

Abstract
Hereditary spastic paraplegia (HSP) is a neurodegenerative disorder that is characterized by retrograde axonal degeneration that primarily affects long spinal neurons. The disease is clinically heterogeneous, and there are >20 genetic loci identified. Here, we show a physical interaction between spastin and atlastin, two autosomal dominant HSP gene products. Spastin encodes a microtubule (MT)-severing AAA ATPase (ATPase associated with various activities), and atlastin encodes a Golgi-localized integral membrane protein GTPase. Atlastin does not regulate the enzymatic activity of spastin. We also identified a clinical mutation in atlastin outside of the GTPase domain that prevents interaction with spastin in cells. Therefore, we hypothesize that failure of appropriate interaction between these two HSP gene products may be pathogenetically relevant. These data indicate that at least a subset of HSP genes may define a cellular biological pathway that is important in axonal maintenance.
Keywords: microtubule, AAA ATPase, microtubule severing
Hereditary spastic paraplegia (HSP) is a genetically and clinically heterogeneous disorder that is characterized clinically by spastic gait, mild vibratory sensory impairment, and urinary urgency and pathologically by a retrograde axonopathy that primarily involves the long neurons of the corticospinal tract and fasciculus gracilis (1). There are >20 genes linked to HSP. No molecular interaction between any of the proteins encoded by these genes has been described.
Broadly speaking, the proven or inferred functions of HSP genes have led to several broad hypotheses about the molecular pathogenesis of HSP. HSP has been suggested to be a mitochondrial disorder, based on the fact that two HSP genes, paraplegin (2) and mtHSP 60 (3), are mitochondrial proteins with roles in protein quality control and folding. Mutations in KIF5a, a conventional kinesin that is highly expressed in neurons, cause HSP (4), suggesting that impaired axonal transport may underlie some forms of the disease. Mutations in spastin (SPG-4), an AAA ATPase (ATPase associated with various activities), cause 40% of the autosomal dominant cases of HSP (5). We and others have shown that spastin is a microtubule (MT)-severing enzyme and that disease mutations impair this activity (6, 7).
Several HSP genes are associated with various membranous organelles and have inferred roles in vesicular trafficking. Spastin and spartin (SPG-20) (8), another HSP gene, both contain an MIT (microtubule interacting and transport) domain (9), which is often found in endosome-associated proteins, including Vps4, an AAA ATPase that disassembles ESCRT (endosomal sorting complex required for transport) protein complexes as part of the multivesicular body pathway (10). Spastin’s MIT binds CHMP1B (11), one of a family of proteins implicated in the multivesicular body pathway. Mutations in atlastin (12), an integral membrane protein GTPase that is primarily localized to the Golgi apparatus (13), also cause HSP. Mutations in alsin, which has Rab 5 and Rac guanine nucleotide exchange domains, suggest that a defect in intracellular trafficking may cause HSP (14, 15).
Clinically, HSP can be variable with respect to age of onset, penetrance, expressivity of phenotype, and the range of neurological abnormalities found in patients. Given the heterogeneity and the myriad hypotheses of the molecular basis of axonal loss in HSP, a central question arises as to what degree HSP is a single disease. In other words, do any or some of these disease genes function together in a single cellular pathway whose function is important in axonal maintenance? The interaction we describe between spastin and atlastin suggests that a subset of HSP gene products may function in this manner.
Results
To identify spastin-interacting proteins, we performed a yeast two-hybrid screen that used full-length spastin as bait. The assay yielded 12 independent clones, one of which encoded a fragment of atlastin-1 (Fig. 1A). Like spastin, atlastin is an autosomal dominant HSP gene (12). Atlastin is a Golgi-localized integral membrane protein GTPase (13). The recovered clone was in-frame and lacked the GTPase domain but encoded the two transmembrane segments and cytoplasmic tail (C-tail) of atlastin. Deletion of the C-tail domain from this clone (496 Stop) in human atlastin-1 abolished interaction in the yeast two-hybrid assay (Fig. 1A).
