

Abstract
We previously demonstrated that TIMP-2 increases the association of Crk with C3G and via subsequent activation of Rap1 enhances the expression of RECK, a membrane-anchored MMP inhibitor. In the present study, we investigate the mechanism of how the TIMP-2 signal is transduced from the α3β1 integrin receptor to the Crk-C3G-Rap1 molecular complex. TIMP-2 treatment of human microvascular endothelial cells (hMVECs) increased the phosphorylation levels of Src at Tyr-527, the negative regulatory site, through enhanced association of Src with Csk. This results in the reduction of Src kinase activity and dephosphorylation of paxillin at Tyr-31/118, the target sites for Src kinase phosphorylation and also the binding sites for the downstream effector Crk. Such TIMP-2 effects accompany the disassembly of paxillin-Crk-DOCK180 molecular complex and, in turn, Rac1 inactivation. On the contrary, levels of paxillin-Crk-C3G complex formation are not reduced, rather slightly increased, which is consistent with our previous finding. Therefore, TIMP-2 mediated inhibition of Src kinase activity leads to the signaling switch from Rac1 to Rap1, thereby leading to enhanced RECK expression.
Keywords: TIMP-2, RECK, Rac1, paxillin, Src
Tissue inhibitors of metalloproteinases (TIMPs) constitute a family of four structurally related proteins (TIMP-1–4), and in addition to their MMP-inhibitory activity, it is now widely appreciated that TIMPs have direct effects on cellular behaviors such as cell growth, apoptosis, migration, and differentiation (Baker et al., 2002; Jiang et al., 2002). It was recently demonstrated that TIMP-2 can inhibit the proliferation of endothelial cells in response to angiogenic stimuli, such as fibroblast growth factor 2 or VEGF-A (Seo et al., 2003). TIMP-2 also promotes the association of Crk with C3G, a guanine nucleotide exchange factor (GEF) of Rap1, and subsequently rap1 activation, which leads to the enhanced RECK gene expression and suppression of endothelial cell migration (Oh et al., 2004). RECK, initially identified with its activity inducing morphological reversion in a v-Ki-ras-transformed NIH3T3 cell line (Takahashi et al., 1998), is a crucial regulator of MMP activity, as demonstrated by examination of RECK homozygous null mouse embryos (Oh et al., 2001). In this study, we focused on the molecular mechanism of how TIMP-2 signal is transduced to the formation of Crk-C3G-Rap1 molecular complex, thereby leading to the increase in RECK expression.
Cell migration is primarily mediated by integrin binding to extracellular matrices, organization of the actin cytoskeleton, and regulated by the formation of focal adhesions (Hood and Cheresh, 2002). Several signaling molecules that localize at focal adhesions include focal adhesion kinase (FAK), Src tyrosine kinase, and paxillin. It has been demonstrated that upon integrin stimulation, tyrosine phosphorylation of FAK and Src kinase is enhanced (Parsons, 2003; Playford and Schaller, 2004). As TIMP-2 signaling requires its selective interaction with integrin α3β1 (Seo et al., 2003; Oh et al., 2004; Perez-Martinez and Jaworski, 2005), we sought to determine the effects of TIMP-2 on the phosphorylation status of FAK and Src. Human microvascular endothelial cells (hMVECs) were treated with 100nM recombinant human TIMP-2 (Wingfield et al., 1999) and cell lysates analyzed by immunoblotting using phosphospecific antibodies. It was found that TIMP-2 increases the phosphorylation levels of FAK at both Tyr-397, the autophosphorylation site, and Tyr-925, known as a docking site for the adaptor Grb2 (Parsons, 2003) (Figure 1a). Src kinase activity is reportedly regulated by phosphorylation of the autophosphorylation site, Tyr416, and dephosphorylation of the negative regulatory site, Tyr527 (Harrison et al., 2003). Studies showed that a phosphorylated, C-terminal Tyr-527 residue of Src leads to an intramolecular interaction with its SH2 domain, promotes an intramolecular SH3 domain-mediated interaction, which inhibits catalytic activity, and loss of Tyr527 phosphorylation is a common mechanism that leads to oncogenic activation of c-Src (Playford and Schaller, 2004). Interestingly, TIMP-2 treatment did not significantly affect phosphorylation levels at Tyr416 but increased those at Tyr527 reproducibly (Figure 1a). As TIMP-2 signaling involves the activity of protein tyrosine phosphatase (PTP) (Seo et al., 2003; Oh et al., 2004), we pretreated hMVECs with orthovanadate, a general inhibitor of PTPs, prior to TIMP-2 addition and found that the pretreatment notably ablates TIMP-2 mediated phosphorylation of FAK Tyr-925 and, to a lesser extent but reproducibly, of Src Tyr-527 (Figure 1a). However, orthovanadate pretreatment did not reverse the TIMP-2 mediated increase in FAK phosphorylation at Tyr397, suggesting that phosphorylation at this site is independent of protein tyrosine phosphatase activity. Studies showed that phosphorylated Tyr-397 residue of FAK serves as a binding site for Src, which in turn promotes Src kinase activation (Playford and Schaller, 2004). Our immunoprecipitation using anti-Src antibody, however, showed no significant changes in the association of FAK-Src upon TIMP-2 treatment (data not shown), suggesting that TIMP-2 may affect Src kinase activity independently of FAK. The implication of TIMP-2 induced phosphorylation of FAK is currently unclear.
