

Abstract
Protein phosphatases have very recently emerged as important targets for chemical biology and medicinal chemistry research, and new phosphatase inhibitor classes are in high demand. The underlying frameworks of natural products represent the evolutionarily selected fractions of chemical space explored by nature so far and meet the criteria of relevance to nature and biological prevalidation most crucial to inhibitor development. We refer to synthesis efforts and compound collection development based on these criteria as biology-oriented synthesis. For the discovery of phosphatase inhibitor classes by means of this approach, four natural product-derived or -inspired medium-sized compound collections were synthesized and investigated for inhibition of the tyrosine phosphatases VE-PTP, Shp-2, PTP1B, MptpA, and MptpB and the dual-specificity phosphatases Cdc25A and VHR. The screen yielded four unprecedented and selective phosphatase inhibitor classes for four phosphatases with high hit rates. For VE-PTP and MptpB the first inhibitors were discovered. These results demonstrate that biology-oriented synthesis is an efficient approach to the discovery of new compound classes for medicinal chemistry and chemical biology research that opens up new opportunities for the study of phosphatases, which may lead to the development of new drug candidates.
Keywords: chemical biology, medicinal chemistry, phosphatase inhibition
Protein phosphatases are key regulators of innumerable biological processes (1, 2). Small-molecule modulators of phosphatase activity have proven to be powerful tools for the study of the chemical biology of these enzymes (3), and, in particular, protein tyrosine phosphatases (PTPs) (4, 5) and dual-specificity phosphatases (6) have recently moved into the focus of a growing number of drug discovery programs, for instance in diabetes and anticancer research. However, although important progress has been made, the development of potent and selective phosphatase inhibitors is still in its early stages, and structurally new phosphatase inhibitor classes are in high demand.
Relevance to nature is one of the most important criteria to be met by compound classes for chemical biology and medicinal chemistry research. The underlying frameworks of natural products (NPs) provide evolutionarily selected chemical structures encoding the properties required for binding to proteins, and their structural scaffolds represent the biologically relevant and prevalidated fractions of chemical space explored by nature so far (7–9). Consequently, it is to be expected that compound collections designed on the basis of NP structure will be enriched in biochemical and biological activity. Based on this reasoning, we have introduced a structural classification of natural products (SCONP) in a tree-like arrangement as an idea- and hypothesis-generating tool for the design and synthesis of compound collections (8). It permits the selection of library scaffolds based on relevance to and prevalidation by nature. We refer to synthesis efforts based on these criteria as biology-oriented synthesis (BIOS).
Here, we describe the discovery of four phosphatase inhibitor classes for four different protein tyrosine phosphatases by means of BIOS and SCONP.
Results
For the identification of new phosphatase inhibitor classes by means of the BIOS concept, we drew from a NP-based compound library and a collection of individual NPs available to us (all compounds used in the initial screen are available from AnalytiCon Discovery). Fig. 1 shows the general structures of two compound collections used (for a brief description of the synthesis of collections 1 and 2, see Supporting Materials and Methods, which is published as supporting information on the PNAS web site), the structures of the guiding NPs, and the number of the library members.
Fig. 1.

Scaffolds, guiding NPs, and substitution patterns of the two investigated NP-derived and -inspired compound collections, size of the collections, and SAR derived for the inhibition of different phosphatases.
These compound collections and additional 354 isolated NP singletons were used in a biochemical screen for inhibition of seven different phosphatases. The choice of the individual NPs and the collections shown in Fig. 1 was based on the criterion to employ a structurally diverse screening set.
The screen included the tyrosine phosphatases PTP1B, Shp-2, VE-PTP, MptpA, and MptpB as well as the dual specificity phosphatases Cdc25A and VHR. PTP1B is involved in insulin signaling and considered a target for the development of new drugs against diabetes and the metabolic syndrome (4, 5). MptpA and MptpB are phosphatases secreted by Mycobacterium tuberculosis that modify host signaling pathways and are considered targets for the development of new antituberculosis drugs (10). Shp-2 is a tyrosine phosphatase that is considered a target in the development of new antiinfective drugs (11). VE-PTP dephosphorylates the Tie-2 receptor tyrosine kinase and enhances the adhesive function of endothelial VE-cadherin (12, 13). Disruption of the VE-PTP gene leads to embryonal lethal defects in angiogenesis (14). Cdc25A is involved in regulation of the cell cycle and is considered an anticancer target (6, 15). VHR influences signaling via dephosphorylation of ERK1/2 and Jun kinases (16).
