

Abstract
The enantioselective synthesis of the (R,R)- and (S,S)-enantiomers of 1 from commercially available 3-chlorocinnamic acid is reported. The Sharpless asymmetric epoxidation was used to establish the stereocenters in the synthesis of both enantiomers of 1.
1. Introduction
Stimulant abuse is a serious problem in the US with 37% of state and local law enforcement agencies identifying cocaine as their greatest drug threat.1 It has been postulated that the addictive properties of cocaine are due to the blockade of dopamine transporters (DAT) in the brain by cocaine and subsequent increase in extracellular dopamine levels (the dopamine hypothesis).2 It is known that cocaine displays similar in vitro affinity for the DAT, serotonin transporter (SERT), and norepinephrine transporter (NET) in nervous tissue.3 Furthermore, a growing body of evidence indicates that the norepinephrine transporter (NET) and serotonin transporter (SERT) also play a role in mediating cocaine s pharmacological effects.4-12 It has been suggested that blockade of DAT by cocaine contributes positively to cocaine s reinforcing effects, while blockade of SERT may have a negative influence.11,12 The influence of NET blockade is, as yet, unclear though it has been suggested that the blockade of NET may mediate the aversive effects of cocaine.7,10
To probe the role of NET in mediating the pharmacological effects of cocaine, high-affinity ligands are required as pharmacological tools. Compounds possessing the aryloxy-propanolamine or aryloxy-methyl-morpholino moiety, such as nisoxetine, tomoxetine, and reboxetine (Fig. 1), have been shown to possess high affinities and selectivities for NET.13 Compounds of these classes have been evaluated as potential treatments for depression and attention-deficit/hyperactivity disorder (ADHD).14-16 In fact, reboxetine (EdronaxTM) is currently marketed in Europe as an antidepressant while tomoxetine (StratteraTM), has been approved by the FDA as a treatment for ADHD.17,18 The enantiomers of these compounds often display significantly different activities at NET. For example, reboxetine is sold commercially as an enantiomeric mixture of (R,R)- and (S,S)-isomers. However, the (S,S)-enantiomer of reboxetine is approximately 24 times more potent than the (R,R)-enantiomer.19 In the case of tomoxetine, almost all activity resides in the (R)-enantiomer.13 Current interest in the development of pharmacotherapies for the treatment of depression and ADHD has necessitated the development of enantioselective routes to these classes of compounds.
Figure 1.
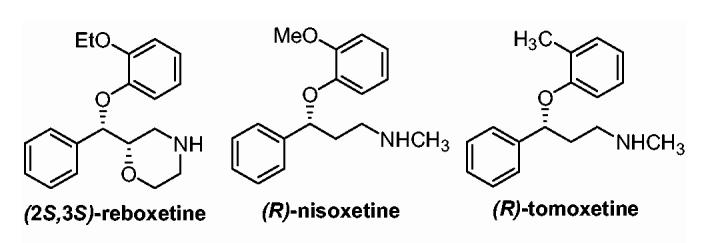
Structures of reboxetine, nisoxetine, and tomoxetine.
Racemic 1 was previously reported to inhibit norepinephrine uptake in rat hypothalamic synaptosomes with high selectivity versus DAT and SERT.20 However, the chiral isomers of 1 have never been evaluated at these monoamine transporters. This, together with the possibility for the development of 1 as a NET-selective PET radiotracer for brain imaging studies, makes it a particularly attractive target for synthesis. The route described herein provides an efficient enantioselective synthesis of (R,R)- and (S,S)-enantiomers of 1.
2. Results and discussion
Initially, we embarked on the racemic synthesis of 1 (Scheme 1) to validate the reaction sequence and to provide a racemic material for biological testing. Thus, DIBAL-H reduction of ethyl 3-chlorocinnamate 221,22 afforded cinnamyl alcohol 323 in 93% yield. Racemic epoxy alcohol 424 was prepared from 3 by reaction with peracetic acid. This epoxide was subsequently cleaved with guaiacol to provide diol 5. Epoxide 6 was prepared via a sequence of reactions from 5 involving the selective protection of the primary and secondary alcohol functionalities as TMS ether and mesylate, respectively, and subsequent cleavage of the primary alcohol followed by ring closure by elimination of the secondary mesyloxy group. The overall sequence of events was conducted without any intermediate purification and the overall transformation was completed in 57% yield from 5. The reaction of 6 with ethanolamine in isopropanol gave amino alcohol 7. Treatment of 7 with tosyl chloride gave 8a, which was subsequently cyclized to 9a under phase transfer catalysis conditions. However, N-detosylation of 9a to give racemic 1 as described in the literature for similar morpholino substrates,25 gave unsatisfactory yields of product. Attempted deprotection of 9a with sodium naphthalide resulted in recovery of starting material. Since the tosyl group proved so difficult to remove we decided to investigate use of the more labile nosyl group. Thus, compound 9b was prepared from 7 according to Scheme 1. However, removal of the nosyl protecting group could not be achieved with benzenethiol and some decomposition of starting material occurred. After this disappointing result, we contemplated the use of other nitrogen protecting groups for compound 7. The Boc group seemed particularly appealing in terms of ease of cleavage. Thus compound 10 was prepared. It was envisaged that cyclization to 1 could be effected by tosylation of the primary alcohol function in 10 to give the corresponding tosylate followed by ring closure under PTC conditions and Boc deprotection. However, tosylation of 10 led to decomposition. At this juncture, with 10 in hand, we sought to effect cyclization to 11 in one pot by treatment of 10 with NaH and tosylimidazole in THF.25 The Boc group was then removed from 11 with trifluoroacetic acid to give racemic 1. The HCl salt of 1 was prepared and found to have a melting point identical to the previously reported value.20
Scheme 1.

