

Abstract
Biological clocks with a period of ≈24 h (circadian) exist in most organisms and time a variety of functions, including sleep–wake cycles, hormone release, bioluminescence, and core body temperature fluctuations. Much of our understanding of the clock mechanism comes from the identification of specific mutations that affect circadian behavior. A widely studied mutation in casein kinase I (CKI), the CKIεtau mutant, has been shown to cause a loss of kinase function in vitro, but it has been difficult to reconcile this loss of function with the current model of circadian clock function. Here we show that mathematical modeling predicts the opposite, that the kinase mutant CKIεtau increases kinase activity, and we verify this prediction experimentally. CKIεtau is a highly specific gain-of-function mutation that increases the in vivo phosphorylation and degradation of the circadian regulators PER1 and PER2. These findings experimentally validate a mathematical modeling approach to a complex biological function, clarify the role of CKI in the clock, and demonstrate that a specific mutation can be both a gain and a loss of function depending on the substrate.
Keywords: kinase, systems biology, phosphorylation, PER, degradation
Circadian rhythms govern key physiologic processes including sleep–wake cycles; glucose, lipid, and drug metabolism; heart rate; stress and growth hormones; and immunity, as well as basic cellular processes such as timing of the cell division cycle (1–6). The disruption of circadian rhythm causes significant physiologic stress, is frequently experienced in jet lag and night-shift work, and has been linked to bipolar disorder (7). Thus, circadian regulation of physiology has important consequences for health. A detailed quantitative model that makes clear, testable, and accurate predictions about the clock and how we may manipulate it can therefore have benefits for human health.
Much of our understanding of clock components and their interactions began with the identification of mutations that affect circadian behavior (8, 9). In mammals, the original and most extensively studied circadian rhythm mutation is the semidominant tau, first described in 1988. Hamsters with this mutation show phase-advanced activity and have a circadian period of 20 h when homozygous mutant animals are isolated from time cues (9). This tau mutation has been identified as a missense mutation within the substrate recognition site of casein kinase Iε (denoted CKIεtau) (10). CKIε and the closely related CKIδ are widely expressed serine–threonine protein kinases implicated in development, circadian rhythms, and DNA metabolism (11). When tested in vitro on multiple substrates, CKIεtau was shown to have a much reduced overall catalytic activity (10, 12, 13). This partial loss-of-function mutation and its phenotype have been difficult to reconcile with our current understanding of the molecular feedback loop that governs timing in mammalian cells (13) and recent empirical observations on clock function (14–16). For example, Dey et al. (15) reported that PER proteins disappeared from the nucleus more rapidly in the tau hamster than in WT controls.
The mammalian clock is established by a transcription–translation negative feedback loop, which establishes stable ≈24-h oscillations in transcription from circadian promoters. These oscillations are then reinforced by additional feedback loops. The core clock is regulated by the negative feedback of PERIOD (PER1 and PER2) and cryptochrome (CRY1 and CRY2) proteins on the transcription activation by heterodimeric transcription factors CLOCK and BMAL1. CLOCK and BMAL1 drive expression both of circadian output genes and, importantly, their own negative regulators PER1, PER2, CRY1, and CRY2. Within this molecular feedback loop, CKI binds to and phosphorylates PER1 and PER2 to (i) promote the degradation of the PER1 and PER2 proteins and (ii) promote or block the nuclear import of the PER1 but not PER2 proteins (14, 17–21).
From these findings, potential molecular explanations proposed for the fast period phenotype of the tau mutation are that (i) decreased phosphorylation causes decreased degradation of the PER1 and PER2, which causes the proteins to accumulate faster in the nucleus of the cell; or (ii) decreased inhibition of nuclear import causes PER1 to accumulate in the nucleus faster. Although these models are intuitive, they may not be correct. For example, increased PERIOD protein accumulation might speed up the clock when PERIOD protein is scarce, but alternatively it could slow down the clock when PERIOD levels are decreasing. It is unclear from qualitative arguments which will have a larger effect.
The field of circadian rhythms has long benefited from modeling studies (e.g., ref. 22). There have been many recent advances in models of the molecular biology of circadian rhythms (23–27). To test the role of the tau mutation in the mammalian circadian clock we simulated the Forger–Peskin model and modifications to this model because it directly and accurately represents much of the detailed experimental data on the mammalian circadian clock (28). Our quantitative analysis predicts that, contrary to published in vitro results, the kinase mutant CKIεtau is a gain-of-function mutation, and we verify this prediction experimentally. Our data show that CKIεtau is a highly specific gain-of-function mutation that increases the in vivo phosphorylation and degradation of the circadian regulators PER1 and PER2. These findings experimentally validate a mathematical modeling approach to a complex biological function and demonstrate that a specific mutation can be both a gain and loss of function depending on the substrate.