Screening of the compound collections shown in Fig. 1 yielded potent inhibitors of the tyrosine phosphatases VE-PTP, Shp-2, PTP1B, and MptpB. Compounds with IC50 < 10 μM were considered hits. Compound collection 1, which embodies the underlying scaffold structure of the alkaloid cytisine (17) and related NPs, delivered inhibitors of VE-PTP with a hit rate of 1.57%. Closer inspection of the structures characteristic for hits and inactive compounds revealed a clear structure–activity relationship (SAR) pattern (Fig. 1 and Table 1; and see Table 2, which is published as supporting information on the PNAS web site).
Table 1.
Selected phosphatase inhibitors identified in the screens
| No. | Structure | IC50,* μM | Phosphatase inhibited† |
|---|---|---|---|
| 1 |  |
2.08 ± 0.29 | VE-PTP |
| 2 |  |
2.45 ± 0.28 | VE-PTP |
| 3 |  |
2.47 ± 0.93 | Shp-2 |
| 4 |  |
3.95 ± 1.49 | Shp-2‡ |
| 5 |  |
1.13 ± 0.63 | MptpB |
| 6 | 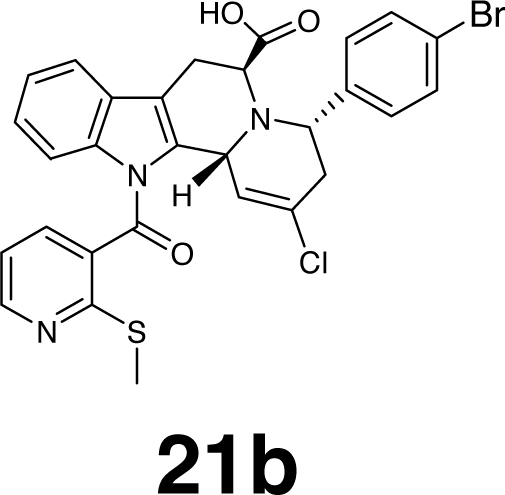 |
2.38 ± 1.17 | MptpB |
| 7 |  |
0.36 ± 0.12 | MptpB |
| 8 |  |
0.43 ± 0.17 | MptpB |
*All IC50 values were calculated from at least three independent determinations.
†The other phosphatases investigated were not inhibited at 100 μM.
‡IC50 for inhibition of PTP1B = 5.86 ± 1.35 μM.
The most potent VE-PTP inhibitors (Table 1) were at least 40-fold selective for VE-PTP. The compounds shown in Tables 1 and 2 are the first inhibitors to be described for the phosphatase VE-PTP. Compounds with the structural scaffold of lupine alkaloid like 1 have not been described as phosphatase inhibitors.
Compound collection 2 (Fig. 1), which was inspired by the furanodictines (see Fig. 1) (18), revealed a previously uncharacterized inhibitor class for the phosphatases PTP1B and Shp-2 with a hit rate of 0.36%. Compounds 2a–2c are the active representatives of a larger class with a clear SAR (Fig. 1 and Table 1; and see Table 3, which is published as supporting information on the PNAS web site). The furofuran hits were at least 20-fold selective for Shp-2 and PTP1B. Remarkably, compound 2a (Table 1) is selective for Shp-2. Furofurans have not been described before as phosphatase inhibitors.
Screening of the two NP-derived compound collections did not yield hits for MptpA, Cdc25A, and VHR.
These results demonstrate that compound collections synthesized by derivatization of basic NP scaffolds may indeed yield new inhibitor classes for phosphatases with an appreciably high hit rate at a comparably small library size, i.e., employing compound collections with member numbers up to ≈1,000 (instead of tens or hundreds of thousands).
The phosphatase inhibition screen of the isolated NPs led to the discovery of further classes of unprecedented phosphatase inhibitors. Surprisingly, from seven investigated yohimbane-type alkaloids, three NPs proved to be weak inhibitors for the phosphatase Cdc25A (Fig. 2). Inspection of the NP structures indicated that the inhibitory activity is influenced by the stereochemistry of the D–E ring linkage and the substitution pattern of the E ring. This initial finding suggested that by means of appropriate structure variation, new phosphatase inhibitor classes with enhanced potency might be identified. For the generation of a working hypothesis, we resorted to the SCONP tree classification of NPs and the design of a NP-inspired compound collection for guidance. The most logical application of SCONP for compound collection development is the selection of NP-derived scaffolds as demonstrated, for instance, above. However, as previously shown for the development of 11-β-hydroxysteroid dehydrogenase inhibitors from a pentacyclic NP belonging to the carbocycle branch of the SCONP tree (8), brachiation within the tree from an outer branch to structurally less complex scaffolds on an inner branch by means of intermediate naturally occurring parent scaffolds may lead to structurally simplified compound classes with retained biological activity.
