]
- Munro S. and Pelham,H.R. (1987) A C-terminal signal prevents secretion of luminal ER proteins. Cell, 48, 899–907. [DOI] [PubMed] [Google Scholar]
- Nilsson T., Jackson,M. and Peterson,P.A. (1989) Short cytoplasmic sequences serve as retention signals for transmembrane proteins in the endoplasmic reticulum. Cell, 58, 707–718. [DOI] [PubMed] [Google Scholar]
- Orci L., Stamnes,M., Ravazzola,M., Amherdt,M., Perrelet,A., Sollner,T.H. and Rothman,J.E. (1997) Bidirectional transport by distinct populations of COPI-coated vesicles. Cell, 90, 335–349. [DOI] [PubMed] [Google Scholar]
- Palokangas H., Ying,M., Vaananen,K. and Saraste,J. (1998) Retrograde transport from the pre-Golgi intermediate compartment and the Golgi complex is affected by the vacuolar H+-ATPase inhibitor bafilomycin A1. Mol. Biol. Cell, 9, 3561–3578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelham H.R. (1996) The dynamic organisation of the secretory pathway. Cell Struct. Funct., 21, 413–419. [DOI] [PubMed] [Google Scholar]
- Pepperkok R., Whitney,J.A., Gomez,M. and Kreis,T.E. (2000) COPI vesicles accumulating in the presence of a GTP restricted arf1 mutant are depleted of anterograde and retrograde cargo. J. Cell Sci., 113, 135–144. [DOI] [PubMed] [Google Scholar]
- Rapoport T.A., Matlack,K.E., Plath,K., Misselwitz,B. and Staeck,O. (1999) Posttranslational protein translocation across the membrane of the endoplasmic reticulum. Biol. Chem., 380, 1143–1150. [DOI] [PubMed] [Google Scholar]
- Rothman J.E. and Wieland,F.T. (1996) Protein sorting by transport vesicles. Science, 272, 227–234. [DOI] [PubMed] [Google Scholar]
- Saito T., Weiss,A., Miller,J., Norcross,M.A. and Germain,R.N. (1987) Specific antigen-Ia activation of transfected human T cells expressing murine Tiαβ–human T3 receptor complexes. Nature, 325, 125–130. [DOI] [PubMed] [Google Scholar]
- Samelson L.E. (1985) An analysis of the structure of the antigen receptor on a pigeon cytochrome c-specific T cell hybrid. J. Immunol., 134, 2529–2535. [PubMed] [Google Scholar]
- Schekman R. and Orci,L. (1996) Coat proteins and vesicle budding. Science, 271, 1526–1533. [DOI] [PubMed] [Google Scholar]
- Springer S., Spang,A. and Schekman,R. (1999) A primer on vesicle budding. Cell, 97, 145–148. [DOI] [PubMed] [Google Scholar]
- Teasdale R.D. and Jackson,M.R. (1996) Signal-mediated sorting of membrane proteins between the endoplasmic reticulum and the Golgi apparatus. Annu. Rev. Cell Dev. Biol., 12, 27–54. [DOI] [PubMed] [Google Scholar]
- Townsley F.M., Wilson,D.W. and Pelham,H.R. (1993) Mutational analysis of the human KDEL receptor: distinct structural requirements for Golgi retention, ligand binding and retrograde transport. EMBO J., 12, 2821–2829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White J. et al. (1999) Rab6 coordinates a novel Golgi to ER retrograde transport pathway in live cells. J. Cell Biol., 147, 743–760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiest D.L., Bhandoola,A., Punt,J., Kreibich,G., McKean,D. and Singer,A. (1997) Incomplete endoplasmic reticulum (ER) retention in immature thymocytes as revealed by surface expression of ‘ER-resident’ molecular chaperones. Proc. Natl Acad. Sci. USA, 94, 1884–1889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao G., Chung,T.F., Fine,R.E. and Johnson,R.J. (1999a) Calreticulin is transported to the surface of NG108-15 cells where it forms surface patches and is partially degraded in an acidic compartment. J. Neurosci. Res., 58, 652–662. [DOI] [PubMed] [Google Scholar]
- Xiao G., Chung,T.F., Pyun,H.Y., Fine,R.E. and Johnson,R.J. (1999b) KDEL proteins are found on the surface of NG108-15 cells. Brain Res. Mol. Brain Res., 72, 121–128. [DOI] [PubMed] [Google Scholar]
- Yang M., Omura,S., Bonifacino,J.S. and Weissman,A.M. (1998) Novel aspects of degradation of T cell receptor subunits from the endo plasmic reticulum (ER) in T cells: importance of oligosaccharide processing, ubiquitination, and proteasome-dependent removal from ER membranes. J. Exp. Med., 187, 835–846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu H., Kaung,G., Kobayashi,S. and Kopito,R.R. (1997) Cytosolic degradation of T-cell receptor α chains by the proteasome. J. Biol. Chem., 272, 20800–20804. [DOI] [PubMed] [Google Scholar]