Fig. 1.

Spastin–atlastin interaction. (A) A yeast two-hybrid screen identified atlastin as a spastin interactor. Full-length human spastin was used as a bait and interacted with a clone encoding amino acids 408–558 of human atlastin-1. This clone and a deletion mutant lacking the C-tail of atlastin (ΔCtail) were used to retransform yeast in the two-hybrid assay. The original clone, but not the ΔCtail clone, interacted with spastin in this assay. (B) Coimmunoprecipitation of spastin and atlastin. HeLa cells were transiently transfected for 24 h with 2HA-spastin and either atlastin-His-6x (lysate 1) or His-6x-tagged β-gal (lysate 2) as a negative control. Cell lysates were subjected to immunoprecipitation with a His-6x antibody followed by anti-HA (Left) or anti-His-6x (Right) Western blotting. Both lysates expressed equivalent amounts of HA-spastin (SM) and spastin coimmunoprecipitated with atlastin but not β-gal. The 50- and 20-kDa bands are Ig heavy and light chains. (C) Coomassie blue-stained gel showing that spastin binds to the atlastin C-tail. The GST was cleaved from GST-spastin with precision protease (PP), and the crude reaction mixture (lane 3) containing 10 μg of spastin was applied to glutathione-Sepharose beads containing 10 μg of GST-atlastin C-tail or GST alone (lanes 4–6) in a total volume of 200 μl. Lanes 1 and 2 show samples of the beads containing only GST-atlastin C-tail or GST. All of the added spastin but none of the cleaved GST or PP bound to the atlastin C-tail, and ATP was not required for binding (compare lanes 4 and 5). In contrast to GST-spastin, the GST-atlastin C-tail protein does not contain a PP cleavage site.
Next, we cotransfected HeLa cells with plasmids encoding 2HA-tagged spastin and either atlastin-6xHis or β-gal-6xHis (Fig. 1B). Clarified lysates were subjected to immunoprecipitation with an anti-6xHis antibody. Immunoprecipitates were probed for spastin by anti-hemagglutinin (HA) Western blotting. Spastin coprecipitated with atlastin but not with β-gal, despite the fact that the control protein was expressed at a higher level than atlastin (Fig. 1B). Western blotting with an anti-HA antibody confirmed that both lysates expressed equivalent amounts of spastin. The HA antibody detected two spastin-related bands. Although we do not know why, we imagine that the upper band may represent a posttranslationally modified form of spastin. Although it appears as though only the lower band associates with atlastin, we cannot exclude the possibility that the upper band binds but that we were unable to detect it because of the lower abundance of this form.
Because the yeast two-hybrid analysis indicated that the C-tail of atlastin was binding spastin, we produced and purified recombinant GST-atlastin C-tail. The GST moiety was cleaved off of recombinant spastin with a site-specific protease. The spastin/GST/protease reaction mixture was added to bead-immobilized GST-atlastin C-tail or GST. The GST-atlastin C-tail fusion protein does not contain a precision protease site and was therefore not cleaved by the protease. Spastin bound to GST-atlastin C-tail, but not to GST, in an ATP-independent manner, demonstrating direct interaction (Fig. 1C). We also performed this experiment with full-length GST-spastin immobilized on beads and pulled down full-length recombinant maltose binding protein-atlastin (in detergent-containing buffers) (see Fig. 6, which is published as supporting information on the PNAS web site).