Fig. 2.

Structures of the yohimbane alkaloids that were identified as Cdc25A inhibitors.
Given the structural complexity of the pentacyclic yohimbane alkaloids and the severe difficulties to be expected in the synthesis of a compound collection of appropriate size with this scaffold structure, we sought to apply the brachiation approach as a hypothesis-generating tool for the development of compound collections with more readily accessible structure. The basic scaffold 7 of the yohimbane alkaloids can be assigned to the indole branch of the SCONP tree (Fig. 3). Brachiation along the line of prevalidation by nature then initially led to the tetracyclic indoloquinolizidine scaffold 8 and via tricyclic tetrahydro-β-carbolines 9 to indoles 10. Based on these guiding trains of thought, we synthesized a collection of tetracyclic indoloquinolizidines carrying different substituents in the D ring. In addition, a collection of 188 compounds consisting of tricyclic indole derivatives with six- to eight-membered carbo- and heterocyclic rings attached to the indole core as well as 2,3- or 2,3,5-substituted indole derivatives, synthesized as described in refs. 19 and 20 (see Fig. 3 for the general structure 11 of the indole-inspired collection), was used for the subsequent biochemical evaluation. The indoloquinolizidine collection was synthesized on the solid phase as shown in Fig. 4A. Polystyrene beads equipped with Wang or 4-(hydroxymethyl)benzoic acid amide (HMBA) resin were loaded with d- or l-fluorenylmethoxycarbonyl-tryptophan (loading 0.87 mmol/g), and after removal of the fluorenylmethoxycarbonyl group by treatment with base, tryptophan imines 13 were formed and subjected to Lewis acid-mediated tandem Mannich–Michael reaction with electron-rich dienes 14 (21, 22) to yield immobilized enaminones 15 that were formed with diastereomer ratios of 65:35 to 90:10. Treatment of the intermediate attached to the HMBA linker with trifluoroacetic acid (TFA) in the presence of trimethylsilylchloride (TMSCl) (23–27) induced the formation of tetracyclic ketones 16, which were released from the polymeric carrier as methyl esters 17 by treatment with NaOMe. Alternatively, treatment of enaminones 15 linked to Wang resin with phosgene and TMSCl (28) induced the formation of solid-phase-bound vinyl chlorides 18. The intermediates either were released from the solid support to yield tetracyclic compounds 19 or were N-acylated or N-sulfonated on resin to give amides 20. These polymer-bound esters were cleaved from the polymeric carrier to yield acids 21 that subsequently could be esterified to give esters 22.
Fig. 3.

Schematic representation of the indole branch of the SCONP tree and general structure of indole-inspired compound collection 11.
Fig. 4.

Solid-phase synthesis of a NP-inspired compound collection. (A) Synthesis of indoloquinolizidine derivatives on solid support. Reagents and conditions were as follows. a: 2,6-dichlorobenzoyl chloride (2 eq), pyridine (3 eq), N,N-dimethylformamide, room temperature, 24 h (two cycles). b: piperidine, N,N-dimethylformamide 20% (vol/vol), room temperature, four times for 5 min. c: R1CHO (5 eq), trimethyl orthoformate/dichloromethane 3:2, room temperature, 12 h. d: dienes 14(5 eq), ZnCl2 1 M in tetrahydrofuran (2 eq), propionitrile, 0°C to room temperature, 15 h. e: TFA, TMSCl, CH2Cl2, room temperature, 30 min. f: NaOMe 0.25 M in MeOH:1,4-dioxane (1:1); 50°C, 18 h, 15–45% overall yield. g: phosgene, TMSCl, CH2Cl2, room temperature, 30 min. h: R4X (X = halogen), lithium hexamethyldisilazane, hexamethylphosphortriamide, −78°C to room temperature, 12 h. i: TFA, H2O, room temperature, 12 h. j: 5 eq EDC·HCl, pyridine/MeOH/CH2Cl2 1:1:1, room temperature, overnight, 29–72% (overall yield). (B) SAR developed for indoloquinolizidines 19, 21, and 22. (C) SAR developed for indole-inspired compound collection 11.
