

Abstract
The hepatitis C virus (HCV) 5′ untranslated region (UTR) has been extensively studied with regard to its internal ribosomal entry site (IRES) activity. In this work we present results suggesting the existence of a strong promoter activity carried by the DNA sequence corresponding to the HCV 5′ UTR. This activity was not detected when the HCV 5′ UTR sequence was replaced by HCV 3′ UTR or poliovirus 5′ UTR sequences. These results were further confirmed by using bicistronic constructions. We demonstrated the presence of an mRNA initiated in this 5′ UTR sequence and located the initiation site by the 5′ RACE method at nucleotide 67. Furthermore, northern experiments and flow cytometry analysis showed the unambiguous activity of such a promoter sequence in stably transfected cells. Our results strongly suggest that the data obtained using bicistronic DNA constructs carrying the HCV 5′ UTR should be analyzed not only at the translational but also at the transcriptional level.
INTRODUCTION
With an estimated 170 million infected individuals worldwide (1), hepatitis C virus (HCV) infection is considered a serious public health problem. HCV is an enveloped positive single-strand RNA virus belonging to the Flaviviridae family. Its RNA genome of approximately 9600 nucleotides codes for a single polyprotein. Functionally important untranslated regions (UTR) are present at each end of the genome. They are both involved, either individually or in concert, in viral translation and replication of the viral genome. The highly-conserved 5′ UTR is directly related to the translation of the viral polyprotein, since it contains an internal ribosomal entry site (IRES) (2–4). This RNA element, comprising most of the 5′ UTR, forms four highly structured domains, each of them being mandatory for translation activity, except domain I located in the first 45 nt of the RNA genome. Deletion experiments suggest a regulatory role of domains II and III in polyprotein translation (3). The activity of the HCV IRES also appears to be dependent on sequences downstream of the initiation codon, extending at least to the first 12 nt of the open-reading frame (ORF) (1). The 3′ UTR is composed of three domains: a variable region following the stop codon of the ORF, a polypyrimidine tract of variable length and a highly conserved 98 nt sequence known as the X region (5). The latter region has been shown to be essential for HCV replication in chimpanzees (6,7). The X region in HCV 3′ UTR enhances IRES-dependent RNA translation in vitro and in vivo. It has been suggested that this effect involves the polypyrimidine tract binding protein, which is found associated with both the 5′ and the 3′ ends of the viral mRNA (8). Similarly, the first 125 nt of the 5′ UTR, have been shown to be essential for the replication of the HCV genome (9).
Although HCV was identified more than 10 years ago (10), a cellular model system allowing the efficient replication of the entire viral genome is not available. The only known animal model supporting replication of most HCV genotypes is the chimpanzee, whose experimental use is rather difficult and controlled. Several cell culture systems have been described based either on infection of primary cell culture or cell lines, or cultivation of primary cells of infected patients (11). Unfortunately, all of them show poor reproducibility and low levels of HCV replication. Alternatively, the recent development of a cellular system able to replicate HCV subgenomic RNAs (12) has allowed the study of the conditions of RNA replication in vivo. These replicons have been used more recently to study the replication of the full-length HCV genome (13) that could be stably propagated in the human hepatoma cells HuH7. However, no evidence for the virus particle production was found. For these reasons, the detailed mechanisms of HCV replication and translation remain unknown.
We describe here results showing that the HCV 5′ UTR, in its DNA form, was able to drive the expression of downstream reporter genes. To assess this property further, we used monocistronic and bicistronic constructions. In this study, we show that the DNA sequence corresponding to the HCV 5′ UTR displays a promoter activity able to generate an RNA transcript specifically initiated inside the 5′ UTR sequence.
Besides the possible implications of this observation in the viral life cycle as well as in the pathologies associated with HCV, it is important to bear in mind that results obtained when using cells transfected with bicistronic DNA constructs carrying the HCV 5′ UTR should be analyzed not only at the translational, but also at the transcriptional, level.