Results: Mathematical Modeling Predicts a Gain of Function for CKIεtau
To move beyond qualitative arguments on the effect of changes in kinase activity on the clock, we simulated the Forger–Peskin model of the mammalian circadian clock (28). In this model three possible phosphorylation states of the PERIOD proteins are mediated by CKIε: no phosphorylation, primary phosphorylation, and secondary phosphorylation (see Fig. 1). Primary phosphorylation decreases PERIOD stability and promotes nuclear localization and PER-CRY binding. In the model, secondary phosphorylation decreases nuclear entry of PER1. When we decreased the rate of primary phosphorylation of PER1 or PER2 we consistently found that the model predicted a longer period. For example, a 50% decrease in the rate of the primary phosphorylation of PER1 and PER2 lengthened the period of the model by 0.13 and 2.43 h, respectively (for further simulations see Fig. 6, which is published as supporting information on the PNAS web site). Decreasing the rate of secondary phosphorylation of PER1 also lengthened the period (by 0.03 h for a 50% decrease; see Fig. 6). This quantitative model predicts that, contrary to in vitro data, the tau mutation must cause increased rather than decreased PER phosphorylation as the explanation for a short period phenotype. This prediction of the Forger–Peskin model is in direct conflict with the observed in vitro decrease in CKIε kinase activity caused by the tau mutation.
Fig. 1.

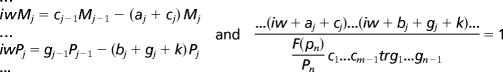 |
We next verified that the predictions of this model were not specific to certain phosphorylation assumptions of the model. Decreasing by 50% the overall rate of degradation, nuclear import of the PER proteins, or nuclear export of the PER proteins or decreasing the rate at which the PER proteins bound to the CRY proteins all lead to a lengthened period (by 0.28, 0.26, 1.37, and 0.16 h, respectively; see Fig. 6). We changed the order of phosphorylation so that all phosphorylation events could occur simultaneously, and again no candidate phosphorylation event that speeds up the clock in the face of a CKI loss-of-function mutation could be found (data not shown). We also considered the possibility that decreased phosphorylation of BMAL1 could decrease its activation of circadian E-box promoters (29). However, decreased BMAL1 activation of the PER1, PER2, CRY1, and CRY2 promoters also lengthens the period (50% decrease increases the period by 0.12 h; see Fig. 6). To further check these predictions, we analytically studied a broad class of models and showed that there is no possible explanation for decreased phosphorylation shortening the period for the class of models considered (see Fig. 1). This analysis, based on Fourier analysis, argues that the predictions of the Forger–Peskin model are based on an underlying “design principle” of the circadian clock and not based on a specific model assumption. In particular, this analysis predicts that decreasing the rate of nuclear localization or decreasing the rate of nuclear export both increase the period of the clock. Thus, there is a clear conflict between the experimental data and model predictions.
If the tau mutation caused a gain of function, and thereby increased the rate of phosphorylation of sites on the PERIOD proteins that are essential to clock function, a shorter period would be expected, and this conflict would be resolved. We therefore tested CKIε function in cell-based assays using NIH 3T3 cells, a cell type known to sustain circadian rhythms in culture (30). PER1 or PER2 was expressed alone or with WT CKIε, CKIεtau, or kinase-inactive CKIε(K38A). When expressed alone, PER1 and PER2 protein levels remain largely unchanged over the course of the assay. Expression of WT CKIε modestly shortened the half-life, as previously reported, whereas CKIεtau markedly decreased PER abundance (Fig. 2A) and shortened the PER1 and PER2 half-lives (Fig. 2 B and C). Exogenous epitope-tagged kinase expression was lower than endogenous kinase levels, so the effects seen are not due to nonphysiologic levels of mutant kinases (Fig. 2B). The kinase loss-of-function mutant CKIε(K38A) had no effect on PER abundance and half-life, notably distinct from the effect of CKIεtau. These results indicate that the tau mutation can act as a gain of function that stimulates PER protein degradation.
Fig. 2.

CKIεtau increases the degradation of PER1 and PER2. (A) Coexpression with CKIεtau reduces the abundance of PER1 and PER2 proteins. Myc epitope-tagged mPER1 or mPER2 was coexpressed with HA epitope-tagged CKIε WT, tau, or K38A mutants in 3T3 cells for 20 h as indicated. The abundance of PER proteins was then determined by immunoblotting (IB) with anti-myc antibodies. (B and C) CKIεtau increases the degradation rate of PER proteins. Twenty hours after transfection, cells were treated with 10 μg/ml cycloheximide (CHX) for 0, 1.5, 3, or 5 h. The abundance of PER1 or PER2 in the cell lysates was determined by immunoblotting. The data shown are representative results of at least three separate experiments. In B, the asterisk indicates HA-tagged CKIε. CKIεtau expression increased the degradation of PER1 3.4-fold and the degradation of PER2 2.9-fold compared with no kinase (P < 0.05 for both) and 1.4-fold and 1.6-fold compared with WT CKIε (P = 0.3 for PER1 and 0.0033 for PER2).