Addition of a 10-fold molar excess of GST-atlastin C-tail but not GST caused a very slight increase in spastin’s ATPase activity in vitro (Fig. 7, which is published as supporting information on the PNAS web site). Next, we asked whether this slight increase in ATPase activity results in enhanced MT severing. To this end, we incubated GST-spastin with taxol-stabilized, rhodamine-labeled MTs in the absence or presence of a 10-fold molar excess of GST-atlastin C-tail. At various time points, aliquots of each reaction were removed, and severing was stopped by addition of glutaraldehyde. MT length was measured from micrographs, and mean lengths were calculated. Fig. 2A shows that the average length of MTs decreases over time. We did not detect any atlastin-induced alterations in the extent or apparent rate of MT shortening in this assay. Although we did not detect any atlastin effect, the assay likely has a large margin of error, and we cannot exclude a role for atlastin in modulating spastin activity. In agreement with the in vitro data, cooverexpression of atlastin-FLAG along with yellow fluorescent protein (YFP)-spastin in Cos-7 cells did not prevent spastin-mediated severing (Fig. 2B).
Fig. 2.

Atlastin C-tail does not inhibit spastin-mediated MT severing. (A) Taxol-stabilized, rhodamine-labeled MTs and spastin were used, severing assays without or with recombinant atlastin C-tail as described in Materials and Methods. At the indicated time points, reactions were stopped by fixation in glutaraldehyde. MTs were photographed, and average lengths were calculated as described in Materials and Methods. The bar graph shows the mean lengths ± standard error. Representative photomicrographs show the extent of MT severing. There was no appreciable difference between reactions incubated with or without C-tail. The Coomassie blue-stained gel shows the relative amounts of each protein (tubulin, 0.1 mg/ml; spastin, 0.02 mg/ml). The C-tail was used at 10-fold molar excess compared with spastin. (B) Cos-7 cells were cotransfected with YFP-spastin and atlastin-FLAG. After 24 h, cells were fixed and stained with anti-FLAG and anti-tubulin antibodies. Spastin (green) and atlastin (red) colocalize. Transfected cells show decreased MT (blue) content compared with neighboring cells. (Scale bar, 15 μm.)
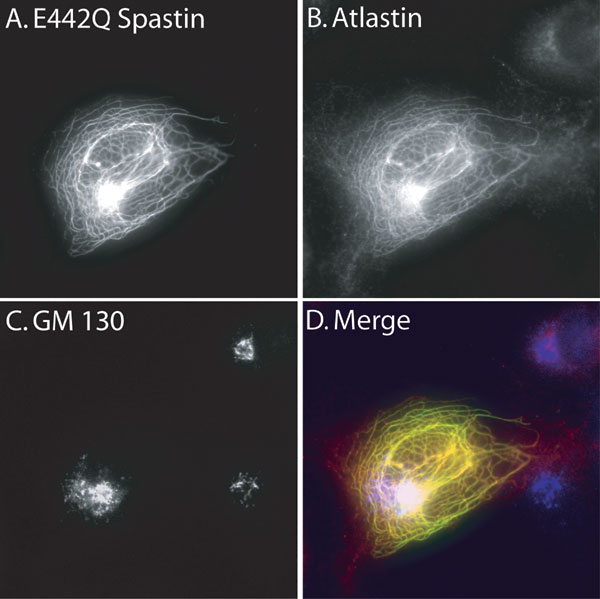
However, we did notice that in the cotransfected cells, the atlastin-FLAG staining pattern appeared altered (Fig. 2B) compared with cells transfected only with atlastin-FLAG (Fig. 8, which is published as supporting information on the PNAS web site). Whereas most endogenous atlastin is found in the Golgi (13), and overexpressed atlastin-FLAG mostly colocalizes with a Golgi marker (Fig. 8) in the cotransfected cells, atlastin-FLAG was codistributed in puncta with spastin. As is the case with MT disruption induced by many treatments, spastin-induced MT loss results in dispersion of the Golgi (data not shown). Although overexpressed spastin and atlastin are colocalized in this experiment (Fig. 2B), neither protein shows a staining pattern that is typical of either endogenous protein (see Fig. 4A).
Fig. 4.