MATERIALS AND METHODS
Plasmid constructs
The Escherichia coli DH5α strain was used for plasmid transformation and propagation. The following plasmids, described schematically in Figure 1, were used. (i) The monocistronic pEGFP vectors (Fig. 1A) were based on the pEGFP backbone (Clontech), in which the SV40 polyadenylation signal (SV40 polyA) was inserted in the 3′ multiple cloning site (MCS) using NotI and SpeI sites, the SpeI site in SV40 polyA having been previously obtained by mutagenesis. Sequences corresponding to HCV 5′ UTR (nucleotides 1–341) or 3′ UTR (nucleotides 9377–9599) were obtained by PCR from the H77 strain (14; EMBL AF011753). The sequence corresponding to the 5′ UTR of the poliovirus (nucleotides 1–753) was obtained by PCR from the type 3 Sabin strain and that of the cytomegalovirus (CMV) early promoter was obtained by PCR from the pcDNA3.1 vector (Clontech). The fragments HCV-5′ UTR, HCV-3′ UTR, Polio-5′ UTR and CMV-p were ligated between the HindIII and SalI sites in the 5′ MCS of pEGFP to generate pEGFP-HCV-5′ UTR, pEGFP-HCV-3′ UTR, pEGFP-Polio-5′ UTR and pEGFP-CMVp vectors, respectively. (ii) The pEGFP-C3 vector (Clontech) contains the CMV promoter upstream from the EGFP gene and the SV40 polyA signal downstream (Fig. 1B), as well as the selectable marker neomycin. (iii) The pIRF bicistronic reporter vector (Fig. 1C) contains the cDNAs of firefly luciferase (FLuc), HCV 5′ UTR (nucleotides 1–371 of the HCV strain 1a) and Renilla luciferase (RLuc) inserted under the control of the CMV promoter in the pcDNA3.1/Zeo vector (Invitrogen) (15), which carries the selectable marker zeocin. (iv) The pIRF-delCMV vector was obtained from pIRF by excision of the CMV promoter sequence between the sites MluI and HindIII (Fig. 1D). (v) The bicistronic vector pLuc-IEX (Fig. 1E) was constructed from the pIRF as follows: the RLuc sequence was excised from pIRF after digestion by PstI and NotI and replaced by a 36 bp PstI–NotI fragment of the d2EGFP sequence obtained by digestion of the pd2EGFP-N1 vector (Clontech) by PstI and by NotI. The 861 bp PstI–PstI EGFP fragment resulting from that digestion was then inserted in the PstI site of pIRF, so that EGFP was in the ORF of the AUG of the HCV IRES, 75 nt downstream. Finally, the HCV 3′ UTR region (nucleotides 9377–9599) obtained by PCR was inserted in the modified pIRF vector between NotI and XhoI.
Figure 1.

Schematic representation of plasmid constructions. The various vectors were constructed as described in Materials and Methods. (A and B) monocistronic vectors; (C–E) bicistronic vectors.
Cell culture and transient transfection
The human hepatoma cell lines HepG2 and HuH7 were cultured in Dulbecco’s modified Eagle medium supplemented with 10% heat-inactivated fetal calf serum, 2 mM l-glutamine and 1% non-essential amino acids, at 37°C in a 5% CO2 atmosphere. HeLa cells (human cervical carcinoma cells) were cultured in the same medium without non-essential amino acids. All culture reagents were purchased from Gibco-BRL (Invitrogen). For transient transfection, 105 cells were seeded into 24-well plates. Transfection of HeLa cells was performed the next day by addition of a mixture of 2.5 µg of DNA in 25 µl of water, 10 µl of Plus Reagent (Life Technologies) and 25 µl of Lipofectin (Life Technologies) in 100 µl of serum-free culture medium per well. After a 3 h incubation at 37°C, 160 µl of culture medium supplemented with 20% fetal calf serum was added per well. Transfection of hepatoma cells was performed with 2.5 µg of DNA mixed with 7.5 µl of FuGENE (Roche) in 100 µl of serum-free culture medium, which were then added to the well containing 300 µl of culture medium. The transfected cultures were processed for analysis 48 h later.
Stably transfected cell lines
A total of 106 HuH7 cells split in 10 wells of a 24-well plate were transfected with DNA-FuGENE complexes, as described above. Stable transfectants of HuH7 cells were obtained by transfection of the bicistronic pFLuc-IEX vector, which also carries the selectable marker zeocin. Two days after transfection, cells were split 1:10 and fed with growth medium supplemented with 0.5 mg/ml zeocin (Invitrogen). Selection with zeocin was continued for 2–3 weeks until zeocin-resistant colonies developed. These were then isolated and screened for their luciferase activity and green fluorescence. Stable 5′ UTR-EGFP transfectants of HuH7 cells were obtained by co-transfection of 2.25 µg of pEGFP-HCV-5′ UTR vector and 0.25 µg of pcDNA3.1 carrying the selectable marker neomycin (Invitrogen). Stable pEGFP-C3 transfectants of HuH7 cells were obtained by transfection of 2.5 µg of pEGFP-C3. Two days after transfection, cells were split 1:10 and fed with growth medium supplemented with 3 mg/ml geneticin (Invitrogen), a neomycin analog. Selection with geneticin was continued for 2–3 weeks until geneticin-resistant colonies developed that were analyzed for their green fluorescence. In every transfected antibiotic-selected cell population, the genomic integration of EGFP was verified by Southern blot using EGFP probe (see below), and EGFP-expressing cells were further enriched by fluorescence-activated cell sorting in a Coulter Elite FACS.