Reagents and conditions: (a) DIBALH, THF, 93%; (b) CH3CO3H, CH2Cl2, 80%; (c) guaiacol, NaOH, CH2Cl2, 39%; (d) TMSCl, Et3N, EtOAc; (e) MsCl, Et3N, EtOAc; (f) 2 M HCl, EtOAc; (g) NaOH, toluene, methyltributylammonium chloride, 57% from 5; (h) ethanolamine, i-PrOH, reflux, 48%; (i) tosyl chloride or nosyl chloride, Et3N, CH2Cl2, 80-85%; (j) NaOH, toluene, methyltributylammonium chloride, 65%; (k) Nanaphthalide for 9a; (l) Benzenethiol for 9b; (m) Boc2O, CH2Cl2, 92%; (n) NaH, TsIm, 60%; (o) TFA, CH2Cl2, 90%.
Racemic 1 was then reevaluated for its ability to inhibit uptake of [3H]NE.20 The racemate displayed high potency in inhibiting uptake of norepinephrine (Ki = 0.46 nM) with good selectivity versus dopamine and serotonin uptake in vitro (Ki = 1732 and 39.4 nM, respectively).
Having established a route to racemic 1 and bolstered by its high NET potency, we next focused our attentions on preparing chiral material. Racemic syntheses of compounds of the reboxetine class have been previously described in the literature.20,26,27 Chiral products have been accessed by optical resolution20,26,27 or by asymmetric synthesis.28-30 The attractiveness of the present enantioselective route lies in its amenability to the preparation of all four possible stereoisomers of 1.
Our enantioselective route to (R,R)- and (S,S)-enantiomers of 1 started with commercially available 3-chlorocinnamic acid. This was esterified to provide ethyl3-chlorocinnamate (2)21,22 and the ester subsequently reduced with DIBAL-H to give the cinnamyl alcohol 3 in 98% yield.23 The key step in our sequence to (S,S)-1 (Scheme 2) is the Sharpless asymmetric epoxidation (SAE) of 3. We envisaged that SAE of 3 with D-DET should provide (R,R)-4 and thus the (S,S)-enantiomer of 1, while in a similar fashion, L-DET should provide (R,R)-1. Attempts to effect the SAE of 3 under conditions reported for similar substrates31 [Ti(i-OPr)4, tert-butylhydroperoxide, D-DET, -20 °C] resulted in poor and variable enantiomeric excesses (ees). It is known that the SAE can suffer from poor ees due to the presence of adventitious water in the reaction.31 However, even when rigorous attempts were made at keeping the reaction anhydrous the maximum ee obtained was 45%. Significant improvements in the absolute value and variability of ees was achieved by substitution of tert-butylhydroperoxide with cumene hydroperoxide. Thus, both (R,R)-4 and its enantiomer were prepared by SAE in greater than 84% ee when cumene hydroperoxide was used as oxidant. The epoxide (R,R)-4 was cleaved with guaiacol to give diol (S,R)-5. Differences in the reactivities of the primary and secondary alcohol functions of (S,R)-5 were exploited in the selective protection of the primary alcohol functionality as the TMS ether 14a. Subsequently, the secondary hydroxyl group was mesylated and the primary hydroxyl function exposed by treatment of the silyl ether with acid. The intermediate mesylate was converted to the epoxide (S,S)-6 under phase transfer catalysis conditions in basic medium. Mechanistically, this reaction proceeds via an SN2 displacement of the mesyloxy group thus providing a predictable stereochemical outcome. The smooth conversion of intermediates from (S,R)-5 to (S,S)-6 obviated the need for any purification and a 60% overall yield was realized for this transformation.
Scheme 2.

Reagents and conditions: (a) Ti(i-OPr)4, cumene hydroperoxide, D-DET, CH2Cl2, 4Å mol. sieves, 72%, 85% ee; (b) Guaiacol, NaOH, CH2Cl2, 99%; (c) TMSCl, Et3N, EtOAc; (d) MsCl, Et3N, EtOAc; (e) 2 N HCl; (f) NaOH, toluene, methyltributylammonium chloride, 60% from 5; (g) ethanolamine, i-PrOH, reflux, 70%; (h) Boc2O, CH2Cl2, 83%; (i) NaH, TsIm, THF, 61%; (j) TFA, CH2Cl2, 93%.
Compound (S,S)-7 was prepared by reaction of (S,S)-6 with ethanolamine at reflux in toluene. It is envisioned that treatment of (S,R)-5 with 1 equiv of p-toluenesulfonyl chloride followed by reaction with ethanolamine under basic conditions will afford (S,R)-7.32 Boc-protection of the secondary amine in (S,S)-6 gave (S,S)-10. As in the racemic synthesis, ring closure to (S,S)-11 was achieved in one pot by reaction of the N-Boc diol (S,S)-10 with NaH and tosylimidazole.25 Finally, NBoc deprotection of (S,S)-11 in trifluoroacetic acid furnished (S,S)-1. An X-ray diffraction analysis of (S,S)-1 (Fig. 2) confirmed the absolute configuration to be (2S,3S). In similar fashion, (R,R)-1 was also prepared from 3 utilizing an SAE reaction using L-DET.
Figure 2.
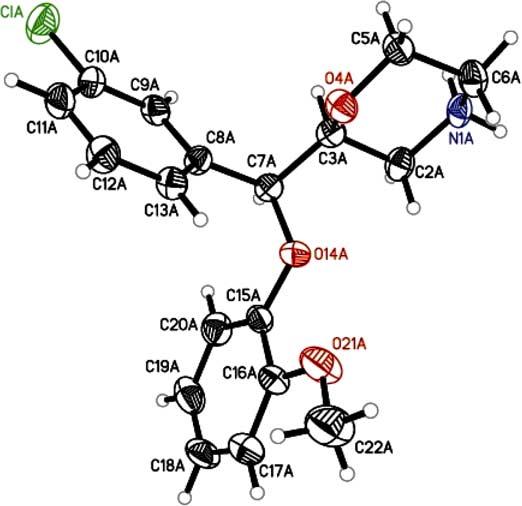
Displacement ellipsoid of (S,S)-1 drawn at 20% probability levels.