All experiments were quantified and normalized to loading controls. We then calculated the average rate of degradation for each sample using the formula −log(fraction left with respect to time 0)/(time elapsed). Without any kinase transfection the average rate of degradation of PER1 and PER2 was 0.019 h−1 and 0.063 h−1, respectively. When WT kinase was coexpressed, the average rates of degradation of PER1 and PER2 were 0.084 and 0.12 h−1 (half-life of 11.9 and 8.58 h). When CKIεtau was coexpressed with PER1 or PER2, the average rates of degradation were 0.114 and 0.183 h−1 (half-life of 8.72 and 5.45 h). In contrast, when kinase-inactive CKIε(K38A) was coexpressed, the average rates of degradation were negligibly different (no change in PER1 and 0.064 h change for PER2). By using a one-sided, unpaired t test the tau mutant had a statistically significant (P < 0.05) higher average rate of degradation than all other cases considered, except when compared on PER2 with the WT kinase [where the confidence (P = 0.176) was slightly lower]. Thus, we find that the tau mutation increases the degradation rate of PER1 by 37% and of PER2 by 57% when compared with expression of WT kinase.
To extend these findings we examined the downstream consequences of PER phosphorylation. CKIε regulates PER1 and PER2 stability by promoting their interaction with the SCFβ-TrCP E3 ubiquitin ligase (14, 21), which leads to their degradation by the ubiquitin/proteasome pathway. When the proteasome proteolytic activity was inhibited with MG132, the CKIεtau-dependent degradation of PER1 was nearly completely abrogated (Fig. 3A). Consistent with these results, a dominant-negative form of β-TrCP also reversed the CKIεtau-dependent degradation of PER1 and PER2 (31) (Fig. 3B and Fig. 7A, which is published as supporting information on the PNAS web site). Taken together, these results confirm that CKIεtau accelerates the previously demonstrated β-TrCP-dependent proteasomal degradation of PER1 and PER2 (14).
Fig. 3.

CKIεtau increases degradation of PER proteins through the ubiquitin–proteasome pathway. (A) Inhibition of the proteasome prevents CKIεtau-induced PER1 degradation. NIH 3T3 cells cotransfected with myc-PER1, HA-CKIε, and CKIεtau were pretreated with vehicle or the proteasome inhibitor MG132 (30 μM) for 5 h and then treated with cycloheximide and analyzed as in Fig. 2. (B) Dominant-negative β-TrCP blocks the CKIεtau-induced degradation of PER1 and PER2. Myc-tagged PER1 was coexpressed with HA-tagged CKIεtau or CKIε(K38A) and the dominant-negative β-TrCP(ΔFbox) as indicated. The abundance of PER was visualized by immunoblotting (IB). Each study was performed at least twice, and in each repetition, MG-132 and dominant-negative β-TrCP inhibited degradation by at least 50%.
CKIεtau has been demonstrated to be a partial loss-of-function mutation (10). This finding is at odds with the data in Fig. 2 that the tau mutation enhances the phosphorylation-dependent degradation of PER. We therefore confirmed that CKIεtau expressed in cells is less active than WT CKIε on the nonspecific substrate casein (Fig. 7B). To test whether the CKIεtau mutation specifically increased the cellular phosphorylation of the PER proteins, the incorporation of [32P]orthophosphate into PER2 in cells coexpressing WT or mutant CKIε was assessed. As Fig. 4A shows, expression of CKIεtau caused a marked increase in PER2 phosphorylation in cells compared with WT CKIε, whereas the catalytically inactive CKIε(K38A) had little effect on PER2 phosphorylation (Fig. 4A). To test whether in cells CKIεtau is a general gain of function, its effect on BMAL1 phosphorylation was assessed. We have previously reported that BMAL1 is a cellular substrate of CKIε and that expression of CKIε(K38A) decreases cellular BMAL1 phosphorylation (29). CKIεtau, like CKIε(K38A), inhibits the cellular phosphorylation of BMAL1 in cells (Fig. 4B). Thus, CKIεtau appears to be a PER-specific gain-of-function mutation in vivo. To extend this result, we tested whether CKIεtau was a loss or gain of function in vivo in a distinct signaling pathway (Fig. 7C). CKIε is a positive regulator of the Wnt signaling pathway and activates β-catenin- and TCF/Lef-1-dependent transcription (32–34). WT CKIε caused a 2.5-fold increase in β-catenin-dependent transcription. Neither kinase-inactive CKIε(K38A) nor CKIεtau was able to activate the Wnt pathway. This finding demonstrates that the CKIεtau mutation produces a highly selective and substrate-specific gain of function. Consistent with this finding, if CKIεtau was a gain of function in the Wnt pathway, significant developmental abnormalities in the tau hamster would have been expected.
Fig. 4.