Flow cytometry analysis
Parental or transfected cells were dissociated by trypsination and suspended at a concentration of 0.5 × 106–1 × 106 cells/ml in phosphate-buffered saline, 2 mM EDTA. Samples were analyzed by flow cytometry using an argon laser to excite cells at 488 nm and a 530 ± 15 nm band-pass filter to detect EGFP expression, in a FACS Calibur (Becton-Dickinson).
Measurement of luciferase activity
The enzymatic activities of FLuc and RLuc were measured by using the Dual-Luciferase Reporter assay system (Promega), 48 h after transfection. The cells were lysed for 15 min in the culture dishes by ice-cold 1× lysis solution (0.5 ml per 106 cells). The lysates were clarified by centrifugation at 10 000 g for 5 min at 4°C. The activity of each luciferase was measured sequentially from a single 20 µl sample of cell lysate, as recommended by the manufacturer. Luminescence was measured with a Lumat LB 9501 luminometer (Wallax Berthold Bioanalyticals).
Northern blot analysis
Total cellular RNA was isolated using the RNeasy kit (Qiagen) and treated with RNase-free DNase 1 (Promega). RNA (10 µg) obtained from stably transfected cells was denatured in a mixture of 50% formamide (Sigma), 17 mM morpholinepropane sulfonic acid (MOPS) and 2.2 M formaldehyde for 15 min at 55°C and chilled on ice. RNA was separated by electrophoresis in a 1% agarose gel containing 2.2 M formaldehyde and 20 mM MOPS and transferred by capillarity in 20× SSC (3 M NaCl, 0.3 M sodium citrate) onto a nylon membrane (Hybond-N, Amersham), on which they were cross-linked by UV.
Membranes were pre-hybridized for 1 h at 42°C in 50% formamide, 5× Denhard solution, 5× SSC, 1% sodium dodecyl sulfate (SDS) and 100 µg/ml sheared double-stranded salmon sperm DNA. They were then incubated overnight at 42°C in the same solution in the presence of one of the following 32P-labeled probes (∼109 c.p.m./ml): EGFP probe obtained by digestion of the pd2EGFP-N1 vector (Clontech) by BamHI and by BsrG, and FLuc probe obtained by digestion of pGEM-luc vector (Promega) by HindIII and XhoI. The probes were labeled with [α-32P]dCTP by random priming, using the RadPrim DNA labeling system (Invitrogen). Membranes were washed under stringent conditions: twice for 5 min in 2× SSC, 0.1% SDS at room temperature, twice for 5 min in 0.2× SSC, 0.1% SDS at room temperature, 30 min in 0.1× SSC, 0.1% SDS at 65°C, and finally 5 min in 2× SSC at room temperature. The membranes were exposed to Kodak X-OMAT AR films at –80°C with intensifying screens.
Rapid amplification of 5′ end RNA
The 5′ end of the EGFP transcript was analyzed using the 5′ RACE method (GeneRacer kit, Invitrogen). Briefly, 106 HuH7 cells were transfected with 10 µg of pEGFP-5′ UTR and total RNAs were extracted by the RNeasy kit (Qiagen). Total RNA (5 µg) was dephosphorylated with 10 U of calf intestinal phosphatase and precipitated by ethanol after phenol:chloroform treatment. The cap structure was subsequently removed with 40 U of tobacco acid pyrophosphatase to produce a phosphorylated RNA at the 5′ end, to which the RNA oligonucleotide (5′-CGACUGGAGCACGAGGACACUGA CAUGGACUGAAGGAGUAGAAA-3′) was ligated. The resulting RNA was reverse-transcribed using an EGFP+2 specific primer (5′-GCTCCTCGCCCTTGCTCACC-3′) and 5 U of AMV reverse transcriptase, and then treated with 2 U of RNase H. Tailed cDNA was amplified by PCR using the GeneRacer 5′ primer (CGACTGGAGCACGAGGACAC TGA), the EGFP-specific primer and 2.5 U of AmpliTaq Gold DNA polymerase (Roche). A nested PCR was performed using the GeneRacer 5′ primer (GGACACTGACATGG ACTGAAGGAGTA) and the 5′ UTR341–322 primer (5′-GGTGCACGGTCTACGAGACC-3′). The resulting cDNA was sequenced using the BigDye DNA sequencing kit (Applied Biosystems) and analyzed on an automated sequencer (ABI Prism 310 Genetic Analyzer, Applied Biosystems).