This six- to eight-step solid phase synthesis sequence is compatible with the use of aromatic, aliphatic, and α,β-unsaturated aldehydes and differently substituted dienes. It gives access to the desired vinyl chlorides 19, 21, and 22 and ketones 17 with overall yields of 55–76% and 12–43% yield, respectively; i.e., it proceeds with very high overall efficiency. The stereoselectivity for cyclization to vinyl chlorides 18 is ≈2:1, and tetracyclic indoloquinolizidines 19, 21, and 22 are formed as mixtures of four diastereomers, which were readily separated by HPLC. Ketones 17 were isolated as single diastereomers possibly due to equilibration to the thermodynamically most stable isomer via retro Mannich reaction under the basic conditions for release from the solid support. The sequence gave access to 450 diastereomerically pure tetracyclic alkaloid analogues that were >99% pure. Their configuration was determined by means of nuclear Overhauser effect-difference spectroscopy and characteristic signal shifts in their 1H-NMR spectra (see Supporting Materials and Methods).
Screening of the indoloquinolizidines for inhibition of phosphatases revealed that the compound collection contains two weak inhibitors of Cdc25A (see Table 4, which is published as supporting information on the PNAS web site) with an activity comparable to the guiding NPs. Thus, in principle, brachiation from the pentacyclic scaffold to a tetracyclic scaffold retaining activity for the same enzyme appears possible. However, very gratifyingly, the compound collection contained 11 compounds displaying an IC50 value of <10 μM (hit rate of 2.4%) for inhibition of MptpB (Table 1; and see Table 5, which is published as supporting information on the PNAS web site). Nine of these compounds did not inhibit any other phosphatase at a concentration of 100 μM. Inhibition of MptpB was characterized by a clearly recognizable SAR (Fig. 4B and Table 5).
The indoloquinolizidines shown in Table 1 are the first inhibitors to be described for MptpB. Indoloquinolizidines have not been identified as phosphatase inhibitors before.
Even more encouraging results were obtained by screening the 188 structurally further simplified tricyclic and bicyclic indole derivatives. Eighteen compounds displayed an IC50 value of <10 μM; eight of them had an IC50 value of 340–860 nM (Table 1; and see Table 6, which is published as supporting information on the PNAS web site). Six compounds were at least 30-fold selective. This compound class displayed a clear SAR as well (Fig. 4C and Table 6).
The collection contained one moderate inhibitor of Cdc25A (9e IC50 = 18.9 μM) and two inhibitors of PTP1B (IC50 = 3.74 and 10.25 μM, respectively; see 10c and 10f and Table 6); i.e., their potency is comparable to the activity of the originally guiding NPs. Taken together the results obtained in the investigation of the indoloquinolizidines, the tetrahydro-β-carbolines, and the substituted indoles clearly indicate that brachiation along the main branch of the indole part of the SCONP tree, together with introduction of focused chemical diversity by means of BIOS, is a valid approach for the identification of new phosphatase inhibitors.
Discussion
The structural scaffolds of NP classes are endowed with relevance to nature and provide evolutionarily selected starting points in chemical structure space for compound collection design and development. Because they emerge via biosynthesis by proteins and fulfill multiple functions via interaction with proteins, NP classes encode structural properties required for binding to these biomacromolecules. BIOS builds on these arguments and employs core structures delineated from NPs as scaffolds of compound collections. Such scaffolds can be identical with the underlying core structures of the guiding NPs. In this case the synthesis typically builds on genuine NPs and consists of a series of transformations that introduce different substituents at sites of the NP scaffold predetermined by nature. We refer to compound collections obtained by means of this subsequent modification of a given scaffold as “NP-derived.” Alternatively the scaffolds used in the synthesis are not identical but closely related to the core structure of a NP class. In the synthesis the scaffold typically is built up, and in this process different functional groups and substituents are introduced not necessarily at the sites of substitution found in the guiding NP. Notably, also stereochemistry may be varied. We refer to compound collections obtained by means of this approach that approximates a true NP synthesis as “NP-inspired.”
In both strategies, focused diversity around a biologically prevalidated starting point in vast structural space is generated. BIOS, therefore, builds on the diversity created by nature in evolution and aims at its local extension in areas of proven biological relevance. Consequently BIOS offers a conceptual alternative to other guiding strategies for library design that, for instance, are based on mechanistic considerations, sequence or structure homology, or the creation of chemical diversity (29).
As a hypothesis- and idea-generating tool for BIOS, we have introduced a SCONP that arranges the scaffolds of NP classes according to structural genealogy in a tree-like arrangement and thereby systematically orders NP diversity (8). SCONP can be used to choose the scaffold structures of NP-inspired or -derived compound collections, or brachiation along the branches of the NP tree may be used as a hypothesis generator to possibly arrive at simpler NP scaffolds retaining biological relevance and activity. Because of this guidance by nature the application of SCONP and BIOS should yield new opportunities for the discovery of unprecedented protein ligand classes with high hit rates at comparably small library sizes.