Colocalization of endogenous spastin and atlastin. (A) Cos cells were stained with antibodies to atlastin and spastin. Signals were detected by using a Cy-5-conjugated or Alexa Fluor 488 secondary antibody. Spastin signal was detected by using Alexa Fluor 488. Note the partial colocalization of the two proteins. Atlastin staining intensity was variable, and the spastin signal is higher in cells expressing more atlastin. (B) Spastin RNAi does not appreciably alter atlastin staining. Spastin expression (Left) was partially suppressed by transfection of a spastin-specific siRNA but not a scrambled siRNA. Transfection with a rhodamine-labeled siRNA showed that >90% of the cells are transfected (data not shown). Duplicate coverslips prepared from the same transfections were stained for endogenous atlastin (Right), which appeared unaltered. (C) Effects of atlastin RNAi on spastin. Cells were fixed 48 h after transfection, and duplicate coverslips were stained for either spastin or atlastin. In this experiment, background staining of nuclei was sometimes observed. Transfected cells, identified by GFP expression (Insets), are indicated by arrowheads. Nuclei were stained with DAPI (blue). Compared with nontransfected cells, atlastin staining is partially suppressed in atlastin RNAi but not in control RNAi cells. Compared with control transfected cells, a smaller percentage of cells subjected to atlastin RNAi showed the perinuclear concentration of spastin (29% vs. 45%, n = 135 cells from each transfection). Quantitative Western blot analysis of equal numbers of transfected cells obtained by flow cytometry demonstrates that atlastin suppression was 60% and that spastin levels were also reduced to a similar degree. Signals were normalized to actin intensity. (Scale bars, 15 μm.)
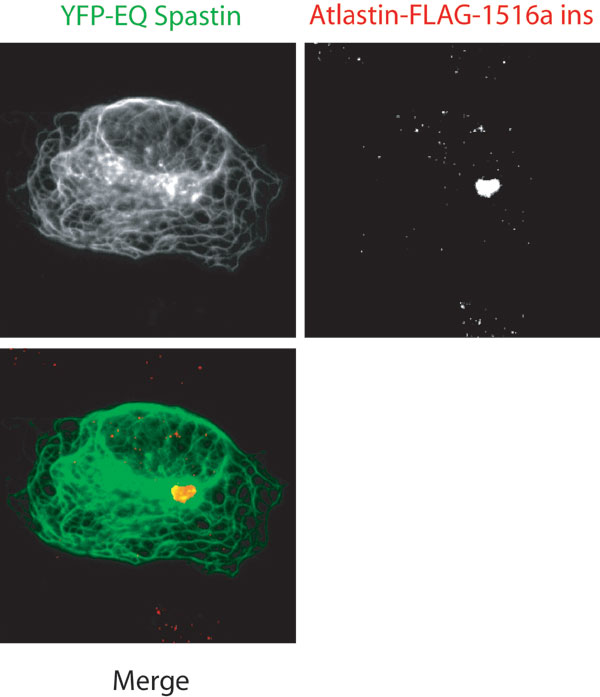
Nevertheless, this experiment suggested that cotransfection experiments might be a convenient way to identify domains of spastin and atlastin required for coassociation in cells. To circumvent the fact that WT spastin destroys MTs, which in turn might alter the subcellular localization of proteins, we used E442Q mutant spastin, which binds MTs but cannot release or sever (6) and consequently decorates MTs in transfected cells. Surprisingly, cotransfection with E442Q spastin and atlastin-FLAG resulted in atlastin being distributed along with the spastin on the MTs (Fig. 3). In the same cells, the Golgi morphology as assessed by GM130 staining appeared relatively normal (Fig. 9, which is published as supporting information on the PNAS web site). Because atlastin is an integral membrane protein, we imagine that vesicles carrying newly synthesized atlastin-FLAG from the endoplasmic reticulum to the Golgi or post-Golgi vesicles carrying atlastin must have been recruited to the MT-bound spastin. From this assay, we conclude only that the two proteins interact in cells in a manner such that one influences the localization of the other. In affected patients where mutant spastin is not overexpressed, no stable association of mutant spastin with MTs has been observed (16).