RESULTS
The HCV 5′ UTR cDNA displays a promoter activity
Our original aim to study the ex vivo activity of the HCV RNA-dependent RNA polymerase (RdRp) led us to design reporter plasmids containing the HCV 5′ UTR or 3′ UTR, in which the untranslated cDNA regions of the HCV genome were inserted upstream of the EGFP cDNA. HeLa cells constitutively expressing RdRp were transfected with the reporter plasmids and cell cultures were analyzed by fluorescence microscopy to detect EGFP expression. An intense green fluorescence was observed in the cells transfected with plasmid containing 5′ UTR, but not with 3′ UTR (data not shown). Unexpectedly, the control group performed with parental HeLa cells showed the same level of fluorescence, although they did not express the HCV RdRp. To explain this surprising observation, we used flow cytometry to analyze the expression of EGFP in HuH7 cells transiently transfected with the following plasmids described in Figure 1A: pEGFP-HCV-5′ UTR, pEGFP-HCV-3′ UTR, pEGFP-polio-5′ UTR or pEGFP-CMVp. As shown in Figure 2, the presence of pEGFP-HCV-5′ UTR led to a level of EGFP synthesis very similar to that produced by the CMV early promoter. On the other hand, neither pEGFP-HCV-3′ UTR nor pEGFP-polio-5′ UTR were able to generate the fluorescent protein. These results strongly suggested that the cDNA corresponding to the HCV 5′ UTR seems to behave as a eukaryotic promoter in both HuH7 and HeLa cells.
Figure 2.

Analysis of the promoter activity of different DNA sequences. The EGFP expression in transient transfected HuH7 cells was analyzed by flow cytometry. The mean fluorescence was calculated from positive fluorescent cells. The results are the mean of four independent experiments.
To test the hypothesis of a possible cryptic promoter within the pEGFP sequence which could have been activated by the HCV 5′ UTR cDNA, we analyzed the activity of the latter sequence with the aid of pIRF, the bicistronic reporter vector described in Figure 1C. This construction had been designed to analyze the IRES activity present in the 5′ UTR of the HCV genome (15). As shown in Figure 3, the transfection of HuH7 cells with pIRF led to the expression of both enzymatic activities, FLuc and RLuc. In contrast, transfection with the pIRF-delCMV, in which the CMV promoter had been deleted, resulted in the disappearance of the FLuc activity without a noticeable effect on RLuc activity.
Figure 3.

Promoter activity associated with the DNA sequence of HCV 5′ UTR. HuH7 cells were transfected with pIRF or pIRF-delCMV vectors and analyzed 48 h later. Luciferase activity (RLU) was measured with a luminometer as described in Materials and Methods. RLuc and FLuc represent Renilla luciferase and firefly luciferase, respectively. The results are the mean of three experiments in duplicate.
The disappearance of FLuc activity after the deletion of the CMV promoter points to the absence of a cryptic promoter upstream of this gene. The persistence of RLuc activity, despite the deletion of the CMV promoter, lends further weight to the presence of an active promoter in the sequence of the HCV 5′ UTR inserted between the genes FLuc and RLuc.
Gene expression from the HCV 5′ UTR
We have shown above that the cDNA corresponding to the HCV 5′ UTR could act as a eukaryotic promoter present in the bicistronic construct used in this work. We further investigated whether this promoter activity could be detected if the DNA form of the HCV 5′ UTR was integrated in the nuclear genome. To address this point, a transfection of either pEGFP-HCV-5′ UTR + pcDNA3.1/Neo or pEGFP-C3, which contains the CMV promoter upstream the EGFP gene and the neomycin-resistance gene, was performed in HuH7 cells. Stably transfected cells were obtained after geneticin selection. After 3 weeks of very stringent selection, 35% of cells were fluorescent. At this stage, the homogeneity and the proportion of fluorescent cells were improved by cell sorting and the genomic integration was verified by Southern blot (results not shown).
Cells containing either pEGFP-HCV-5′ UTR or pEGFP-C3 were analyzed by flow cytometry (Fig. 4). In both cases, transfected cells were fluorescent and the observed EGFP expression depending on the HCV 5′ UTR was similar to that obtained using the CMV early promoter.
Figure 4.