In this article, we describe the joint application of BIOS and SCONP for the discovery of phosphatase inhibitors. Protein phosphatases have very recently emerged as important targets for chemical biology and medicinal chemistry research, and new phosphatase inhibitor classes are in high demand (4–6).
For the phosphatase inhibition screen, a NP-derived and a NP-inspired compound collection were used (Fig. 1) that represent typical examples from the N- and O-heterocycle branches of the NP tree (8). The libraries 1 and 2 were obtained by derivation of the structural scaffold of the NP cytisine (1) (17) and based on the scaffold embedded in the furanodictines (2) (18). They contain 1,271 (1) and 1,112 (2) members; i.e., they can be regarded as medium-sized compound collections.
The compound collections were screened for inhibitors of seven different protein-tyrosine and dual-specificity phosphatases of different origin and which modulate different biological processes. Compound collection 1 with an N-heterocyclic scaffold yielded potent inhibitors of the tyrosine phosphatase VE-PTP at a hit rate of 1.57%. This phosphatase is involved in the regulation of VE-cadherin (14) and angiogenesis. VE-PTP inhibitors have not been described before, and compound class 1 has not been described as being capable of phosphatase inhibition before. The most potent inhibitors display IC50 values in the low micromolar range (Table 1) and are at least 40-fold selective among the group of enzymes screened. These compounds may find application for the study of the biological function of VE-PTP by means of a chemical biological approach.
Compound collection 2 with an O-heterocyclic scaffold yielded potent inhibitors of the tyrosine phosphatases Shp-2 and PTP1B with a hit rate of 0.36%. Shp-2 is considered a target in the development of new antiinfectives, and inhibition of PTP1B is an actively pursued approach for the development of drugs against diabetes type II, obesity, and the metabolic syndrome (4, 5). The most potent inhibitors displayed IC50 values in the low micromolar range (Table 1) and were at least 20-fold selective. Members of compound class 2 have not been identified as phosphatase inhibitors before.
To provide an opportunity for the development of new NP-inspired compound collections, 354 isolated NPs additionally were chosen based on structural diversity and assayed for phosphatase inhibition. Surprisingly, three yohimbane alkaloids, a NP class that has not been linked to phosphatase inhibition before, were discovered as weak inhibitors of the dual-specificity phosphatase Cdc25A (Fig. 2). According to the most logical application of the SCONP tree, this result would suggest the synthesis of a compound collection embodying the pentacyclic yohimbane alkaloid framework as scaffold. However, because of the foreseeable complexity of this undertaking, as an alternative, brachiation along the indole branch of the SCONP tree toward simpler yet biologically prevalidated scaffolds was considered to generate hypotheses for the synthesis of compound collections with less complex scaffold structures yet retained biological activity. This strategy had proven to be valid and efficient before for one example stemming from the carbocycle branch of the tree (8). Successful application of brachiation through the N-heterocycle branch of the SCONP tree has not been demonstrated before. Brachiation from the pentacyclic scaffold 7 toward smaller scaffolds initially led to the tetracyclic indoloquinolizidine scaffold 8 (Fig. 3). A collection of 450 compounds embodying this scaffold was synthesized on the solid phase by means of a six- to eight-step synthesis sequence employing as key steps a (i) Lewis acid mediated Mannich–Michael reaction between immobilized d- or l-tryptophan imines 13 and electron rich silyloxy dienes 14, (ii) subsequent acid- or phosgene-mediated cyclization of enaminones 15 to tetracyclic ketones 16 and vinyl chlorides 17 followed by (iii) derivatization, and (iv) base- or acid-mediated release of indoloquinolizidines 17, 19, 21, and 22 from the solid phase (Fig. 4A). The target compounds were obtained in high overall yield, and isomer mixtures were separated by HPLC to >99% purity for subsequent screening. The screen revealed two compounds with potency for Cdc25A inhibition similar to the guiding NPs. Gratifyingly, the NP-inspired collection also yielded potent inhibitors of the tyrosine phosphatase MptpB at a hit rate of 2.4%. The most potent inhibitors displayed IC50 values in the low micromolar range and were selective for MptpB. These results validated the brachiation approach for the example under investigation.