Figure 1.

TIMP-2 inactivates Src kinase. Human microvascular endothelial cells (hMVECs) (Clonetics, San Diego, CA) were grown in EGM-2 MV BulletKit medium containing 5% fetal bovine serum and growth factors (Clonetics). (a) After serum starvation for 24 h, cells were treated with 100 nM TIMP-2 for 15 min with or without the pretreatment of 1 μM orthovanadate (V) for 5 min. After the lysis, cell lysates were analyzed by western blot analysis using antibodies indicated. (b) The same cell lysates obtained in (a) were immunoprecipitated with anti-Src antibody, assayed for the kinase activity using a kit as per manufacturer’s instruction (Chemicon International). Relative kinase activity was determined using untreated cells as control. The results are representative of three independent experiments; bars, ±SD. ** P < 0.01 compared with untreated controls. (c-d) Cell lysates were immunoprecipitated with anti-Src (c) or anti-paxillin antibody (d) and analyzed by immunoblotting with antibodies indicated. Immunoblot figures are representatives of at least two independent experiments. Relative band intensities were measured by NIH image software and are noted at the bottom of the blot. Antibodies against FAK, phosphospecific Tyr-397-FAK, Csk, and paxillin were purchased from BD Transduction Laboratories ; antibodies against phosphospecific Tyr-925-FAK, Src, SHP-1, and SHP-2 from Santa Cruz Biotechnology; antibodies against phosphospecific Tyr-416-Src, Tyr-527-Src, and PTP-PEST from Cell Signaling Technology.
To examine whether TIMP-2 signaling indeed downregulates Src kinase activity as suggested by immunoblot assay (Figure 1a), TIMP-2 treated cell lysates were immunoprecipitated with anti-Src antibody and assayed for the kinase activity. Src kinase activity was ~35% decreased by TIMP-2, but restored to the control level when pretreated with orthovanadate (Figure 1b). It has been demonstrated that phosphorylation level of Src Tyr527 is dictated by the activities of C-terminal Src kinase (Csk) and protein tyrosine phosphatases, including SHP-1, SHP-2, PTP1B, and PTPα(Roskoski, 2005). To investigate the molecular mechanism underlying the TIMP-2-mediated phosphorylation of Src Tyr-527, TIMP-2 treated cell lysates were immunoprecipitated with anti-Src antibody and analyzed. Immunoblotting shows that Src associates with SHP-1, SHP-2, and Csk, among which TIMP-2 affects only Src-Csk interaction in an orthovanadate-sensitive manner (Figure 1c). This enhanced association between Src and Csk would account for the increased phosphorylation of Src Tyr-527 and subsequent reduction of Src kinase activity. We could not, however, detect the association of Src with PTP1B and PTPα (data not shown). Though the dependency of PTP activity in TIMP-2 signaling remains to be further characterized, it is interesting to note that TIMP-2 signaling suppresses the tyrosine kinase activity of Src, whose oncogene expression was reported to downregulate the RECK gene expression (Takahashi et al., 1998).