Fig. 3.

Spastin–atlastin interaction in cells. In Cos-7 cells cotransfected with YFP-E442Q spastin and atlastin-FLAG, atlastin is “recruited” to the MTs along with the spastin mutant, indicating that the two proteins interact in cells (A–C). Deletion of the N-terminal region (residues 1–132) of spastin (G–I), but not the MIT (residues 116–194) (J–L) or exon 4 (residues 195–227) (M–O) domains, prevented recruitment of atlastin. Deletion of the C-tail from atlastin prevented recruitment (D–F).
Atlastin lacking the C-tail was not recruited to the E442Q spastin (Fig. 3), consistent with the in vitro binding data that showed that spastin binds to the atlastin C-tail. To define the domain of spastin that is responsible for atlastin interaction, we made a series of single domain deletions in spastin. Broadly speaking, spastin is a bipartite molecule with a C-terminal AAA ATPase domain (amino acids 344–616). The N-terminal region of spastin (amino acids 1–343) contains four distinct subdomains. The first 115 amino acids of spastin contain a proline-rich region and a hydrophobic domain. Amino acids 116–194 comprise the MIT domain (9). Exon 4 (amino acids 195–227) is alternatively spliced, and little is known about this domain (17). The N-terminal domain, but neither the MIT domain nor exon 4, is required for atlastin recruitment (Fig. 3). Together, these results suggest that the N-terminal domain of spastin binds to the C-tail of atlastin. Two polymorphisms (S44L and P45Q), which may act as genetic modifiers (18), and a disease-associated mutation (dupA102,S103) (19) are located in this N-terminal region of spastin. None of these changes in spastin prevented atlastin recruitment in the cotransfection assay (Table 1, which is published as supporting information on the PNAS web site). Using transfection assays and working with purified MTs in a videomicroscopic severing assay (6), we found that Δ1–227 spastin (lacking the N-terminal region, the MIT domain, and exon 4) can still sever MTs and has an ATPase activity similar to that of full-length protein (data not shown and unpublished data).
We examined several mutations in both spastin and atlastin by using this assay (see Tables 1 and 2, which are published as supporting information on the PNAS web site). We analyzed two clinical mutations in the atlastin C-tail and found that R495W (20), but not ins1688A, interacted with spastin (Fig. 10, which is published as supporting information on the PNAS web site).
We raised antibodies to both spastin and atlastin and examined whether endogenous spastin and atlastin colocalize in Cos cells. Western blots of cell lysates using affinity-purified antibodies are shown (Fig. 11, which is published as supporting information on the PNAS web site). The atlastin antibody showed a perinuclear staining pattern (Fig. 4) similar to that published by Zhu et al. (13). Staining intensity varied among cells. The intensity of endogenous spastin staining was proportional to the amount of atlastin expressed (Fig. 4A). Also, there was partial colocalization of the two endogenous proteins.
We asked whether RNA interference (RNAi)-mediated silencing of either spastin or atlastin affected the other protein. Spastin suppression, achieved by using a short interfering RNA (siRNA), resulted in decreased but not abolished spastin staining (Fig. 4B) but did not appreciably alter either the atlastin staining pattern or intensity. In the Western blot analysis, suppression of spastin by 50% by RNAi did not result in altered atlastin levels (data not shown). To suppress atlastin, we transfected cells with a plasmid containing a short hairpin RNA (shRNA) specific to atlastin. Transfected cells coexpress GFP along with the shRNA. Western blotting of transfected cells obtained by flow cytometry (Fig. 4C) showed 60% suppression of atlastin. Interestingly, atlastin RNAi also resulted in a similar decrease in spastin levels. Atlastin RNAi resulted in variably decreased atlastin staining compared with control cells (Fig. 4C). With regard to spastin staining, 45% of control transfected cells showed a perinuclear region with concentrated spastin staining (see Fig. 4C). In contrast, only 29% of the atlastin RNAi transfected cells showed this feature. The decrease in the perinuclear spastin staining could result from either decreased spastin levels or the inability to concentrate spastin to atlastin-containing areas.