Further brachiation toward structurally less complex compound classes led to tricyclic and bicyclic indole derivatives 9 and 10. Accordingly, a collection of 188 compounds containing examples for both scaffold structures and synthesized as described earlier (19, 20) was investigated for inhibition of the seven phosphatases in the screen. Again, two compounds with potency for Cdc25A inhibition similar to the guiding NPs and the tetracyclic compounds were identified. In addition, the NP-inspired collection yielded seven compounds with IC50 values for inhibition of MptpB in the nanomolar range. Six compounds were at least 100-fold selective for MptpB (compounds 9a, 9b, 10a, 10b, 10d, and 10e; Table 6). MptpB is considered a target in the search for new antimycobacterial drugs, but inhibitors for this enzyme have not been described before. Neither the tetracyclic indoloquinolizidines nor the 2,3-disubstituted indole derivatives identified in the screen have been described as inhibitors of protein phosphatases before.
Together with the first example from the carbocycle branch of the SCONP tree (8), the successful brachiation through the N-heterocycle branch of the NP tree indicates that the brachiation approach may be viable for the identification of new inhibitor classes with reduced structural complexity. However, although this finding is supportive, generality cannot be and is not claimed based on two successful examples.
Our results demonstrate that the application of BIOS and SCONP, which build on biological prevalidation and relevance to nature, is a valid and efficient approach to the discovery of unprecedented protein phosphatase inhibitors. The four NP-derived and -inspired compound collections investigated in this study yielded four new classes of inhibitors for four different phosphatases. This finding together with earlier results (8, 9) lends proof to the notion that compound collections obtained by means of BIOS may yield high hit rates at comparatively small library size.
The efficient synthesis of the indoloquinolizidine collection furthermore demonstrates that the stereoselective synthesis of NP-inspired medium-sized compound collections is within the reach of current synthesis methodology (30). In our experience for such multistep synthesis sequences, solid-phase methodology is advantageous because it allows for efficient removal of the reagents used in the various steps and thereby facilitates purification of the final products. These arguments favorably counterbalance the development effort typically required to establish such multistep sequences on the solid phase.
Conclusion
BIOS employs the core structures of compound classes that are relevant to nature as guiding scaffolds for the design and synthesis of compound collections for chemical biology and medicinal chemistry research. In this approach the scaffolds of NP classes and their structural relationships provide important hypothesis- and idea-generating sources for the design of NP-derived and -inspired compound collections.
Based on this approach, we have discovered four inhibitor classes for four different tyrosine and dual-specificity phosphatases. The medium-sized compound collections synthesized and used yielded potent and selective phosphatase inhibitors with high hit rates at comparably small library size. The use of both NP-derived and -inspired compound collections proved to be efficient. The identification of structurally less complex inhibitors by means of brachiation from the pentacyclic yohimbane scaffold to the scaffolds of substituted indoles via indoloquinolizidines provides a convincing example and argument for the notion that brachiation along the N-heterocyclic branches of the SCONP tree is a viable approach to phosphatase inhibitor development. The inhibitor classes discovered and the identification of the first inhibitors for two phosphatases open up new avenues of research and opportunity for the study of the biological role of these important enzymes and possibly the development of new drug candidates.
Materials and Methods
Synthesis of Compound Collection 1.
Cytisine derivatives were synthesized by means of the synthesis pathways shown in Scheme 1 of Supporting Materials and Methods. Cytisine was obtained from Zhejiang Wonderful Pharma & Chem Co. (Zhejiang, China). All compounds are available from AnalytiCon Discovery.
Synthesis of Compound Collection 2.
Compound collection 2 was synthesized according to the synthesis pathways shown for representative examples in the five schemes summarized in Scheme 2 of Supporting Materials and Methods. The furofuran starting materials were obtained from GLYCON Biochemicals (Luckenwalde, Germany). All compounds are available from AnalytiCon Discovery.
Synthesis of the Indoloquinolizidine Collection.
Acid-mediated cyclization on HMBA-amino Merrifield (AM) support.
The resin (200 mg) was suspended in a solution of CH2Cl2/TMSCl/TFA (3:1:0.5; 10 ml) and stirred at room temperature for 30 min. The resin was filtered; successively washed with CH2Cl2 (three times), methanol (three times), N,N-dimethylformamide (three times), and CH2Cl2 (three times); and dried in vacuo. Final cleavage of the product from the HMBA-AM resin was achieved by treatment with 0.25 M NaOMe in 1:1 methanol/dioxane solution at 50°C for 12 h. The remaining residue obtained after EtOAc extraction and removal of the solvent in vacuo was purified by means of RP-HPLC. All other tetracyclic ketones were prepared by analogy.