Src and its negative regulator Csk bind to paxillin via SH3 and SH2 domain, respectively (Schaller, 2001). Paxillin also interacts with protein tyrosine phosphatases (PTPs), such as SHP-2 and PTP-PEST, which have been reported to affect the phosphorylation levels of paxillin and thereby the accessibility of Csk to Src (Turner, 2000; Ren et al., 2004). To test the possibility that these PTPs are involved in the TIMP-2 mediated inhibition of Src kinase activity, we examined their molecular interaction with paxillin by immunoprecipitation, but found no significant changes (Figure 1d). Recent studies also showed that SHP-2 can control Csk access to Src by dephosphorylating Csk-associated protein, PAG/Cbl (Zhang et al., 2004), but TIMP-2 did not affect the association of Csk with PAG/Cbl (data not shown). We also examined the possibility that TIMP-2 might upregulate Csk activity, by checking the phosphorylation levels of VE-cadherin, as VE-cadherin was recently found to be a downstream target of Csk (Baumeister et al., 2005). Western blot analysis shows no significant change (data not shown), indicating that TIMP-2 enhances the association of Csk with Src by a yet-to-be identified mechanism. Further study is needed to characterize the protein tyrosine phosphatase(s) involved in this TIMP-2 enhanced association of Src-Csk.
In response to motility signals, paxillin undergoes tyrosine phosphorylation primarily at two sites, Tyr-31/118 (Nakamura et al., 2000; Petit et al., 2000), and Src kinase has been implicated in regulating tyrosine phosphorylation of paxillin (Thomas et al., 1995; Richardson et al., 1997). As TIMP-2 suppresses cell migration (Oh et al., 2004; Ahn et al., 2004) and we showed in this study that TIMP-2 signaling downregulates Src kinase activity (Figure 1b), the effects of TIMP-2 on paxillin phosphorylation was examined. As expected, TIMP-2 considerably attenuates paxillin phosphorylation at both tyrosine residues, and orthovanadate pretreatment reverses TIMP-2 effects, albeit not completely (Figure 2a). It was reported that these two phosphorylation sites of paxillin serve as docking sites for the downstream effector Crk (Turner, 2000). Crk, known as an adaptor protein containing both SH2 and SH3 domains, mediates various signaling pathways through its SH3 binding partners, such as C3G and DOCK180 (Feller, 2001). To determine how TIMP-2 signaling affects the molecular interaction of paxillin, we immunoprecipitated TIMP-2 treated cell lysates with anti-paxillin antibody and analyzed by immunoblotting. It appears that the total amount of Crk associated with paxillin is not significantly changed by TIMP-2, but the complex formation with DOCK180, a GEF of Rac1 (a member of Rho family GTPase), greatly diminishes in an orthovanadate-sensitive manner (Figure 2b). On the contrary, the complex formation of paxillin with Crk and C3G, an activator of Rap1, is not reduced, rather slightly but reproducibly increased by TIMP-2. This result is consistent with our previous finding (Oh et al., 2004) and also with the recent reports that phosphorylation of paxillin at Tyr-31/118 promotes its association with Crk and DOCK180 and the activation of Rac1 (Valles et al., 2004; Chen et al., 2004). The potential involvement of p130CAS, another major target protein of FAK/Src kinase, in TIMP-2 signaling was examined by immunoprecipitation followed by immunoblotting. We could, however, detect only modest interaction of p130CASwith FAK and Crk, which does not significantly change upon TIMP-2 treatment (Figure 2c), suggesting that p130CASmay not be involved in TIMP-2 signaling.
Figure 2.