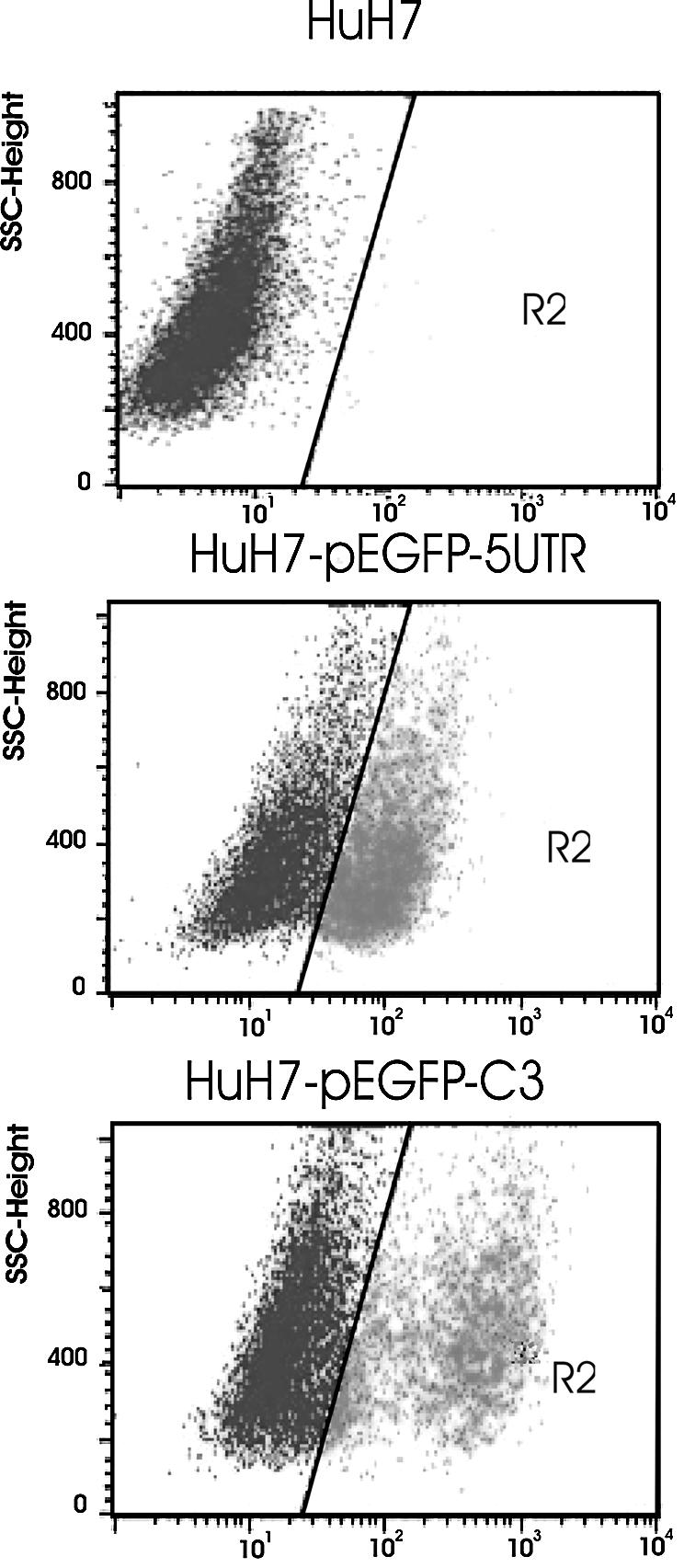
Gene expression in stably transfected cells. Flow cytometry analysis of EGFP expression. HuH7 cells were transfected with pEGFP-HCV-5′ UTR (middle) or pEGFP-C3 (bottom). Untransfected cells (top).
The promoter activity of the UTR was further assessed by analyzing the transcription product from both types of transfected cells. Northern blot analysis with an EGFP probe revealed a transcript from each of the pEGFP-5′ UTR or pEGFP-C3 transfectants (data not shown) suggesting that the promoter carried by the DNA sequence corresponding to the HCV 5′ UTR could also be active when this sequence is present in the nuclear genome.
A transcript is initiated from the HCV 5′ UTR
The results reported above suggested the existence of two transcripts in cells transfected with a bicistronic expression vector containing the HCV 5′ UTR cDNA, one starting from the CMV promoter, the other from the region corresponding to the 5′ UTR of HCV. To avoid unlikely cross-hybridization between two luciferase probes, we investigated this hypothesis with the bicistronic expression vector pLuc-IEX (Fig. 1E) carrying the FLuc and d2EGFP genes with HCV 5′ UTR inserted in between. As a control, we used the monocistronic vector pEGFP-C3.
These vectors were transfected in HuH7 cells and several clones were obtained after antibiotic selection. Results in Figure 5A show the luciferase activity (FLuc) and the fluorescence due to EGFP expression. With the bicistronic vector, as expected, the level of expression of FLuc and EGFP in these stably transfected cells was lower than that observed in transient transfection experiments, although FLuc and EGFP activities were significantly detected (Fig. 5A, column 3).
Figure 5.

Expression from the HCV 5′ UTR sequence in bicistronic vector pLuc-IEX. Cells were transfected with pEGFP-C3 (column and lane 2), pLuc-IEX (column and lane 3) or no construction (column and lane 1), and analyzed for protein expression and transcript synthesis after G418 selection. (A) Luciferase activity (RLU) and d2EGFP expression obtained from flow cytometry experiments; the luciferase activity was expressed for 106 cells; the mean of fluorescence was calculated from positive fluorescent cells. (B) Schematic representation of expected transcripts; transcript 1 is transcribed from the CMV promoter, transcript 2 from the HCV 5′ UTR. (C) Northern blot analysis of transcripts with 10 µg of RNA in the presence of d2EGFP (right) or FLuc (left) 32P-labeled probes, as described in Materials and Methods.