CKIεtau is a PER-specific gain of function in cells. (A) CKIεtau is a gain of function on PER2 in an in vivo kinase assay. Myc-PER2 and HA-CKI (as indicated) were coexpressed in NIH 3T3 cells. In vivo phosphorylation of PER2 was assessed by 3 h of metabolic labeling with [32P]orthophosphoric acid (150 μCi per well). Myc-PER2 was immunopurified and analyzed by SDS/PAGE, transferred to a poly(vinylidene difluoride) membrane, autoradiographed, and immunoblotted (IB). Phosphorylation-stimulated proteolysis was inhibited by inclusion of 30 μM MG-132 starting 2 h before labeling. Equal expression of WT and mutant kinases was verified in parallel nonradioactive experiments. This experiment was repeated at least three times with similar results. (B) CKIεtau is a loss of function on the intracellular circadian substrate BMAL1. Myc epitope-tagged BMAL1 was expressed in 3T3 cells alone or with the indicated CKI constructs. Metabolic labeling was performed as above and as previously described (29), with the inclusion of the CKI inhibitor IC261 where indicated. The CKIεtau mutant represses BMAL1 phosphorylation, similar to the effect of CKIε(K38A) and IC261. (C) CKIεtau phosphorylates a specific site(s) on PER2. PER2 was metabolically labeled with [32P]orthophosphate as above, except that 1 mCi per well was used. Immunopurified myc-PER2 was analyzed by combined trypsin/chymotrypsin digest, and two-dimensional phosphopeptide mapping was performed as previously described (44). CKIε expression enhanced phosphorylation of peptides 1, 2, and 4, and CKIεtau expression specifically increased phosphorylation of peptides 3, 4, and 5.
We considered two potential mechanisms for how the tau mutation could cause increased in vivo phosphorylation of PER2. CKIεtau may increase phosphorylation of all of the CKI sites in the PER proteins, or it may selectively phosphorylate a subset of CKI sites in PER that specifically regulates its stability. To differentiate between these possibilities, the cellular phosphorylation sites on PER2 (Fig. 4C) and PER1 (Fig. 7D) were examined by two-dimensional phosphopeptide mapping after metabolic labeling of cells. Expression of WT CKIε increases the prominence of a number of phosphopeptides (Fig. 4C Center, 1, 2, and 4), consistent with increased phosphorylation of all CKI sites within PER2. Coexpression of CKIεtau, however, increases the relative intensity of only three specific phosphopeptides (designated peptides 3, 4, and 5 in Fig. 4C). A similar increase in a limited number of phosphopeptides was seen when CKIεtau was expressed with PER1 (Fig. 7D). We conclude that the tau mutation causes a site-specific increase in PER protein phosphorylation at sites that stimulate degradation.
Mutations in two other members of the CKI family also cause short circadian periods and a decrease in vitro CKI kinase activity (12, 35, 36). Our detailed quantitative model suggests, however, that all short period mutations in CKI family members will produce a gain of function on circadian substrates. To assess this hypothesis, the human short-period mutant CKIδ(T44A) and the Drosophila double-time short-period mutant P47S (dbtS) (engineered into CKIε) were coexpressed with PER1 or PER2. Consistent with the model predictions and similar to the effect of CKIεtau, both of these short-period mutants in CKI showed increased activity on the PER proteins, as reflected in a kinase-dependent decrease in electrophoretic mobility (Fig. 5). These results are consistent with an accelerated mobility shift of Drosophila PER seen with coexpression of dbtS in S2 cells (16). Coexpression of the kinase-inactive K38A mutant or an engineered Drosophila long-period mutant dbtL [CKIε(M80I)] did not lead to a consistent change in PER electrophoretic mobility. Thus, all short-period mutations in CKI appear to enhance the phosphorylation of PERIOD proteins in cells. But notably, unlike CKIεtau, not all of the other short-period mutations in CKI accelerated the degradation of the PER proteins. We speculate that these gain-of-function mutations may increase phosphorylation of PERIOD at distinct regulatory sites.
Fig. 5.

Short-period mutations are active on PER proteins in vivo. PER1 (Left) or PER2 (Right) was expressed with the indicated kinase in 3T3 cells, and the effect on electrophoretic mobility was assessed by SDS/PAGE and immunoblotting (IB). The CKIε(T44A) mutation was in human CKIε, whereas the Drosophila dbtS and dbtL mutations were made in the same highly conserved residues in human CKIε. Protein expression was confirmed by immunoblot using the Myc or HA epitope tags.
Discussion
Mechanistic models of signaling pathways rely heavily on in vitro and cell-based analysis of the behavior of molecular components. One goal of these studies is to predict the behavior of these pathways in complex systems. Circadian rhythms have fascinated biologists and mathematicians alike because of the precise self-regulating oscillator and the availability of a number of molecularly characterized circadian rhythms mutants. A mathematical model that accurately describes many aspects of circadian behavior (23, 28) predicts the output of the clock but made a nonintuitive prediction about the nature of the tau mutation in CKIε that is in conflict with published results that CKIεtau is a partial loss-of-function mutation that accelerates the clock. The quantitative model makes the opposite prediction: that all loss-of-function mutations in CKI can only slow, not accelerate, the circadian period. This prediction was tested in mammalian cells, and we found that, contrary to what was observed in vitro, the three known short-period circadian rhythm mutations, in hamster, human, and Drosophila CKI, efficiently increase phosphorylation of a biologically critical substrate in cells. In cells we find that CKIεtau is a gain-of-function mutation that generates a short period by increasing the rate of phosphorylation of specific residues on the PERIOD proteins, resulting in their more rapid degradation. Consistent with these data, pharmacologic inhibition of CKI activity or the proteasome also increases circadian period length (14). This finding supports the conclusion that nonintuitive predictions provided by quantitative analysis can provide important insights into basic biological mechanisms. It also provides a cautionary tale regarding extrapolation of in vitro phosphorylation sites to in vivo activities.