Enantiomeric purities of (R,R)- and (S,S)-1 were determined by a 1H NMR experiment in which the corresponding ureas were prepared from enantiomerically pure (R)-(+)-1-phenylethyl isocyanate and the spectra analyzed.33 In this experiment the urea prepared from racemic 1, exhibited a quartet of peaks at δ 5.20, 5.20, 5.19, and 5.18, which proved to be diagnostic in assessing purity. The 1H NMR spectrum of the individual isomeric ureas each showed a doublet in this region with the (S,S)-urea derivative having peaks at δ 5.20 and 5.18 and the (R,R)-derivative with peaks at δ 5.20 and 5.19. To determine the limits of detection, in a separate experiment, increasing amounts (2% increments by weight) of purified (R,R)-derivative were added to a known amount of purified (S,S)-urea derivative and the δ 5.2-5.1 region of the 1H NMR spectrum examined. Addition of up to 2% by weight of (R,R) derivative did not cause any detectable change in the appearance of the spectrum. However, upon addition of 4% by weight of the (R,R)-derivative a noticeable difference in the baseline appearance of the diagnostic region of the spectrum was observed (Fig. 3). Similarly, a 4% detection limit was observed for the (R,R)-derivative. Thus, we can conclude that the isomers prepared by this route are at least 96% enantiomerically pure (ee 92%).
Figure 3.
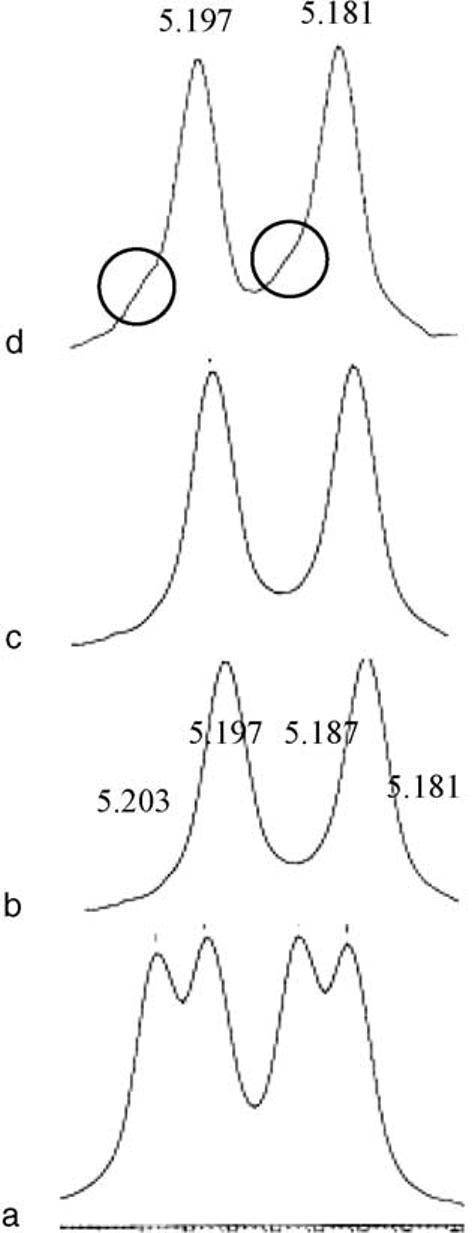
Diagnostic 1H NMR spectral region for ureas prepared from reaction of (R)-(+)-1-phenylethyl isocyanate and compound 1 for (a) racemic 1; (b) (S,S)-1; (c) (R,R)-1; and (d) (S,S)-1 +4% (R,R)-1.
3. Conclusions
We have demonstrated the utility of the Sharpless asymmetric epoxidation in the enantioselective synthesis of 1. Higher ees of the key intermediate epoxide 4 were obtained by using cumene hydroperoxide as the oxidant component than with tert-butylhydroperoxide. The enantiomeric excess of (S,S)-1 was determined by NMR analysis of the urea formed from (R)-(+)-1-phenylethyl isocyanate and the stereostructure confirmed by single crystal X-ray crystallographic analysis of the dibenzoyl-D-tartarate salt. The NMR experiment described here is a rapid method for determining ees of 1 and should also prove useful for other similar morpholino substrates.
4. Experimental
4.1. General
Unless otherwise indicated, all reagents were purchased from commercial suppliers and used without further purification. All melting points were determined on a Thomas-Hoover capillary melting apparatus and are uncorrected. NMR spectra were recorded at 300 MHz on a Bruker Avance-300 spectrometer using CDCl3 as solvent, δ values in parts per million (TMS as internal standard), and J (Hz) assignments of 1H resonance coupling unless otherwise stated. Thin-layer chromatography (TLC) was performed on 0.25 mm plates Analtech GHLF silica gel plates. Spots on TLC were visualized with vanillin/H2SO4 in ethanol. Column chromatography was performed with silica gel (32-63 l particle size) from Bodman Industries (Atlanta, GA). Optical rotations were performed on a Jasco P-1020 polarimeter at room temperature. Elemental analyses were performed by Atlantic Microlabs, Norcross, GA and were within ±0.4% of the theoretical values.