Next, these clones were used to analyze the RNA transcripts coding for FLuc and EGFP. The expected transcripts are shown schematically in Figure 5B. With the pEGFP-C3 vector, only one RNA band appeared after being probed with EGFP (Fig. 5C, lane 2). With the bicistronic pLuc-IEX vector, two bands (transcripts 1 and 2) were observed after hybridization with the EGFP probe (Fig. 5C, EGFP probe, lane 3) and a single one after hybridization with the FLuc probe (Fig. 5C, FLuc probe, lane 3). The larger fragment (transcript 1), which contains both the FLuc and the EGFP mRNAs, has the expected size (3.2 kb) if the transcription of the two cistrons is driven by the CMV promoter, while the smaller fragment (transcript 2), revealed only by the EGFP probe, again showed the promoter ability of the HCV 5′ UTR. Compared to the control EGFP (lane 2), the difference in size of the small transcript in lane 3, which is somewhat larger, may be due to the following: (i) the larger size of the destabilized d2EGFP gene (see Materials and Methods) as compared with the EGFP-coding sequence; (ii) the recruitment of a part of the 5′ UTR sequences if the initiation site of the transcription is internal (1–341 nt); (iii) the presence of the 3′ UTR sequence (221 nt). This smaller transcript (transcript 2) carrying only the EGFP-coding sequence could be initiated inside the 5′ UTR sequence.
Localization of the initiation site of the transcription
The presence of a promoter within the HCV 5′ UTR DNA sequence raises the question of the initiation site of the transcription. Does the transcript possess a fragment of the sequence carrying the IRES activity? To address this question, we determined the localization of the initiation site by using the 5′ RACE method that selects capped mRNA. HuH7 cells transiently transfected with pEGFP-5′ UTR were harvested after 48 h of culture, the capped mRNAs were extracted, amplified by RT–PCR with the 5′ UTR341–322 primer and then sequenced. The HCV 5′ UTR sequence was obtained as expected and the junction between the GeneRacer RNA oligonucleotide and the EGFP sequence was resolved (Fig. 6A), showing clearly that the transcript driven by the HCV 5′ UTR was initiated at nucleotide 67 (Fig. 6B). This nucleotide is located on the stem–loop II domain of the 5′ UTR structure proposed by Honda et al. (16).
Figure 6.

Delineation of the transcription initiation site. (A) The upper sequence (5′ UTR) corresponds to the HCV 5′ UTR sequence. The EGFP mRNA sequence corresponds to that obtained from capped mRNA transcripts derived from HCV 5′ UTR. Bold letters represent the RNA oligonucleotide used in the 5′ RACE method. The vertical arrowhead indicates the position of the initiation site for EGFP transcription. The horizontal arrow indicates the location (nucleotides 321–341) of the primer used for the reverse transcriptase reaction. (B) Position of the initiation site in the IRES structure proposed by Honda et al. (16).
DISCUSSION
In this work, we show that a DNA sequence corresponding to the HCV 5′ UTR possesses a promoter activity able to drive the expression of genes inserted downstream. This activity was found independently of the plasmid backbone into which these sequences were inserted, since it was present when either vectors pEGFP or pcDNA3.1 were used. The promoter activity was also observed when the fragment 5′ UTR-EGFP was inserted into the pBK plasmid having a pBR322 backbone (results not shown). Moreover, the promoter activity of the 5′ UTR cDNA was also independent of the reporter gene utilized, because it was also able to drive the expression of genes coding for EGFP or RLuc.
The promoter activity was observed in various cell lines: HuH7, HepG2, HeLa and Cos-7, and not only in those of hepatic origin (results not shown). Nevertheless, the promoter activity was variable among these cell lines.
Both in stably or transiently transfected cells, the level of expression obtained from the HCV 5′ UTR was of the same order of magnitude as that obtained when the CMV early promoter was used (Fig. 2). Northern blot analysis (Fig. 5C) showed that the two RNA species transcribed from a bicistronic construction were obtained with similar yields, thus confirming that the DNA sequence carrying the HCV 5′ UTR could act as a promoter which is as active as the CMV early promoter. However, while the EGFP expression from the CMV promoter was detected in 52% of the transfected cells, the expression from the 5′ UTR was observed in 12–20% (results not shown). This cellular variability may be due to the possible regulation of this promoter activity by cellular factors.