Importantly, CKIεtau acts as a gain-of-function mutation only when mPER is the substrate; when tested in the Wnt signaling pathway, CKIεtau behaves similarly to the loss-of-function mutation CKIεK38A. The fact that the tau mutation selectively increases phosphorylation of PER proteins may be under strong genetic selection. A broad gain-of-function mutation in CKI is unlikely to be tolerated because CKI regulates the activity of multiple pathways. If CKIεtau was a gain of function in the Wnt pathway, significant developmental abnormalities in the tau hamster would have been expected.
Why would the short-period CKI mutations produce a gain of function in the degradation pathway yet decrease overall PER phosphorylation in vitro? In vitro CKI appears to phosphorylate a large number of nonphysiologic sites. Supporting this finding, we and others have mapped multiple in vitro CKI phosphorylation sites on PER2 (data not shown and ref. 37); the great majority of these in vitro phosphorylation sites have upstream acidic residues, making them good artificial CKI sites. CKIε can efficiently phosphorylate substrates with acidic character (either acidic residues or phosphoryl groups) immediately upstream of the target site, presumably because of interactions with the positively charged arginine at the CKI position 178 (10, 13). However, these in vitro sites are nonconserved even between mammalian PER2s, strongly suggesting that they are irrelevant to cellular PER2 phosphorylation and function. If the CKIεtau mutation hinders phosphorylation of these nonphysiologic sites, that could explain why in vitro it has decreased overall activity. In vivo, the phosphorylation of irrelevant sites may be suppressed by (i) modification of CKI activity due to changes in its phosphorylation state and (ii) the fact that PER2 is in a multiprotein complex interacting with other circadian regulators, including cellular phosphatases. Our experimental data demonstrate that the tau mutation increases the rate of degradation of PER proteins by increasing site-specific phosphorylation, and the modeling studies indicate that this increase can be the cause of a faster circadian period. We note that our data do not exclude the possibility that in vivo the CKIεtau mutation could indirectly, rather than directly, increase the phosphorylation of PER proteins, for example, by decreasing the activity of a phosphatase or increasing the activity of another kinase.
Since its identification in 1988, the circadian period mutant tau has been a critical tool in dissecting physiological and molecular mechanisms of mammalian circadian biology (9). Modeling can lead to unexpected and testable predictions about mechanism. Here, modeling leads to the unexpected finding that, although the tau mutation creates a partial loss of function on standard substrates, it is a gain of function in the circadian clock that speeds up the clock by increasing the phosphorylation-dependent degradation of PER proteins. Despite the rich history of experimental results in circadian rhythms that were inspired by, interpreted through, and incorporated into mathematical models (see ref. 22), model-based experimental approaches are almost never used in studies of the genetic and molecular biology of circadian rhythms [a notable exception is Locke et al. (25)]. The current study makes clear that incorporating modeling as part of a systematic approach to circadian research can strengthen both experimental design and conceptual analysis. One goal of the study of human circadian rhythms is the development of tools to manipulate the clock to relieve human suffering, in the form of depression, insomnia, and chronic fatigue (36, 38). Inhibitors of CKI activity have been proposed as interventions in these disorders, but predictions of drug effects have been based on the models incorporating the hypothesis that kinase loss-of-function mutations speed up the clock. Our data suggest that CKI inhibitors may have effects on human circadian rhythms opposite from those predicted by previous models.
Materials and Methods
Plasmids and Antibodies.
The hemagglutinin (HA)-tagged CKIε was cloned in pCEP4 (Invitrogen) as previously described (39, 40). Myc epitope-tagged PER1 and PER2, myc-CKIδ, and βTrCP(ΔFbox) were subcloned into pCS2+MT as described (14, 19). The mutations K38A and R178C in CKIε and T44A in CKIδ were generated by using a QuikChange site-directed mutagenesis kit (Stratagene) according to the manufacturer’s directions. Anti-HA (F-7) and anti-myc (9E10) antibodies were obtained from Santa Cruz Biotechnology. Monoclonal anti-CKIε antibody was obtained from Transduction Laboratories, and the anti-actin (A2066) antibody was from Sigma.
Cell Culture, Maintenance, and Transient Transfection.
NIH 3T3 cells and HEK 293 STF cells were grown in DMEM or DMEM (Invitrogen) supplemented with 10% calf serum and 10% FBS, respectively, and antibiotics (100 units/ml penicillin and 100 μg/ml streptomycin). Cultures were maintained in a humidified incubator at 37°C and 5% CO2. When they reached 80–90% confluence, cells were transfected with Lipofectamine 2000 (Invitrogen) according to the manufacturer’s instructions. The total amount of transfected DNA was adjusted to 2 μg with empty vector when necessary.