4.2. Preparation of 3-chlorocinnamyl alcohol, 3
A solution of ethyl 3-chlorocinnamate 2 (6.5 g, 30.9 mmol) in THF (30 mL) was cooled to 0 ° C and DIBAL-H (73 mL, 1 M in THF) added dropwise over 30 min. The mixture was then stirred at 0 ° C for 5 h. H2O (3.6 mL), 10% NaOH (3.6 mL) and Celite were added to the solution and the mixture filtered. The solid material was washed with THF and the filtrate was dried over Na2SO4 and concentrated in vacuo to a pale yellow syrup. The crude oil was purified by flash column chromatography (eluent: 30% ethyl acetate/hexanes) to give 5.1 g (98%) of 323,24 as a colorless oil: 1H NMR (CDCl3): δ 1.93 (br s, 1H), 4.33 (d, 2H, J = 5.1 Hz), 6.34 (dt, 1H, J = 5.1, 15.9 Hz), 6.53 (dt, 1H, J = 1.5, 15.9 Hz), 7.2 (m, 1H), 7.36 (s, 1H); 13C NMR (CDCl3): 63.3, 124.9, 126.6, 127.8, 129.6, 130.0, 130.3, 134.7, 138.8; HRMS calcd 168.0342, found 168.0333.
4.3. (2R,3R)-2,3-Epoxy-3-(3-chlorophenyl)-1-propanol, (R,R)-4
Diethyl-D-tartarate (856 μL, 4.99 mmol), titanium isopropoxide (1.22 mL, 4.17 mmol), and cumene hydroperoxide (12 mL) were added sequentially to a mixture of 4Å molecular sieves (4 g) and CH2Cl2 (180 mL) at 35 ° C under a nitrogen atmosphere. The mixture was then stirred at -35 ° C for 2 h. A solution of 3 (7.0 g, 41.7 mmol) in CH2Cl2 (20 mL) was added dropwise over 1 h and the resulting mixture stirred for an additional 8h at -35 ° C. The solution was filtered and washed with 10% NaOH (2 × 80 mL) and brine (100 mL). The organic layer was dried over Na2SO4 and the solvent removed under reduced pressure to afford a crude residue. The residue was purified by flash column chromatography (eluent: 2% ethyl acetate-CH2Cl2) to give 5.5 g (72%, 85% ee) of (R,R)-4 as a colorless oil. The ee was determined by the preparation of the mosher ester and examination of the terminal methylene proton signals at δ 4.00-4.35 of the 1H NMR spectrum in C6 D6.16,18 [α]D = +21.3 (c 1.93, CHCl3); 1H NMR (CDCl3): δ 2.30 (br s, 1H), 3.20 (dt, 1H, J = 2.4, 3.6 Hz), 3.81 (br d, 1H, J = 12.6 Hz), 3.93 (d, 1H, J = 2.4 Hz), 4.06 (br d, 1H, 12.6 Hz), 7.18 (m, 1H), 7.28 (m, 3H); 13C NMR (CDCl3): δ 55.0, 61.2, 62.8, 124.2, 125.9, 128.7, 130.0, 134.8, 139.1; HRMS calcd 184.0291, found 184.0292.
4.4. (2S,3S)-2,3-Epoxy-3-(3-chlorophenyl)-1-propanol, (S,S)-4
Diethyl-L-tartarate (658 μL, 3.83 mmol), titanium isopropoxide (0.94 mL, 3.20 mmol), and cumene hydroperoxide (12 mL) were added sequentially to a mixture of 4Å molecular sieves (3 g) and CH2Cl2 (150 mL) at 35 ° C under a nitrogen atmosphere. The mixture was then stirred at -35 ° C for 2 h. A solution of 3 (5.3 g, 32.0 mmol) in CH2Cl2 (20 mL) was added dropwise over 1 h and the resulting mixture stirred for an additional 8h at -35 ° C. The solution was filtered and washed with 10% NaOH (2 × 80 mL) and brine (100 mL). The organic layer was dried over Na2SO4 and the solvent removed under reduced pressure to afford a crude residue. The residue was purified by flash column chromatography (eluent: 2% ethyl acetate-CH2Cl2) to give 4.78 g (81%, 82% ee) of (S,S)-434 as a colorless oil. The ee was determined by the preparation of the mosher ester and examination of the terminal methylene proton signals at δ 4.00-4.35 of the 1H NMR spectrum in C6 D6.31,33 [α]D = -24.1 (c 1.66, CHCl3); H NMR (CDCl3): δ 2.30 (br s, 1H), 3.20 (dt, 1H, J = 2.4, 3.6 Hz), 3.81 (br d, 1H, J = 12.6 Hz), 3.93 (d, 1H, J = 2.4 Hz), 4.06 (br d, 1H, 12.6 Hz), 7.18 (m, 1H), 7.28 (m, 3H); 13C NMR (CDCl3): δ 55.0, 61.2, 62.8, 124.2, 125.9, 128.7, 130.0, 134.8, 139.1.
4.5. (2R,3S)-3-(3-Chlorophenyl)-3-(2-methoxyphenoxy)-propane-1,2-diol, (R,S)-5
A mixture of (R,R)-4 (4.7 g, 25.7 mmol), CH2Cl2 (150 mL), H2O (18 mL), 50% NaOH (1.9 mL), methyltributyl ammonium chloride (6.6 mL), and guaiacol (5.9 mL, 53.7 mmol) was stirred at room temperature for 2 h. CH2Cl2 was distilled over 1 h and the mixture heated to 65 ° C for 3 h. The mixture was cooled to room temperature, toluene (25 mL) added and the mixture stirred for 5 min. The layers were separated and the organic layer washed with 1 M NaOH (2 × 20 mL) and water (2 × 30 mL), dried over Na2SO4, and the solvent removed under reduced pressure to afford a viscous oil. The crude product was purified by flash column chromatography (eluent: 50% ethyl acetate/hexanes) to give 7.8 g (99%) of (R,S)-5 as a colorless oil: [α]D = +11.4 (c 5.45, CHCl3); 1H NMR (CDCl3): δ 3.29 (br s, 1H), 3.72 (dd, 1H, J = 4.5, 12.3 Hz), 3.92 (s, 3H), 3.96 (m obscured, 2H), 5.20 (d, 1H, J = 4.5 Hz), 6.65 (dd, 1H, J = 1.5, 8.1 Hz), 6.76 (ddd, 1H, J = 2.1, 6.9, 8.1 Hz), 6.94 (m, 2H), 7.31 (m, 3H), 7.44 (s, 1H); 13C NMR (CDCl3): δ 56.0, 62.5, 74.5, 85.0, 112.0, 116.6, 121.2, 123.0, 125.0, 127.0, 128.6, 130.2, 135.0, 140.4, 147.0, 150.0; HRMS calcd 308.015, found 308.0816.