The IRES structure embedded in the HCV 5′ UTR plays an important role in viral protein expression. In the past few years, several lines of evidence regarding the IRES function using transfected DNA corresponding to the HCV 5′ UTR either as bicistronic constructs (17–19) or in transgenic mice (20) have been reported. Thus, our work shows that careful consideration must be taken when interpreting the results obtained from bicistronic constructs involving the HCV IRES sequence. We show here that bicistronic constructs like pIRF express the second cistron in two ways: first, by an RNA generated by the CMV promoter, and second, by the 5′ UTR promoter. The longest RNA possesses the IRES activity, while the shortest, beginning at nucleotide 67 in the 5′ UTR sequence, does not possess the complete IRES sequence and in addition has a 5′ cap end. It is unclear whether the RNA initiated from the 5′ UTR would be translated in a cap-dependent or an IRES-dependent way. The variations observed in gene expression using these constructs are usually explained at the translational level. On the basis of our results, they should also be analyzed at the transcriptional level. Consequently, the use of bicistronic vectors including DNA sequences corresponding to the HCV 5′ UTR should be carefully considered when analyzing the translational activity of the IRES structure by DNA transfection.
It is generally admitted that the RNA-dependent RNA replication of the HCV genome excludes the presence of DNA intermediates. However, our results showing the presence of the promoter activity in double-stranded DNA corresponding to the HCV 5′ UTR region, also point to the synthesis of DNA carrying HCV sequences during the replication cycle of this virus. This hypothesis, although apparently heretical, is supported not only by our own results but also by three recent observations from other groups. First, the lack of an efficient cellular system to replicate HCV led to the description selectable replicons derived from a full-length consensus HCV genome (12,21,22). When reporting such new tools, the authors discussed the presence of relatively frequent integrative events of the subgenomic HCV sequences (12). Moreover, the compulsory expression of the neo gene inserted downstream of the HCV 5′ UTR was observed in some constructions defective for replication, thus indicating the presence of a eukaryotic promoter. The latter observations suggest that in addition to the possible occasional integration of subgenomic regions of HCV, the DNA form of the 5′ UTR may be active within the nuclear genome. Secondly, recent results showed that the NS3 viral protein is a poor helicase on RNA, while it is highly processive on DNA (23). Therefore, it is tempting to speculate that this preferential DNA helicase activity, if functional in vivo, might be related to the presence of HCV sequences present as double-stranded DNA in the cell nucleus (see below). Thirdly, although extensive DNA sequences corresponding to the HCV genome have not been described to date, fragments of the 5′ UTR have been detected in the human genome (24). Altogether, these results point to the need for further studies to elucidate the possible functions of these HCV DNA intermediates and to characterize the enzyme activities involved in these events.
Acknowledgments
ACKNOWLEDGEMENTS
We thank Laura Tarrago-Litvak, UMR 5097 CNRS Bordeaux, and Jean-Jacques Toulmé, U386 INSERM Bordeaux, for critical reading of the manuscript and helpful discussions. This work was supported by grants from the Association de Recherches sur le Cancer (ARC), the Ligue Régionale contre le Cancer, the Réseau Hépatite C, the CNRS and the University Victor Segalen Bordeaux 2.
REFERENCES
- 1.WHO (2000) Hepatitis C Fact Sheet No. 164, revised October 2000.
- 2.Reynolds J.E., Kaminski,A., Carroll,A.R., Clarke,B.E., Rowlands,D.J. and Jackson,R.J. (1996) Internal initiation of translation of hepatitis C virus RNA: the ribosome entry site is at the authentic initiation codon. RNA, 2, 867–878. [PMC free article] [PubMed] [Google Scholar]
- 3.Honda M., Ping,L.H., Rijnbrand,R.C., Amphlett,E., Clarke,B., Rowlands,D. and Lemon,S.M. (1996) Structural requirements for initiation of translation by internal ribosome entry within genome-length hepatitis C virus RNA. Virology, 222, 31–42. [DOI] [PubMed] [Google Scholar]
- 4.Odreman-Macchioli F., Baralle,F.E. and Buratti,E. (2001) Mutational analysis of the different bulge regions of the HCV domain II and their influence on IRES translational ability. J. Biol. Chem., 276, 41648–41655. [DOI] [PubMed] [Google Scholar]
- 5.Tanaka T., Kato,N., Cho,M.J. and Shimotohno,K. (1995) A novel sequence found at the 3′ terminus of hepatitis C virus genome. Biochem. Biophys. Res. Commun., 215, 744–749. [DOI] [PubMed] [Google Scholar]