Degradation Assays.
Approximately 20 h after transfection, cells were treated with cycloheximide (10 μg/ml; Biomol) diluted in DMEM. At the indicated times, cells were washed once with PBS and lysed in lysis buffer (150 mM NaCl/20 mM Hepes, pH 7.5/0.1% Nonidet P-40/1 mM EDTA/2 mM DTT) supplemented with 1× Complete protease inhibitor mixture (Roche Applied Science). The cells were then mechanically sheared, and the lysates were centrifuged at 16,000 × g for 10 min at 4°C. One hundred micrograms of total protein was resolved in SDS/PAGE, and then proteins were transferred to a nitrocellulose membrane (Hybond-C Extra; Amersham Pharmacia Biosciences) and further analyzed by immunoblotting with the indicated antibodies.
Lef1-Luciferase Reporter Gene Assay.
HEK 293 STF cells with an integrated SuperTop Flash TCF-luciferase reporter (41) plated in 12-well plates were transfected with 600 ng of pCEP4–4HA-CKI (WT), HA-CKIεTau, HA-CKIε(K38A), or empty vector along with 2 ng of a Renilla luciferase control plasmid. At 24 h after transfection, cells were lysed, and firefly and Renilla luciferase were measured with the Dual-Luciferase reporter assay system (Promega) and a microtiter plate luminometer (Dynatech Laboratories) according to the manufacturers’ instructions. To standardize for transfection efficiency, the firefly luciferase activities of all transfected cells were divided by the Renilla luciferase activities. The fold increase of Lef-1-dependent activity was defined by comparing the normalized level observed from cells transfected with empty vector pCEP4.
Immunoprecipitation Kinase Assay.
NIH 3T3 cells were transiently transfected with 1 μg of HA-tagged CKI expression constructs as indicated in the figure. Eighteen hours after transfection, cells were lysed in 200 μl of RIPA buffer (50 mM Hepes, pH 7.5/1% Nonidet P-40/0.1% SDS/150 mM NaCl/1 mM EDTA) supplemented with 1 mM DTT, 1× Complete protease inhibitor mixture, and the phosphatase inhibitors sodium fluoride (50 mM), β-glycerophosphate (20 mM), and sodium orthovanadate (20 mM). HA epitope-tagged proteins were immunoprecipitated from 300 μg of soluble cell-free extract with 0.5 μg of anti-HA antibody for 1 h. Immune complexes were then mixed with 25 μl of protein A–agarose beads (Invitrogen) for 30 min at 4°C with gentle agitation. The isolated beads were collected by gentle centrifugation at 185 × g and subsequently washed once each with lysis buffer and lysis buffer lacking both SDS and DTT, and then twice in protein storage buffer/kinase buffer (10% sucrose/50 mM Hepes, pH 7.2/15 mM NaCl/1 mM DTT/0.5 mM EDTA). For determining CKI activity, 1.2 μg of casein was added to the immunoprecipitates, and the reactions were incubated for 15 min at 37°C in kinase reaction mixture (30 mM Hepes, pH 7.5/1 mM MgCl2/0.1 mM DTT) containing 5 μCi (1 Ci = 37 GBq) of [γ-32P]ATP. The reactions were stopped by boiling samples in 25 μl of 5× SDS/PAGE loading buffer. Proteins were separated by 9% SDS/PAGE, transferred to a nitrocellulose membrane, and then visualized by autoradiography. Next, to confirm the equal presence of CKI in the immunoprecipitate, immunoblotting with anti-HA antibody was performed. Quantification of [32P]casein was performed by using a Storm 860 gel imaging system (Molecular Dynamics). Fold activation of the kinase was normalized to the CKIε expression levels estimated by immunoblotting analysis.
Metabolic Labeling of Cells.
3T3 cells were transiently transfected with 1.5 μg of mPER2 plasmid alone or with 0.5 μg of HA-CKIε, HA-CKIεTau, or HA-CKIε(K38A). Eighteen hours after transfection, cells were pretreated with 20 μM MG132 for 3 h and then labeled for 3 h with [32P]orthophosphoric acid (150 μCi/ml) in phosphate-free DMEM containing MG132. After labeling, cells were washed and then lysed in RIPA buffer supplemented with 2 mM DTT, 10 mM sodium fluoride, 10 mM β-glycerol phosphate, 1 mM NaVO4, and 1× Complete protease inhibitor mixture. Myc epitope-tagged PER2 was immunoprecipitated from 500 μg of lysate with 2 μg of anti-myc antibodies and protein A–agarose. After washing, bound proteins were eluted in 5× SDS/PAGE loading buffer and examined by SDS/PAGE and PhosphorImager analysis.
Phosphopeptide Mapping.
Metabolic labeling and immunoprecipitation were performed as described above, except that 2 mCi/ml was used. After resolving samples in SDS/PAGE, the gel was dried and examined by PhosphorImager analysis. The selected bands were then excised from the gel, rehydrated in 50 mM ammonium bicarbonate (pH 8.0), and digested with a mixture of trypsin/chymotrypsin. Peptides were separated as previously described (42, 43).