We wondered whether atlastin overexpression would result in recruitment and perhaps an increased steady-state level of endogenous spastin. Also, to test whether the atlastin C-tail is necessary and sufficient for these phenomena, we created two additional constructs. Among the reported disease-causing mutations in atlastin is one that deletes most of the C-tail (21). This mutation, an insertion of an adenine at position 1688, changes the reading frame 10 aa into the C-tail, adds nonnative sequence, and introduces a premature stop codon. Like the ΔC-tail construct used in Fig. 3, this mutation prevented spastin interaction in the cotransfection assay (Fig. 10). We also created a construct (atlastin C-tail-Golgi) with the atlastin C-tail-FLAG moiety fused to a Golgi-localization moiety (the residues of galactosyl transferase encoding amino acids that are responsible for Golgi localization).
We transfected cells with plasmids encoding WT-atlastin-FLAG, ins1688A-atlastin-FLAG, or atlastin C-tail-Golgi. Cells were fixed and double stained for FLAG and endogenous spastin (Fig. 5). In cells overexpressing atlastin lacking the C-tail (atlastin-ins1688A), endogenous spastin was not colocalized with the overexpressed atlastin. In these cells, spastin staining was also less intense (the image shown in Fig. 5E was obtained by using a longer exposure time to make the spastin staining visible). Also, the spastin staining in both the transfected cell and the nontransfected cell appear similar in intensity and distribution. In contrast, in cells overexpressing WT atlastin-FLAG or atlastin C-tail-Golgi, endogenous spastin was colocalized with overexpressed atlastin. In comparing the three cells depicted in Fig. 5 G and H, the amount of endogenous spastin staining is proportional to the amount of atlastin C-tail Golgi expressed. These results suggest that the C-tail of atlastin is necessary and sufficient for spastin recruitment. Together with the data in Fig. 4A, they also suggest that atlastin and spastin levels correlate. We measured atlastin-FLAG fluorescence and fluorescence from endogenous spastin in randomly selected transfected and nontransfected cells. A plot of atlastin-FLAG intensity vs. spastin intensity shows that there is a good correlation between levels of atlastin and spastin on a cell-by-cell basis (Fig. 5).
Fig. 5.

The atlastin C-tail is necessary and sufficient to recruit endogenous spastin to the Golgi. Cos cells were transfected with WT atlastin-FLAG (A–C), ins1688A-atlastin-FLAG (D–F), or atlastin C-tail Golgi (G–I) for 48 h. Cells were stained by using FLAG antibodies (Cy-5 secondary antibody in A, D, and G, shown in red) and stained for endogenous spastin (Alexa Fluor 488 detection in B, E, and H, shown in green). Note the partial colocalization of spastin with WT and atlastin C-tail Golgi but not with ins1688A-atlastin. Spastin intensity is increased in cells expressing more atlastin C-tail Golgi (compare cells in G and H). The image in E was obtained with an increased exposure time to better visualize the spastin staining. Note that spastin levels are the same in the transfected and nontransfected cells and that spastin is not localized with atlastin. The Western blot shows that cells transfected with plasmids encoding atlastin-YFP or atlastin C-tail-Golgi, but not YFP, show increased levels of endogenous spastin (shown in green). The numbers above each lane show the relative increase in spastin levels [compared with YFP transfected cells and normalized for actin expression (red)]. The graph shows spastin fluorescence intensity as a function of the atlastin-FLAG fluorescence intensity in randomly selected cells. Cells not overexpressing atlastin-FLAG had an average spastin intensity of 95 arbitrary units.