4.6. (2S,3S)-1,2-Epoxy-3-[(3-chlorophenyl)-(2-methoxyphenoxy)]propane, (S,S)-6
Chlorotrimethylsilane (3.2 mL, 25.4 mmol) was added to a solution of (R,S)-5 (7.83 g, 25.4 mmol) and NEt3 (3.53 mL, 88.9 mmol) in ethyl acetate (85 mL) at 20 ° C. After stirring for 15 min, methanesulfonyl chloride (2.3 mL, 29.6 mmol) and triethylamine (5.69 mL, 40.8 mmol) were added and the mixture stirred for 15 min. HCl (2 M, 50 mL) was added and the mixture warmed to room temperature and stirred for 45 min. The phases were separated and the organic phase washed with NaHCO3 (2 × 70 mL) and brine (100 mL) and dried over Na2SO4. The solvent was removed under reduced pressure to give 7.5 g (76%) of a crude oil, which was used without further purification. The oil was dissolved in toluene (160 mL) and NaOH (50%, 20 mL) and a catalytic amount of methyltributylammonium chloride were added and the resulting mixture stirred at room temperature overnight. The layers were separated and the aqueous layer extracted with toluene (2 × 40 mL). The combined organic extract was washed with brine (100 mL), dried over Na2SO4, and the solvent removed under reduced pressure to afford a crude residue. The residue was purified by flash column chromatography (eluent: 40% ethyl acetate/hexanes) to give 4.4 g (60%) of (S,S)-6 as a colorless oil: [α]D = +14.5 (c 0.57, CHCl3); 1H NMR (CDCl3): δ 2.73 (dd, 1H, J = 2.6, 4.8 Hz), 2.85 (dd, 1H, J = 4.0, 4.8 Hz), 3.47 (ddd, 1H, 2.6, 4.0, 5.7 Hz), 4.92 (d, 1H, J = 5.7 Hz), 6.82 (m, 2H), 6.94 (m, 2H), 7.32 (m, 3H), 7.49 (d, 1H, J = 1.5 Hz); 13C NMR (CDCl3): δ 44.8, 54.9, 56.2, 82.5, 112.7, 117.6, 121.0, 123.0, 125.1, 127.1, 128.8, 130.1, 134.8, 139.8, 147.2, 150.6; HRMS calcd 290.0710, found 290.0715.
4.7. (2S,3S)-[3-(3-Chlorophenyl)-2-hydroxy-3-(2-methoxyphenoxy)]-(1-hydroxyethylamino)propane, (S,S)-7
Ethanolamine (3.7 mL, 6.1 mmol) was added to a solution of (S,S)-6 (4.4 g, 15.3 mmol) in isopropanol (175 mL) and the mixture heated at reflux overnight. The solvent was removed under reduced pressure and EtOAc (120 mL) added to the residue. The EtOAc solution was washed with saturated NaHCO3 (2 × 50 mL), brine (70 mL), dried over Na2SO4, and concentrated in vacuo. The crude product was purified by flash column chromatography (eluent: 10% MeOH/CH2Cl2) to give 3.78 g (70%) of (S,S)-7 as a colorless oil: 1H NMR (CDCl3): δ 2.57 (d, 2H, J = 5.7 Hz), 2.67 (dd, 2H, J = 5.4, 11.1 Hz), 3.60 (t, 2H, J = 5.4 Hz), 3.88 (s, 3H), 4.11 (dt, 1H, J = 5.7, 7.2 Hz), 4.93 (d, 1H, J = 7.2 Hz), 6.66 (dd, 1H, J = 1.7, 8.3 Hz), 6.74 (ddd, 1H, J = 1.7, 7.1, 7.1 Hz), 6.88 (dd, 1H, J = 1.8, 8.1 Hz), 6.95 (ddd, 1H, J = 1.3, 7.1, 7.1 Hz), 7.29 (m, 3H), 7.45 (s, 1H); 13C NMR (CDCl3): δ 50.5, 51.4, 55.8, 60.5, 73.8, 85.2, 112.1, 117.8, 121.0, 122.9, 125.5, 127.4, 128.5, 129.9, 134.6, 140.7, 147.4, 150.2; HRMS calcd 352.1316, found 352.1329.
4.8. (2S,3S)-[3-(3-Chlorophenyl)-2-hydroxy-3-(2-methoxyphenoxy)propyl]-(2-hydroxyethyl)carbamic acid tert-butyl ester, (S,S)-10
A solution of (S,S)-7 (5.4 g, 15.2 mmol) and di-tert-butyl dicarbonate (9.9 g, 45.7 mmol) in CH2Cl2 (170 mL) was stirred at rt for 1 h. The solvent was removed under reduced pressure and the crude product subjected to flash column chromatography (eluent: 50% EtOAc/hexanes) to afford 5.7 g (83%) of (S,S)-10 as a colorless oil: 1H NMR (CDCl3): δ 1.29 (s, 9H), 3.20 (m, 1H), 3.27- 3.42 (m, 3H), 3.60-3.85 (m, 2H), 3.92 (s, 3H), 4.20- 4.40 (m, 1H), 4.72 (d, 1H, J = 5.4 Hz), 6.63 (dd, 1H, J = 8.4, 8.4 Hz), 6.75 (ddd, 1H, J = 1.8, 7.2, 7.2 Hz), 6.85-7.00 (m, 2H), 7.28-7.31 (m, 3H), 7.42 (s, 1H). HRMS calcd 452.1840, found 452.1842.