Supplementary Material
Acknowledgments
We thank Charles Peskin, Justin Blau, Nadeem Moghal, and Andrew Thorburn for discussions and review of the manuscript. This work was supported by National Institutes of Health (NIH) Grant R01 GM060387 (to D.M.V.), the Huntsman Cancer Institute, and NIH Grant P30CA42014. D.B.F. was supported by a Sloan Foundation fellowship in computational molecular biology.
Abbreviations
- CKI
casein kinase I
- HA
hemagglutinin.
Footnotes
Conflict of interest statement: No conflicts declared.
References
- 1.Lowrey P. L., Takahashi J. S. Annu. Rev. Genom. Hum. Genet. 2004;5:407–441. doi: 10.1146/annurev.genom.5.061903.175925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Reppert S. M., Weaver D. R. Nature. 2002;418:935–941. doi: 10.1038/nature00965. [DOI] [PubMed] [Google Scholar]
- 3.Chang D. C., Reppert S. M. Neuron. 2001;29:555–558. doi: 10.1016/s0896-6273(01)00230-6. [DOI] [PubMed] [Google Scholar]
- 4.Fu L., Pelicano H., Liu J., Huang P., Lee C. Cell. 2002;111:41–50. doi: 10.1016/s0092-8674(02)00961-3. [DOI] [PubMed] [Google Scholar]
- 5.Matsuo T., Yamaguchi S., Mitsui S., Emi A., Shimoda F., Okamura H. Science. 2003;302:255–259. doi: 10.1126/science.1086271. [DOI] [PubMed] [Google Scholar]
- 6.Turek F. W., Joshu C., Kohsaka A., Lin E., Ivanova G., McDearmon E., Laposky A., Losee-Olson S., Easton A., Jensen D. R., et al. Science. 2005;308:1043–1045. doi: 10.1126/science.1108750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mansour H. A., Monk T. H., Nimgaonkar V. L. Ann. Med. 2005;37:196–205. doi: 10.1080/07853890510007377. [DOI] [PubMed] [Google Scholar]
- 8.Konopka R. J., Benzer S. Proc. Natl. Acad. Sci. USA. 1971;68:2112–2116. doi: 10.1073/pnas.68.9.2112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ralph M. R., Menaker M. Science. 1988;241:1225–1227. doi: 10.1126/science.3413487. [DOI] [PubMed] [Google Scholar]
- 10.Lowrey P. L., Shimomura K., Antoch M. P., Yamazaki S., Zemenides P. D., Ralph M. R., Menaker M., Takahashi J. S. Science. 2000;288:483–492. doi: 10.1126/science.288.5465.483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vielhaber E. L., Virshup D. M. Int. Union Biochem. Mol. Biol. Life. 2001;51:273–278. [Google Scholar]
- 12.Preuss F., Fan J. Y., Kalive M., Bao S., Schuenemann E., Bjes E. S., Price J. L. Mol. Cell. Biol. 2004;24:886–898. doi: 10.1128/MCB.24.2.886-898.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Eide E. J., Virshup D. M. Chronobiol. Int. 2001;18:389–398. doi: 10.1081/cbi-100103963. [DOI] [PubMed] [Google Scholar]
- 14.Eide E., Woolf M., Kang H., Woolf P., Camacho F., Vielhaber E., Giovanni A., Virshup D. Mol. Cell. Biol. 2005;25:2795–2807. doi: 10.1128/MCB.25.7.2795-2807.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dey J., Carr A. J., Cagampang F. R., Semikhodskii A. S., Loudon A. S., Hastings M. H., Maywood E. S. J. Biol. Rhythms. 2005;20:99–110. doi: 10.1177/0748730404274264. [DOI] [PubMed] [Google Scholar]
- 16.Ko H. W., Jiang J., Edery I. Nature. 2002;420:673–678. doi: 10.1038/nature01272. [DOI] [PubMed] [Google Scholar]
- 17.Allada R., Emery P., Takahashi J. S., Rosbash M. Annu. Rev. Neurosci. 2001;24:1091–1119. doi: 10.1146/annurev.neuro.24.1.1091. [DOI] [PubMed] [Google Scholar]
- 18.Lowrey P. L., Takahashi J. S. Annu. Rev. Genet. 2000;34:533–562. doi: 10.1146/annurev.genet.34.1.533. [DOI] [PubMed] [Google Scholar]
- 19.Vielhaber E., Eide E., Rivers A., Gao Z.-H., Virshup D. M. Mol. Cell. Biol. 2000;20:4888–4899. doi: 10.1128/mcb.20.13.4888-4899.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Takano A., Isojima Y., Nagai K. J. Biol. Chem. 2004;279:32578–32585. doi: 10.1074/jbc.M403433200. [DOI] [PubMed] [Google Scholar]
- 21.Shirogane T., Jin J., Ang X. L., Harper J. W. J. Biol. Chem. 2005;280:26863–26872. doi: 10.1074/jbc.M502862200. [DOI] [PubMed] [Google Scholar]