Next, we transfected cells with YFP, atlastin-YFP, or atlastin C-tail Golgi. After 24 h, levels of spastin were examined by quantitative Western blotting (Fig. 5). Examination of the YFP and YFP-expressing cells before lysis revealed equal transfection efficiencies of ≈70%. Overexpression of atlastin-YFP, but not YFP alone, resulted in an ≈3-fold increase in the spastin signal, whereas overexpression of atlastin C-tail-Golgi resulted in a 7.8-fold increase in spastin.
Discussion
We showed that the N-terminal domain of spastin binds directly to the C-terminal cytoplasmic domain of atlastin. This interaction between two HSP disease gene products suggests that some of the >20 HSP genes may define a cellular biological pathway that is important in axon maintenance. We also showed the HSP-associated mutant ins1688A-atlastin does not interact with spastin, suggesting that HSP could result from a lack of normal interaction between spastin and atlastin.
Neither deletion of nor missense mutations in atlastin’s N-terminal GTPase domain prevented recruitment (Tables 1 and 2). Thus, the role of the GTPase activity, if any, in regulating atlastin–spastin interaction is unclear.
While this paper was under revision, Sanderson et al. (22) reported that spastin and atlastin interact in a yeast two-hybrid assay. In contrast to our findings, they reported that the N-terminal portion of atlastin binds spastin. This conclusion was based on an experiment where lysates from cells transfected with spastin were incubated with GST-1–447 atlastin or with GST alone. Because the endogenous atlastin was also present in these cells and because atlastin is capable of homooligomerization (13), it is possible that the observed interaction was indirect. Using the yeast two-hybrid assay (Fig. 1), the recruitment assay where the C-tail but not the first 447 amino acids of atlastin are required for recruitment (Figs. 3 and 5 and Tables 1 and 2), and purified recombinant proteins (Fig. 2) and by examining endogenous spastin (Fig. 5), we show that the atlastin C-tail binds directly to spastin and is both necessary and sufficient for spastin interaction.
Because atlastin binds to a region of spastin that is dispensable for MT severing (data not shown) and because we found that recombinant atlastin C-tail did not alter spastin-mediated MT severing in vitro (Fig. 2), we believe that atlastin may function to localize spastin. Current evidence suggests that atlastin is found in the Golgi and is associated with vesicles in axons and in growth cones in neurons (23), raising the possibility that some atlastin from the Golgi gets packaged into Golgi-derived vesicles. Atlastin could recruit spastin to the Golgi or possibly to post-Golgi vesicles under as yet undetermined circumstances. Consistent with this notion, cells expressing higher amounts of either endogenous atlastin (Fig. 4A) or overexpressed atlastin show increased steady-state levels of spastin and enhanced colocalization with spastin. Partial suppression of atlastin also resulted in decreased steady-state levels of spastin and in a mildly altered cellular distribution in some cells.
Materials and Methods
Culture of HeLa and Cos-7 Cells and Transfections.
Cell cultures and transfections were as described in ref. 6. Deletion of the original YFP-spastin constructs (6) was accomplished with the QuikChange mutagenesis system (Stratagene), and mutations were confirmed by DNA sequencing. The ΔN construct deleted amino acids 1–132, the ΔMIT construct deleted amino acids 116–194, and the Δexon 4 construct deleted amino acids 195–227. An atlastin IMAGE clone was obtained and FLAG- or His-6x-tagged at the C terminus. To construct the atlastin C-tail-Golgi construct, an initiator Met codon was added to the DNA encoding the atlastin C-tail-FLAG. The sequence responsible for localizing galactosyl transferase to the Golgi (the 81 N-terminal residues) was fused 3′ to the FLAG sequence.
Yeast Two-Hybrid Screening.