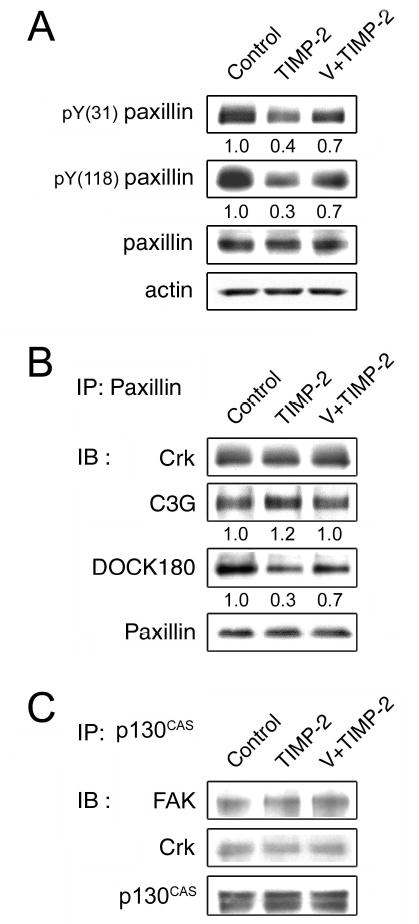
TIMP-2 dephosphorylates paxillin. (a) hMVECs were treated with 100 nM TIMP-2 for 15 min with or without the pretreatment of 1 μM orthovanadate (V) for 5 min. Cells were lysed and analyzed by western blot analysis using antibodies indicated. (b-c) The same cell lysates obtained in (a) were immunoprecipitated with anti-paxillin (b) or anti-p130CAS (c) antibody and analyzed by immunoblotting using antibodies indicated. Antibodies against Crk and p130CAS were purchased from BD Transduction Laboratories; antibodies against actin, C3G, DOCK180 from Santa Cruz Biotechnology; antibodies against phosphospecific Tyr-31-paxillin and Tyr-118-paxillin from BioSource International.
To confirm that TIMP-2 mediated disassembly of paxillin-Crk-DOCK180 complex leads to the inactivation of Rac1, levels of GTP-bound Rac1 (activated Rac1) was measured by pull-down assay using a GST fusion of the p21-binding domain of PAK. It was found that TIMP-2 indeed reduces Rac1 GTP-loading in an orthovanadate-sensitive manner (Figure 3a). To test the possibility that TIMP-2 may affect other GEFs of Rac1, such as Tiam1 and Vav2, contributing to inactivation of Rac1, we examined their phosphorylation levels, but no significant change was found upon TIMP-2 treatment (Figure 3b). Also, the gene expression profile in hMVECs treated with TIMP-2 for 6h was analyzed by cDNA microarray. It revealed that TIMP-2 does not affect the expression levels of Tiam1 and Vav2 (data not shown). Thus, in addition to our previous finding that TIMP-2 promotes the association of Crk-C3G and subsequent Rap1 activation (Oh et al., 2004), these results further support the findings of Valles et al. which show that inhibition of cell migration through C3G/Rap1 activation is concomitant with the disassembly of the paxillin-Crk-DOCK180 molecular complex and subsequent Rac1 inactivation.
Figure 3.
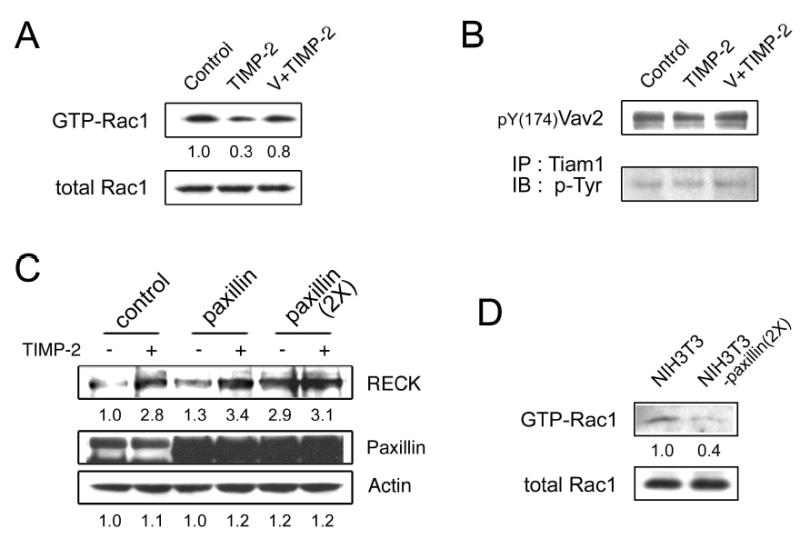
TIMP-2 downregulates Rac1 signaling. (a) hMVECs were treated with 100 nM TIMP-2 for 15 min with or without the pretreatment of 1 μM orthovanadate (V) for 5 min. Cells were lysed and analyzed by immunoblotting using anti-Rac1 antibody to detect total Rac1. To detect GTP-bound Rac1, cell lysates were precipitated with GST-PAK-1-PBD, separated by SDS-PAGE, and immunoblotted with anti-Rac1 antibody, as described in the manufacturer’s guide (Upstate). (b) The same cell lysates obtained in (a) were analyzed to detect the phosphorylation levels of Vav2, by immunoblotting, and Tiam1, by immunoprecipitation followed by immunoblotting. (c) NIH3T3 cells were transfected with the control vector (pBabe) or expression vectors containing cDNA for paxillin or its mutant (2X). The control and transfectant cells were treated with 100 nM TIMP-2 for 24 h and cell lysates analyzed by western blot analysis. (d) Cell lysates from NIH3T3 cells and its paxillin (2X) transfectants were analyzed to detect levels of total Rac1 and GTP-bound Rac1.