- 22.Winfree A. T. The Geometry of Biological Time. Berlin: Springer; 1980. [Google Scholar]
- 23.Forger D. B., Peskin C. S. Proc. Natl. Acad. Sci. USA. 2005;102:321–324. doi: 10.1073/pnas.0408465102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Leloup J. C., Goldbeter A. Proc. Natl. Acad. Sci. USA. 2003;100:7051–7056. doi: 10.1073/pnas.1132112100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Locke J. C. W., Southern M. M., Kozma-Bognár L., Hibberd V., Brown P. E., Turner M. S., Millar A. J. Mol. Syst. Biol. 2005;1:E1–E9. doi: 10.1038/msb4100018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tyson J. J., Hong C. I., Thron C. D., Novak B. Biophys. J. 1999;77:2411–2417. doi: 10.1016/S0006-3495(99)77078-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Smolen P., Hardin P. E., Lo B. S., Baxter D. A., Byrne J. H. Biophys. J. 2004;86:2786–2802. doi: 10.1016/S0006-3495(04)74332-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Forger D. B., Peskin C. S. Proc. Natl. Acad. Sci. USA. 2003;100:14806–14811. doi: 10.1073/pnas.2036281100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Eide E. J., Vielhaber E. L., Hinz W. A., Virshup D. M. J. Biol. Chem. 2002;277:17248–17254. doi: 10.1074/jbc.M111466200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nagoshi E., Saini C., Bauer C., Laroche T., Naef F., Schibler U. Cell. 2004;119:693–705. doi: 10.1016/j.cell.2004.11.015. [DOI] [PubMed] [Google Scholar]
- 31.Hart M., Concordet J. P., Lassot I., Albert I., del los Santos R., Durand H., Perret C., Rubinfeld B., Margottin F., Benarous R., Polakis P. Curr. Biol. 1999;9:207–210. doi: 10.1016/s0960-9822(99)80091-8. [DOI] [PubMed] [Google Scholar]
- 32.Swiatek W., Tsai I. C., Klimowski L., Pepler A., Barnette J., Yost H. J., Virshup D. M. J. Biol. Chem. 2004;279:13011–13017. doi: 10.1074/jbc.M304682200. [DOI] [PubMed] [Google Scholar]
- 33.Peters J. M., McKay R. M., McKay J. P., Graff J. M. Nature. 1999;401:345–350. doi: 10.1038/43830. [DOI] [PubMed] [Google Scholar]
- 34.Sakanaka C., Leong P., Xu L., Harrison S. D., Williams L. T. Proc. Natl. Acad. Sci. USA. 1999;96:12548–12552. doi: 10.1073/pnas.96.22.12548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kloss B., Price J. L., Saez L., Blau J., Rothenfluh A., Wesley C. S., Young M. W. Cell. 1998;94:97–107. doi: 10.1016/s0092-8674(00)81225-8. [DOI] [PubMed] [Google Scholar]
- 36.Xu Y., Padiath Q. S., Shapiro R. E., Jones C. R., Wu S. C., Saigoh N., Saigoh K., Ptacek L. J., Fu Y. H. Nature. 2005;434:640–644. doi: 10.1038/nature03453. [DOI] [PubMed] [Google Scholar]
- 37.Schlosser A., Vanselow J. T., Kramer A. Anal. Chem. 2005;77:5243–5250. doi: 10.1021/ac050232m. [DOI] [PubMed] [Google Scholar]
- 38.Bunney W. E., Bunney B. G. Neuropsychopharmacology. 2000;22:335–345. doi: 10.1016/S0893-133X(99)00145-1. [DOI] [PubMed] [Google Scholar]
- 39.Rivers A., Gietzen K. F., Vielhaber E., Virshup D. M. J. Biol. Chem. 1998;273:15980–15984. doi: 10.1074/jbc.273.26.15980. [DOI] [PubMed] [Google Scholar]
- 40.McCright B., Rivers A. M., Audlin S., Virshup D. M. J. Biol. Chem. 1996;271:22081–22089. doi: 10.1074/jbc.271.36.22081. [DOI] [PubMed] [Google Scholar]
- 41.Xu Q., Wang Y., Dabdoub A., Smallwood P. M., Williams J., Woods C., Kelley M. W., Jiang L., Tasman W., Zhang K., Nathans J. Cell. 2004;116:883–895. doi: 10.1016/s0092-8674(04)00216-8. [DOI] [PubMed] [Google Scholar]
- 42.Gietzen K. F., Virshup D. M. J. Biol. Chem. 1999;274:32063–32070. doi: 10.1074/jbc.274.45.32063. [DOI] [PubMed] [Google Scholar]
- 43.Firulli B. A., Virshup D. M., Firulli A. B. Biol. Procedures Online. 2004;6:16–22. doi: 10.1251/bpo69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gao Z.-H., Seeling J. M., Hill V., Yochum A., Virshup D. M. Proc. Natl. Acad. Sci. USA. 2002;99:1182–1187. doi: 10.1073/pnas.032468199. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.