Phosgene-mediated cyclization on Wang resin.
The resin (320 mg) was suspended in a solution of 3 ml of CH2Cl2 and 1.4 ml of a solution of phosgene in toluene (40%), and 0.7 ml of TMSCl was added. The mixture was shaken for 2 h at room temperature. Resin 18 was filtered and washed three times with MeOH and CH2Cl2 and dried for 2 h in vacuo. Release from Wang resin 18 was achieved by stirring in a mixture 10 ml of TFA/water 95:5 overnight. After evaporation of the solvent a solution of 78.4 mg (0.41 mmol, 5 eq), EDC·HCl [EDC is N-dimethylaminopropyl-N′-ethylcarbodiimide] in 3 ml of pyridine/MeOH/CH2Cl2 1:1:1 was added to the crude mixture, which was stirred for 12 h. Water was added to the reaction mixture. After extraction with CH2Cl2 and evaporation of the solvent, the residue was subjected to RP-HPLC.
All tetracyclic vinyl chlorides were prepared according to this procedure or without esterification in solution. The diastereomeric mixtures were separated by HPLC, and the determination of isomer ratios was performed gravimetrically. Determination of the relative configuration was achieved by nuclear Overhauser effect-difference spectroscopy and by means of characteristic shifts in the 1H-NMR spectra due to the Perlin effect.
Protein phosphatase assays.
All enzyme assays were performed by means of an automated system consisting of a Zymark SciClone ALH 500 in conjunction with a Twister II and a BioTek PowerWave 340 reader. The reaction volume was 10 μl. The reaction was started by the addition of 5 μl of p-nitrophenyl phosphate to 5 μl of a solution containing the respective enzymes that had been preincubated for 10–15 min with different concentrations from twofold dilution series of inhibitors.
Reaction velocity was determined from the slope of the absorbance change at 405 nm and related to control values in the absence of the inhibitor. IC50 values were calculated from linear extrapolations of reaction velocity as a function of the logarithmic of concentration.
For further details, see Supporting Materials and Methods.
Supplementary Material
Acknowledgments
This work was supported by the Deutsche Forschungsgemeinschaft, the Fonds der Chemischen Industrie, the Max Planck Gesellschaft, and the state of Hessen through a grant to the Center for Biomolecular Magnetic Resonance. A.N.-M. is a Novartis research fellow. I.R.-C. is grateful to the Humboldt Foundation for a research scholarship.
Abbreviation
- BIOS
biology-oriented synthesis
- HMBA
4-(hydroxymethyl)benzoic acid amide
- NP
natural product
- SAR
structure–activity relationship
- SCONP
structural classification of natural products
- TFA
trifluoroacetic acid
- TMSCl
trimethylsilylchloride.
Footnotes
Conflict of interest statement: No conflicts declared.
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Hunter T. Cell. 2000;100:113–127. doi: 10.1016/s0092-8674(00)81688-8. [DOI] [PubMed] [Google Scholar]
- 2.Alonso A., Sasin J., Bottini N., Freidberg I., Osterman A., Godzik A., Hunter T., Dixon J., Mustelin T. Cell. 2004;117:699–711. doi: 10.1016/j.cell.2004.05.018. [DOI] [PubMed] [Google Scholar]
- 3.Hinterding K., Alonso-Díaz D., Waldmann H. Angew. Chem. Int. Ed. 1998;37:688–749. doi: 10.1002/(SICI)1521-3773(19980403)37:6<688::AID-ANIE688>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- 4.Bialy L., Waldmann H. Angew. Chem. Int. Ed. 2005;44:2–27. doi: 10.1002/anie.200461517. [DOI] [PubMed] [Google Scholar]
- 5.Johnson T. O., Ermolieff J., Jirousek M. R. Nat. Rev. Drug Discovery. 2002;1:696–709. doi: 10.1038/nrd895. [DOI] [PubMed] [Google Scholar]