We have shown that TIMP-2 signal propagates through dephosphorylation of paxillin tyrosine residues 31 and 118. To confirm this in regard to RECK expression, we established NIH3T3 cell lines stably expressing paxillin or its mutant (2X) in which Tyr-31/118 residues were both replaced by phenylalanine, and measured the RECK protein levels with or without TIMP-2 treatment. The NIH3T3 cell line was chosen for these studies as it was used in the screening strategy used to isolate the RECK gene (Takahashi et al., 1998) and is more readily amenable to these transfection studies than endothelial cells. Western blot analysis shows that RECK levels are increased ~3-fold in the TIMP-2-treated control cells (Figure 3c). Forced expression of paxillin produced slight increase in RECK levels of both the untreated and TIMP-2 treated cells, as compared with the control (mock-transfected) cells. Overexpression of paxillin mutant (2X) lacking Crk binding sites enhanced RECK expression, which is unaffected by TIMP-2 treatment (Figure 3c). Pull-down assay revealed that levels of GTP-bound Rac1 is ~60% decreased in the paxillin (2X) expressing cells (Figure 3d). These results suggest that regulation of paxillin phosphorylation at residues 31/118 is critical determinants that can switch cell between a migratory phenotype in which Rac1 activation dominates and an adhesive phenotype in which case Rap1 signaling (Bos, 2005) dominates with increased RECK gene expression.
In this study, we have demonstrated that TIMP-2 propagates its signal through inhibition of Src kinase activity, dephosphorylation of paxillin Tyr-31/118, leading to the switch from Rac1 to Rap1 signaling (Figure 4). An intriguing question to be addressed is why the reduced phosphorylation of paxillin at Tyr-31/118 ablates its complex formation with DOCK180 but not with C3G. Considering the recent report that in NBT-II cells, both Crk-C3G and Crk-DOCK180 complexes are present constitutively (Valles et al., 2004), it can be postulated that the complex formation of paxillin with Crk-DOCK180 and its disassembly are tightly controlled by the paxillin tyrosine phosphorylation. Constitutive phosphorylation of paxillin on residues 31/118, as is observed in untreated cells (Figure 2a), results in paxillin-Crk-DOCK180 complex formation and subsequent Rac1 activation that prevails over Rap1 signaling. Rac1 controls actin cytoskeletal dynamics and integrin adhesions that are essential for lamellipodium extension during cell migration, whereas Rap1 acts as a negative regulator of cell migration (Bos, 2005; Ohba et al., 2001). We showed that TIMP-2 signaling downregulates Rac1 signaling but upregulates Rap1 signaling, leading to the enhanced RECK expression and reduced cell motility. But the questions of how Rac1 and Rap1, members of the Ras and Rho family GTPases, respectively, affect each other and what the downstream effector of Rap1 is for TIMP-2 signaling remain to be addressed.
Figure 4.
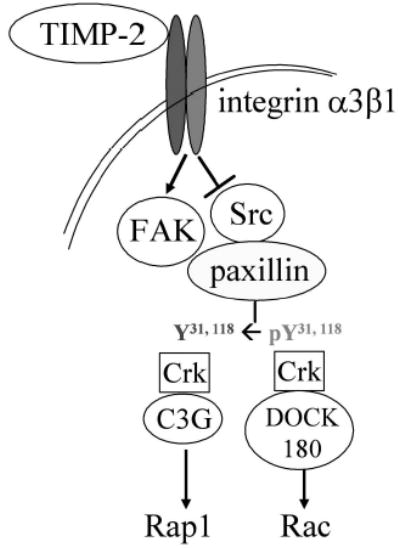
A schematic diagram for the TIMP-2 signaling.