4.9. (2S,3S)-2-[(3-Chlorophenyl)-(2-methoxyphenoxy)-methyl]morpholine-4-carboxylic acid tert-butyl ester, (S,S)-11
A solution of (S,S)-10 (2.3 g, 5.1 mmol) in THF (60 mL) was added in a dropwise manner to a suspension of NaH (60% dispersion in oil, 0.5 g, 21.3 mmol) in THF (250 mL) at 0 ° C. The resulting suspension was stirred for 5 min and then allowed to reach room temperature over 1 h. The mixture was cooled to 0 ° C and 1-(p-toluenesulfonyl)imidazole (1.1 g, 5.1 mmol) was added in one portion. The mixture was stirred for 15 min at 0 ° C and then was allowed to warm to room temperature and stirred for 19 h. The reaction mixture was then cooled to 0 ° C and then saturated NH4Cl solution (80 mL) was added cautiously. The mixture was warmed to room temperature and then extracted with EtOAc (2 × 200 mL). The combined EtOAc portion was washed with a 1:1 mixture of brine/saturated NaHCO3 (2 × 200 mL), dried over Na2SO4, and the solvent was removed under reduced pressure to afford a crude oil. The crude product was purified by flash column chromatography (eluent: 50% EtOAc/hexanes) to give 1.4 g (61%) of 11 as a colorless oil: 1H NMR (CDCl3): δ 1.45 (s, 9H), 2.86 (t, 1H, J = 11.7 Hz), 2.97 (t, 1H, J = 11.7 Hz), 3.55 (dt, 1H, 2.7, 11.7 Hz), 3.80-3.85 (m, 3H), 3.87 (s, 3H), 3.97 (dd, 1H, J = 2.1, 11.4 Hz), 5.14 (d, 1H, J = 5.1 Hz), 6.74-6.77 (m, 2H), 6.86-6.95 (m, 2H), 7.26-7.30 (m, 3H), 7.45 (d, 1H, J = 1.5 Hz); 13C NMR (CDCl3): δ 28.6, 56.1, 66.9, 76.7, 78.0, 78.0, 80.3, 112.6, 117.8, 120.9, 122.8, 125.7, 127.6, 128.6, 129.8, 134.5, 139.9, 147.2, 150.8, 155.0; HRMS calcd 433.1656, found 433.1656.
4.10. (2S,3S)-2-[(3-Chlorophenyl)-(2-methoxyphenoxy)-methyl]morpholine, (2S,3S)-1
4.11. (2S,3S)-2-[(3-Chlorophenyl)-(2-methoxyphenoxy)-methyl]morpholine dibenzoyl-D-tartarate, (S,S)-1 dibenzoyl-D-tartarate
(2S,3S)-1 (0.8 g, 1.9 mmol) was dissolved in ethanol (20 mL) and a solution of dibenzoyl-D-tartaric acid (753 mg, 2.10 mmol) in ethanol (10 mL) added. The salt precipitated on standing overnight and was collected, washed with ethanol (100 mL) and dried to afford 1.2 g (89%) of (S,S)-1 dibenzoyl-D-tartarate: mp 160- 162 ° C; [α]D = +30.0 (c 0.1, acetic acid); Anal. Calcd for (C54H54Cl2N2O14.C2H6O): C, 62.74; H, 5.64; N, 2.61. Found: C. 62.43; H, 5.68; N, 2.57.
4.12. (2R,3R)-2-[(3-Chlorophenyl)-(2-methoxyphenoxy)-methyl]morpholine, (2R,3R)-1
A solution of (R,R)-11 (830 mg, 1.91 mmol) in CH2Cl2 (60 mL) was treated with trifluoroacetic acid (3.2 mL, 41.7 mmol) and stirred for 2 h at room temperature. The mixture was washed with 2 M NaOH (2 × 50 mL) and H2O (2 × 50 mL), dried over Na2SO4, filtered, and the solvent removed under reduced pressure to afford a crude residue. The crude product was purified by flash column chromatography (eluent: 5% MeOH/CH2Cl2) to give 0.55 g (86%) of (R,R)-1 as a colorless oil: [α]D = -6.8 (c 1.56, CHCl3); 1H NMR (300 MHz, CDCl3): δ 1.83 (br s, 1H), 2.60-2.85 (m, 3H), 2.87 (dt, 1H, J = 3.3, 11.6 Hz), 3.65 (dt, 1H, J = 3.3, 10.8 Hz), 3.85 (s, 3H), 3.91 (ddd, 1H, J = 3.6, 5.7, 9.0 Hz), 3.97 (ddd, 1H, J = 1.5, 3.0, 11.4 Hz), 5.10 (d, 1H, J = 6.0 Hz), 6.70-6.80 (m, 2H), 6.83-6.92 (m, 2H), 7.23-7.30 (m, 2H), 7.44 (d, 1H, J = 1.2 Hz); 13C NMR (CDCl3): δ 44.9, 46.5, 56.1, 67.1, 77.4, 78.0, 82.0, 112.5, 117.8, 120.9, 122.9, 125.7, 127.7, 128.6, 129.8, 134.5, 139.8, 147.2, 150.7.