- 6.Kolykhalov A.A., Mihalik,K., Feinstone,S.M. and Rice,C.M. (2000) Hepatitis C virus-encoded enzymatic activities and conserved RNA elements in the 3′ nontranslated region are essential for virus replication in vivo. J. Virol., 74, 2046–2051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lanford R.E., Lee,H., Chavez,D., Guerra,B. and Brasky,K.M. (2001) Infectious cDNA clone of the hepatitis C virus genotype 1 prototype sequence. J. Gen. Virol., 82, 1291–1297. [DOI] [PubMed] [Google Scholar]
- 8.Ito T., Tahara,S.M. and Lai,M.M.C. (1998) The 3′-untranslated region of hepatitis C virus RNA enhances translation from an internal ribosomal entry site. J. Virol., 72, 8789–8796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Friebe P., Lohmann,V., Krieger,N. and Bartenschlager,R. (2001) Sequences in the 5′ nontranslated region of hepatitis C virus required for RNA replication. J. Virol., 75, 12047–12057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Choo Q.L., Kuo,G., Weiner,A.J., Overby,L.R., Bradley,D.W. and Houghton,M. (1989) Isolation of a cDNA clone derived from a blood-borne non-A, non-B viral hepatitis genome. Science, 244, 359–362. [DOI] [PubMed] [Google Scholar]
- 11.Bartenschlager R. and Lohmann,V. (2001) Novel cell culture systems for the hepatitis C virus. Antiviral Res., 52, 1–17. [DOI] [PubMed] [Google Scholar]
- 12.Lohmann V., Korner,F., Koch,J., Herian,U., Theilmann,L. and Bartenschlager,R. (1999) Replication of subgenomic hepatitis C virus RNAs in a hepatoma cell line. Science, 285, 110–113. [DOI] [PubMed] [Google Scholar]
- 13.Pietschmann T., Lohmann,V., Kaul,A., Krieger,N., Rinck,G., Rutter,G., Strand,D. and Bartenschlager,R. (2002) Persistent and transient replication of full-length hepatitis C virus genomes in cell culture. J. Virol., 76, 4008–4021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kolykhalov A.A., Agapov,E.V., Blight,K.J., Mihalik,K., Feinstone,S.M. and Rice,C.M. (1997) Transmission of hepatitis C by intrahepatic inoculation with transcribed RNA. Science, 277, 570–574. [DOI] [PubMed] [Google Scholar]
- 15.Laporte J., Malet,I., Andrieu,T., Thibault,V., Toulme,J.J., Wychowski,C., Pawlotsky,J.M., Huraux,J.M., Agut,H. and Cahour,A. (2000) Comparative analysis of translation efficiencies of hepatitis C virus 5′ untranslated regions among intraindividual quasispecies present in chronic infection: opposite behaviors depending on cell type. J. Virol., 74, 10827–10833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Honda M., Beard,M.R., Ping,L.-H. and Lemon,S.M. (1999) A phylogenetically conserved stem-loop structure at the 5′ border of the internal ribosome entry site of hepatitis C virus is required for cap-independent viral translation. J. Virol., 73, 1165–1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kato J., Kato,N., Yoshida,H., Ono-Nita,S.K., Shiratori,Y. and Omata,M. (2002) Hepatitis C virus NS4A and NS4B proteins suppress translation in vivo. J. Med. Virol., 66, 187–199. [DOI] [PubMed] [Google Scholar]
- 18.Shimazaki T., Honda,M., Kaneko,S. and Kobayashi,K. (2002) Inhibition of internal ribosomal entry site-directed translation of HCV by recombinant IFN-alpha correlates with a reduced La protein. Hepatology, 35, 199–208. [DOI] [PubMed] [Google Scholar]
- 19.Collier A., Tang,S. and Elliott,R. (1998) Translation efficiencies of the 5′ untranslated region from representatives of the six major genotypes of hepatitis C virus using a novel bicistronic reporter assay system. J. Gen. Virol., 79, 2359–2366. [DOI] [PubMed] [Google Scholar]
- 20.Okoshi S., Takeda,Y., Takimoto,M., Shimotohno,K., Asakura,H. and Jay,G. (2001) Molecular dissection of hepatitis C virus expression. Intervirology, 44, 21–28. [DOI] [PubMed] [Google Scholar]
- 21.Blight K.J., Kolykhalov,A.A. and Rice,C.M. (2000) Efficient initiation of HCV RNA replication in cell culture. Science, 290, 1972–1924. [DOI] [PubMed] [Google Scholar]
- 22.Ikeda M., Yi,M., Li,K. and Lemon,S.M. (2002) Selectable subgenomic and genome-length dicistronic RNAs derived from an infectious molecular clone of the HCV-N strain of hepatitis C virus replicate efficiently in cultured Huh7 cells. J. Virol., 76, 2997–3006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pang P.S., Jankowsky,E., Planet,P.J. and Pyle,A.M. (2002) The hepatitis C viral NS3 protein is a processive DNA helicase with cofactor enhanced RNA unwinding. EMBO J., 21, 1168–1176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dennin R.H., Wo,J. and Chen,Z. (1999) Hepatitis C virus-specific DNA sequences in human DNA: differentiation by means of restriction enzyme analysis at the DNA level of healthy, anti-HCV-negative individuals. Clin. Chem. Lab. Med., 37, 623–630. [DOI] [PubMed] [Google Scholar]