Clinicopathological studies with cancer patients have demonstrated that high RECK expression is significantly correlated with good prognosis (Takenaka et al., 2005; van der Jagt et al., 2005; Takenaka et al., 2004; Span et al., 2003; Masui et al., 2003; Furumoto et al., 2001). However, the positive control mechanism of the RECK gene expression has not been studied yet, though the ras-mediated downregulation was studied (Sasahara et al., 2002). Thus, delineation of TIMP-2 signaling pathway provides important insights into the mechanism for inducing RECK expression.
Acknowledgments
We thank Hisataka Sabe (Osaka Bioscience Institute, Japan) for pBabe paxillin and pBabe paxillin (2X). This work was supported by intramural research funds from the National Cancer Institute, Center for Cancer Research (Project # Z01SC 009179).
References
- Ahn SM, Jeong SJ, Kim YS, Sohn Y, Moon A. Cancer Lett. 2004;207:49–57. doi: 10.1016/j.canlet.2003.11.025. [DOI] [PubMed] [Google Scholar]
- Baker AH, Edwards DR, Murphy G. J Cell Sci. 2002;115:3719–3727. doi: 10.1242/jcs.00063. [DOI] [PubMed] [Google Scholar]
- Baumeister U, Funke R, Ebnet K, Vorschmitt H, Koch S, Vestweber D. EMBO J. 2005;24:1686–1695. doi: 10.1038/sj.emboj.7600647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bos JL. Curr Opin Cell Biol. 2005;17:123–128. doi: 10.1016/j.ceb.2005.02.009. [DOI] [PubMed] [Google Scholar]
- Chen HY, Shen CH, Tsai YT, Lin FC, Huang YP, Chen RH. Mol Cell Biol. 2004;24:10558–10572. doi: 10.1128/MCB.24.24.10558-10572.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feller SM. Oncogene. 2001;20:6348–6371. doi: 10.1038/sj.onc.1204779. [DOI] [PubMed] [Google Scholar]
- Furumoto K, Arii S, Mori A, Furuyama H, Gorrin Rivas MJ, Nakao T, et al. Hepatology. 2001;33:189–195. doi: 10.1053/jhep.2001.21048. [DOI] [PubMed] [Google Scholar]
- Harrison SC. Cell. 2003;112:737–740. doi: 10.1016/s0092-8674(03)00196-x. [DOI] [PubMed] [Google Scholar]
- Hood JD, Cheresh DA. Nature Rev Cancer. 2002;2:91–100. doi: 10.1038/nrc727. [DOI] [PubMed] [Google Scholar]
- Jiang Y, Goldberg ID, Shi YE. Oncogene. 2002;21:2245–52. doi: 10.1038/sj.onc.1205291. [DOI] [PubMed] [Google Scholar]
- Masui T, Doi R, Koshiba T, Fujimoto K, Tsuji S, Nakajima S, et al. Clin Cancer Res. 2003;9:1779–1784. [PubMed] [Google Scholar]
- Nakamura K, Yano H, Uchida H, Hashimoto S, Schaefer E, Sabe H. J Biol Chem. 2000;275:27155–27164. doi: 10.1074/jbc.M000679200. [DOI] [PubMed] [Google Scholar]
- Oh J, Takahashi R, Kondo S, Mizoguchi A, Adachi E, Sasahara RM, et al. Cell. 2001;107:789–800. doi: 10.1016/s0092-8674(01)00597-9. [DOI] [PubMed] [Google Scholar]
- Oh J, Seo DW, Diaz T, Wei B, Ward Y, Ray JM, et al. Cancer Res. 2004;64:9062–9069. doi: 10.1158/0008-5472.CAN-04-1981. [DOI] [PubMed] [Google Scholar]
- Ohba Y, Ikuta K, Ogura A, Matsuda J, Mochizuki N, Nagashima K, et al. EMBO J. 2001;20:3333–3341. doi: 10.1093/emboj/20.13.3333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsons JT. J Cell Sci. 2003;116:1409–1416. doi: 10.1242/jcs.00373. [DOI] [PubMed] [Google Scholar]