4.13. (2R,3R)-2-[(3-Chlorophenyl)-(2-methoxyphenoxy)-methyl]morpholine dibenzoyl-L-tartarate, (R,R)-1 dibenzoyl-L-tartarate
(2R,3R)-1 (0.55 g, 1.65 mmol) was dissolved in ethanol (20 mL) and a solution of dibenzoyl-L-tartaric acid (650 mg, 1.82 mmol) in ethanol (10 mL) added. The salt precipitated on standing for 2 h and was collected, washed with ethanol (100 mL) and dried to afford 1.1 g (94%) of (R,R)-1 dibenzoyl-L-tartarate: mp 160- 162 ° C; [α]D = -30.0 (c 0.1, acetic acid); Anal. Calcd for (C54H54Cl2N2O14.C2H6O): C, 62.74; H, 5.64; N, 2.61. Found: C. 62.41; H, 5.69; N, 2.64.
4.14. Single crystal X-ray diffraction analysis of (S,S)-1 dibenzoyl-D-tartarate
C54.50H56Cl2N2O14.50, FW = 1041.91, Tetragonal, P43, a = 8.70440(10) Å, b = 8.70440(10) Å, c = 73.005(2) Å, V = 5531.4(2) Å3, Z =4, ρcalcd= 1.251 mg/m3, μ = 1.603 mm-1, F(000) = 2188, R1 = 0.0972 for 6732 observed (I>2rI) reflections and 0.1148 for all 8700 reflections, Goodness-of-fit = 1.077, 651 parameters.
A thin, colorless needle of dimensions 0.01 × 0.04 × 0.28 mm2 was mounted on glass fiber using a small amount of Epoxy. Data were collected on a Bruker three-circle platform diffractometer equipped with a SMART 6000 CCD detector. The crystals were irradiated using a rotating anode Cu Ka source (λ = 1.54178) with incident beam Göbel mirrors.
Data collection was performed and the unit cell was initially refined using SMART [v5.625].35 Data Reduction was performed using SAINT [v6.36A]36 and XPREP [v6.12].37 Corrections were applied for Lorentz, polarization, and absorption effects using SADABS [v2.03].38 The structure was solved and refined with the aid of the programs in the SHELXTL-plus [v6.10] system of programs.39
The crystal was a merohedral twin, consisting of two components. It was described as such with the appropriate twin law and refined to a relative component ratio of approximately 64:36. A solvent methanol molecule was located in the asymmetric unit, however, it was disordered over two positions. The combined population of these positions equated to the methanol molecule being there half of the time. This is most likely a result of the lack of hydrogen bonding between the solvent and the other molecules. The methanol molecule was refined using a global thermal parameter. For additional stability, the C1S-O1S bond length was fixed at 1.43 Å. The remaining difference peaks were located in the area of the methanol molecule, indicating that there may be additional positions. The variables relating to the solvent molecule were fixed in the final least squares refinements.
For the rest of the model, the full-matrix least-squares refinement on F2 included atomic coordinates and anisotropic thermal parameters for all non-H atoms. The H atoms were included using a riding model for the entire system. The absolute configuration was established by the structure determination of a compound containing a chiral reference molecule of known absolute configuration and confirmed by anomalous dispersion effects in diffraction measurements on the crystal, with a resulting Flack parameter of 0.02(5).40 Coordinates have been deposited with the Cambridge Crystallographic Data Centre (Cambridge University Chemical Laboratory, Cambridge CB2 1EW, UK). CDCC number 268586.
Acknowledgements
The authors (W.W.H., T.E.P.) thank the Biological Sciences Funding Program of the University of Iowa and (W.L.W.) the National Institute on Drug Abuse for financial support. W.L.W. is a recipient of National Institute on Drug Abuse grant K05-DA15343. The X-ray crystallographic work was supported in part by the National Institute on Drug Abuse, NIH, DHHS, and the Office of Naval Research. The authors also thank Vic Parcell and Lynn Teesch of the High Resolution Mass Spectrometry Facility, University of Iowa for mass spectral analysis.
References
- 1.National, D. I. C. National Drug Threat Assessment US Department of Justice. 2004. Report 2004-Q0317-002, 2004.
- 2.Kuhar MJ, Ritz MC, Boja JW. Trends Neurosci. 1991;14:299–302. doi: 10.1016/0166-2236(91)90141-g. [DOI] [PubMed] [Google Scholar]
- 3.Rothman RB, Baumann MH. Eur. J. Pharmacol. 2003;479:23–40. doi: 10.1016/j.ejphar.2003.08.054. [DOI] [PubMed] [Google Scholar]
- 4.Johanson CE, Barrett JE. J. Pharmacol. Exp. Ther. 1993;267:1–8. [PubMed] [Google Scholar]
- 5.Rowlett JK, Platt DM, Spealman RD. J. Pharmacol. Exp. Ther. 2004;310:342–348. doi: 10.1124/jpet.104.065631. [DOI] [PubMed] [Google Scholar]