- 6.Lyon M. A., Ducruet A. P., Wipf P., Lazo J. S. Nat. Rev. Drug Discovery. 2002;1:961–976. doi: 10.1038/nrd963. [DOI] [PubMed] [Google Scholar]
- 7.Koch M. A., Wittenberg L.-O., Basu S., Jeyaraj D. A., Gourzoulidou E., Reinecke K., Odermatt A., Waldmann H. Proc. Natl. Acad. Sci. USA. 2004;101:16721–16726. doi: 10.1073/pnas.0404719101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Koch M. A., Schuffenhauer A., Scheck M., Casaulta M., Odermatt A., Waldmann H. Proc. Natl. Acad. Sci. USA. 2005;102:17272–17277. doi: 10.1073/pnas.0503647102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Koch M. A., Waldmann H. Drug Discovery Today. 2005;10:471–482. doi: 10.1016/S1359-6446(05)03419-7. [DOI] [PubMed] [Google Scholar]
- 10.Koul A., Herget T., Klebl B., Ullrich A. Nat. Rev. Microbiol. 2004;2:189–202. doi: 10.1038/nrmicro840. [DOI] [PubMed] [Google Scholar]
- 11.Pathak M. K., Yi T. J. Immunol. 2001;167:3391–3397. doi: 10.4049/jimmunol.167.6.3391. [DOI] [PubMed] [Google Scholar]
- 12.Nawroth R., Poell G., Ranft A., Kloep S., Samulowitz U., Fachinger G., Golding M., Shima D. T., Deutsch U., Vestweber D. EMBO J. 2002;21:4885–4895. doi: 10.1093/emboj/cdf497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fachinger G., Deutsch U., Risau W. Oncogene. 1999;18:5948–5953. doi: 10.1038/sj.onc.1202992. [DOI] [PubMed] [Google Scholar]
- 14.Bäumer S., Keller L., Holtmann A., Funke R., August B., Gamp A., Wolburg H., Wolburg-Buchholz K., Deutsch U., Vestweber D. Blood. doi: 10.1182/blood-2006-01-0141. in press. [DOI] [PubMed] [Google Scholar]
- 15.Galahtinov K., Lee A. K., Eckstein J., Draetta G., Meckler J., Loda M., Beach D. Science. 1995;269:1575–1577. doi: 10.1126/science.7667636. [DOI] [PubMed] [Google Scholar]
- 16.Alonso A., Saxena M., Williams S., Mustelin T. J. Biol. Chem. 2001;276:4766–4771. doi: 10.1074/jbc.M006497200. [DOI] [PubMed] [Google Scholar]
- 17.Anderson D. J., Arneric S. P. Eur. J. Pharmacol. 1994;253:261–267. doi: 10.1016/0014-2999(94)90200-3. [DOI] [PubMed] [Google Scholar]
- 18.Kikuchi H., Saito Y., Komiya J., Takaya Y., Honma S., Nakahata N., Ito A., Oshima Y. J. Org. Chem. 2001;66:6982–6987. doi: 10.1021/jo015657x. [DOI] [PubMed] [Google Scholar]
- 19.Rosenbaum C., Baumhof P., Mazitschek R., Müller O., Giannis A., Waldmann H. Angew. Chem. Int. Ed. 2004;43:224–228. doi: 10.1002/anie.200352582. [DOI] [PubMed] [Google Scholar]
- 20.Rosenbaum C., Katzka C., Marzinzik A., Waldmann H. Chem. Commun. 2003:1822–1823. doi: 10.1039/b305497g. [DOI] [PubMed] [Google Scholar]
- 21.Waldmann H., Braun M., Drager M. Angew. Chem. Int. Ed. 1990;29:1468–1471. [Google Scholar]
- 22.Waldmann H., Braun M. J. Org. Chem. 1992;57:4444–4451. [Google Scholar]
- 23.Yang L., Guo L. Tetrahedron Lett. 1996;37:5041–5044. [Google Scholar]
- 24.Mohan R., Chou Y.-L., Morrissey M. M. Tetrahedron Lett. 1996;37:3963–3966. [Google Scholar]
- 25.Wang H., Ganesan A. Org. Lett. 1999;1:1647–1649. [Google Scholar]
- 26.Fantauzzi P. P., Yager K. M. Tetrahedron Lett. 1998;39:1291–1294. [Google Scholar]
- 27.Dondas H. A., Grigg R., MacLachlan W. S., MacPherson D. T., Markandu J., Sridharan V., Suganthan S. Tetrahedron Lett. 2000;41:967–970. [Google Scholar]
- 28.Lock R., Waldmann H. Chem. Eur. J. 1997;3:143–151. [Google Scholar]
- 29.Burke M. D., Schreiber S. L. Angew. Chem. Int. Ed. 2004;43:46–58. doi: 10.1002/anie.200300626. [DOI] [PubMed] [Google Scholar]
- 30.Breinbauer R., Vetter I., Waldmann H. Angew. Chem. Int. Ed. 2002;41:2878–2890. doi: 10.1002/1521-3773(20020816)41:16<2878::AID-ANIE2878>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.