- Perez-Martinez L, Jaworski DM. J Neurosci. 2005;25:4917–4929. doi: 10.1523/JNEUROSCI.5066-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petit V, Boyer B, Lentz D, Turner CE, Thiery JP, Valles AM. J Cell Biol. 2000;148:957–970. doi: 10.1083/jcb.148.5.957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Playford MP, Schaller MD. Oncogene. 2004;23:7928–7946. doi: 10.1038/sj.onc.1208080. [DOI] [PubMed] [Google Scholar]
- Ren Y, Meng S, Mei L, Zhao ZJ, Jove R, Wu J. J Biol Chem. 2004;279:8497–8505. doi: 10.1074/jbc.M312575200. [DOI] [PubMed] [Google Scholar]
- Richardson A, Malik RK, Hildebrand JD, Parsons JT. Mol Cell Biol. 1997;17:6906–6914. doi: 10.1128/mcb.17.12.6906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roskoski R. Biochem Biophys Res Commun. 2005;331:1–14. doi: 10.1016/j.bbrc.2005.03.012. [DOI] [PubMed] [Google Scholar]
- Sasahara RM, Brochado SM, Takahashi C, Oh J, Maria-Engler SS, Granjeiro JM, et al. Cancer Detect Prev. 2002;26:435–443. doi: 10.1016/s0361-090x(02)00123-x. [DOI] [PubMed] [Google Scholar]
- Schaller MD. Oncogene. 2001;20:6459–6472. doi: 10.1038/sj.onc.1204786. [DOI] [PubMed] [Google Scholar]
- Seo DW, Li H, Guedez L, Wingfield PT, Diaz T, Salloum R, et al. Cell. 2003;114:171–180. doi: 10.1016/s0092-8674(03)00551-8. [DOI] [PubMed] [Google Scholar]
- Span PN, Sweep CG, Manders P, Beex LV, Leppert D, Linderg RL. Cancer. 2003;97:2710–2715. doi: 10.1002/cncr.11395. [DOI] [PubMed] [Google Scholar]
- Takahashi C, Sheng Z, Horan TP, Kitayama H, Maki M, Hitomi K, et al. Proc Natl Acad Sci USA. 1998;95:13221–13226. doi: 10.1073/pnas.95.22.13221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takenaka K, Ishikawa S, Kawano Y, Yanagihara K, Miyahara R, Otake Y, et al. Eur J Cancer. 2004;40:1617–1623. doi: 10.1016/j.ejca.2004.02.028. [DOI] [PubMed] [Google Scholar]
- Takenaka K, Ishikawa S, Yanagihara K, Miyahara R, Hasegawa S, Otake Y, et al. Ann Surg Oncol. 2005;12:817–824. doi: 10.1245/ASO.2005.09.018. [DOI] [PubMed] [Google Scholar]
- Thomas SM, Soriano P, Imamoto A. Nature. 1995;376:267–271. doi: 10.1038/376267a0. [DOI] [PubMed] [Google Scholar]
- Turner CE. Nature Cell Biol. 2000;2:E231–236. doi: 10.1038/35046659. [DOI] [PubMed] [Google Scholar]
- Valles AM, Beuvin M, Boyer B. J Biol Chem. 2004;279:44490–44496. doi: 10.1074/jbc.M405144200. [DOI] [PubMed] [Google Scholar]
- van der Jagt MF, Sweep FC, Waas ET, Hendriks T, Ruers TJ, Merry AH, et al. (2005). Cancer Lett., on line publication [DOI] [PubMed]
- Wingfield PT, Sax JK, Stahl SJ, Kaufman J, Palmer I, Chung V, et al. J Biol Chem. 1999;274:21362–21368. doi: 10.1074/jbc.274.30.21362. [DOI] [PubMed] [Google Scholar]
- Zhang SQ, Yang W, Kontaridis MI, Bivona TG, Wen G, Araki T, et al. Mol Cell. 2004;13:341–355. doi: 10.1016/s1097-2765(04)00050-4. [DOI] [PubMed] [Google Scholar]