- 6.Gainetdinov RR, Caron MG. Annu. Rev. Pharmacol. Toxicol. 2003;43:261–284. doi: 10.1146/annurev.pharmtox.43.050802.112309. [DOI] [PubMed] [Google Scholar]
- 7.Uhl GR, Hall FS, Sora I. Mol. Psychiat. 2002;7:21–26. doi: 10.1038/sj.mp.4000964. [DOI] [PubMed] [Google Scholar]
- 8.Stephens DN, Mead AN, Ripley TL. Behav. Pharmacol. 2002;13:327–345. doi: 10.1097/00008877-200209000-00004. [DOI] [PubMed] [Google Scholar]
- 9.Rocha BA. Eur. J. Pharmacol. 2003;479:107–115. doi: 10.1016/j.ejphar.2003.08.061. [DOI] [PubMed] [Google Scholar]
- 10.Hall FS, Li XF, Sora I, Xu F, Caron M, Lesch KP, Murphy DL, Uhl GR. Neuroscience. 2002;115:153–161. doi: 10.1016/s0306-4522(02)00379-2. [DOI] [PubMed] [Google Scholar]
- 11.Ritz MC, Kuhar MJ. J. Pharmacol. Exp. Ther. 1989;248:1010–1017. [PubMed] [Google Scholar]
- 12.Roberts DC, Phelan R, Hodges LM, Hodges MM, Bennett B, Childers S, Davies H. Psychopharmacol. (Berl.) 1999;144:389–397. doi: 10.1007/s002130051022. [DOI] [PubMed] [Google Scholar]
- 13.Olivier B, Soudjin W, Wijngaarden I. Prog. Drug Res. 2000;54:60–119. doi: 10.1007/978-3-0348-8391-7_3. [DOI] [PubMed] [Google Scholar]
- 14.Tejani-Butt SM. J. Pharmacol. Exp. Ther. 1992;260:427–436. [PubMed] [Google Scholar]
- 15.Schatzberg AF. J. Clin. Psychiat. 2000;61(Suppl 10):31–38. [PubMed] [Google Scholar]
- 16.Szabadi E, Bradshaw CM, Boston PF, Langley RW. Hum. Psychopharmacol. 1998;13(Suppl 1):S3–S12. [Google Scholar]
- 17.Scates AC, Doraiswamy PM. Ann. Pharmacother. 2000;34:1302–1312. doi: 10.1345/aph.19335. [DOI] [PubMed] [Google Scholar]
- 18.Bymaster FP, Katner JS, Nelson DL, Hemrick-Luecke SK, Threlkeld PG, Heiligenstein JH, Morin SM, Gehlert DR, Perry KW. Neuropsychopharmacology. 2002;27:699–711. doi: 10.1016/S0893-133X(02)00346-9. [DOI] [PubMed] [Google Scholar]
- 19.Strolin Benedetti M, Frigerio E, Tocchetti P, Brianceschi G, Castelli MG, Pellizzoni C, Dostert P. Chirality. 1995;7:285–289. doi: 10.1002/chir.530070416. [DOI] [PubMed] [Google Scholar]
- 20.Melloni P, Carniel G, Torre AD, Bonignori A, Buonamici M, Pozzi V, Ricciardi S, Rossi AC. Eur. J. Med. Chem. 1984;19:235–242. [Google Scholar]
- 21.Huang ZZ, Tang Y. J. Org. Chem. 2002;67:5320–5326. doi: 10.1021/jo025693b. [DOI] [PubMed] [Google Scholar]
- 22.Hagiwara H, Shimizu Y, Hoshi T, Suzuki T, Ando M, Ohkubo K, Yokoyama C. Tetrahedron Lett. 2001;42:4349–4351. [Google Scholar]
- 23.Crilley MML, Golding BT, Pierpoint C. J. Chem. Soc., Perkin Trans. 1988;1:2061–2067. [Google Scholar]
- 24.Von Sprecher A, Beck A. Eur. Patent 89-105427 1989
- 25.Lanman BA, Myers AG. Org. Lett. 2004;6:1045–1047. doi: 10.1021/ol049861t. [DOI] [PubMed] [Google Scholar]
- 26.Orjales A, Mosquera R, Toledo A, Pumar MC, Garcia N, Cortizo L, Labeaga L, Innerarity A. J. Med. Chem. 2003;46:5512–5532. doi: 10.1021/jm0309349. [DOI] [PubMed] [Google Scholar]
- 27.Melloni P, Della Torre A, Lazzari E, Mazzini G, Meroni M. Tetrahedron. 1985;41:1393–1399. [Google Scholar]
- 28.Boot J, Cases M, Clark BP, Findlay J, Gallagher PT, Hayhurst L, Man T, Montalbetti C, Rathmell RE, Rudyk H, Walter MW, Whatton M, Wood V. Bioorg. Med. Chem. Lett. 2005;15:699–703. doi: 10.1016/j.bmcl.2004.11.025. [DOI] [PubMed] [Google Scholar]
- 29.Prabhakaran J, Majo VJ, Mann JJ, Kumar JS. Chirality. 2004;16:168–173. doi: 10.1002/chir.20004. [DOI] [PubMed] [Google Scholar]
- 30.Brenner E, Baldwin RM, Tamagnan G. Org. Lett. 2005;7:937–939. doi: 10.1021/ol050059g. [DOI] [PubMed] [Google Scholar]
- 31.Gao Y, Hanson RM, Klunder JM, Ko SY, Masamune H, Sharpless KB. J. Am. Chem. Soc. 1987;109:5765–5780. [Google Scholar]
- 32.Prisinzano T, Hsin LW, Folk JE, Flippen-Anderson JL, George C, Jacobson AE, Rice KC. Tetrahedron: Asymmetry. 2003;14:3285–3289. [Google Scholar]
- 33.Greiner E, Folk JE, Jacobson AE, Rice KC. Bioorg. Med. Chem. 2004;12:233–238. doi: 10.1016/j.bmc.2003.10.029. [DOI] [PubMed] [Google Scholar]
- 34.Reddy KR, Erion MD, Matelich MC, Kopcho JJ. US Patent 2003229225 A1 2003
- 35.Bruker . SMART v5.625. Bruker AXS Inc.; Madison, WI, USA: 2001. [Google Scholar]
- 36.Bruker . SAINT v6.36A. Bruker AXS Inc.; Madison, WI, USA: 2002. [Google Scholar]
- 37.Bruker . XPREP v6.12. Bruker AXS Inc.; Madison, WI, USA: 2001. [Google Scholar]
- 38.Bruker . SADABS v2.03. Bruker AXS Inc.; Madison, WI, USA: 2000. [Google Scholar]
- 39.Bruker . SHELXTL v6.10. Bruker AXS Inc.; Madison, WI, USA: 2000. [Google Scholar]
- 40.Flack HD. Acta Cryst. A. 1983;39:876–881. [Google Scholar]