The yeast two-hybrid screening was performed with the Clontech Matchmaker system. Human spastin was cloned into the pGBK-T7 plasmid by using the NdeI and BamHI restriction sites, and the plasmid was used to transform strain AH109. These yeasts were mated with strain Y187 that had been pretransformed with a HeLa cDNA library (Clontech) cloned into pGAD T7 rec. Diploids were plated on triple dropout medium (low stringency selection), and positives were replated onto quadruple dropout plates (high stringency) containing X-α-gal. Plasmids were prepared from positive clones and used to transform KC8 Escherichia coli. Recovered plasmids were sequenced and retransformed into strain AH109, and the two-hybrid assay was repeated to confirm that we had isolated the correct plasmids.
Recombinant Proteins.
Spastin expression and purification was as described in ref. 6. A fragment encoding the C-tail of atlastin (amino acids 495–558) was PCR-amplified with primers adding BamHI and EcoRI restriction sites for cloning into pGEX2T. GST-atlastin C-tail was expressed in BL21 RILP cells (Stratagene) by induction of logarithmic-phase cultures with 0.4 mM isopropyl β-d-thiogalactoside at 37°C for 4 h. Washed cells were disrupted, and GST fusion protein purification was the same as for GST-spastin (6). GST was cleaved from recombinant spastin by using precision protease (GE Healthcare).
In Vitro MT Severing.
Rhodamine-labeled, taxol-stabilized MTs prepared as described in ref. 6 (0.1 mg/ml final concentration) were used in severing assays performed at 25°C with spastin (0.02 mg/ml) and ATP (1 mM) in the absence or presence of a 10-fold molar excess of atlastin C-tail. At times indicated in Fig. 2A, aliquots were removed and fixed in glutaraldehyde. Samples were diluted and spotted onto coverslips, and MTs from at least 10 randomly selected microscopic fields were photographed and measured by using metamorph. Approximately 125–250 MTs were photographed and measured for each time point.
Coimmunoprecipitations.
HeLa cells in six-well plates were transfected for 24 h and lysed in RIPA buffer (50 mM Hepes, pH 7.4/100 mM NaCl/2 mM MgCl2/10% glycerol/1% Triton X-100/0.5% deoxycholate/0.1% SDS/protease inhibitor mixture; Roche Applied Science, Indianapolis), and lysates were clarified by centrifugation (20,000 × gav for 20 min at 4°C). Supernatants were precleared with protein G Sepharose, and 2.5 μg of His-6x antibodies and protein G were added for 1 h at 4°C. Beads were washed three times in RIPA buffer and eluted in SDS sample buffer. Lysates were probed for spastin and atlastin by α-HA and α-His Western blotting, respectively. Immunoprecipitates were probed for spastin content by HA blotting.
Antibodies and Immunofluorescence.
Chickens were immunized with GST-spastin, and antibodies were affinity-purified from a yolk-derived IgY fraction by using an affinity matrix where myelin basic protein (MBP)-spastin was immobilized with Affigel. Purified antibodies were recovered at a concentration of 0.2 mg/ml and stored in 50 mM Tris (pH 8.0). To make the atlastin antibody, rabbits were immunized with GST-atlastin C-tail. Antibodies were affinity-purified on an MBP-atlastin matrix.
Detection of endogenous spastin by immunofluorescence microscopy required blocking of methanol fixed cells in BlockHen II (Aves Labs, Tigard, OR) to reduce background staining. Also, antibodies were used at 1:5,000–1:20,000 dilutions, and signal was amplified with an Alexa Fluor 488 or Alexa Fluor 594 Tyramide Signal Amplification Kit (Molecular Probes). Atlastin antibodies were used at a 1:500 dilution or at 1:5,000–1:10,000 when used with tyramide signal amplification. Antibodies were used at the following dilutions: Yl1/2 (Chemicon) for tubulin at 1:500, rabbit anti-FLAG (Affinity BioReagents, Neshanic Station, NJ) at 1:250, anti-His-6x (Clontech) at 1:250, mouse anti-GM130 (BD Biosciences) at 1:250, and rat anti-HA (Roche Applied Sciences) at 1:250. Quantitative Western blotting was performed with the use of infrared dye-conjugated secondary antibodies in conjunction with a Licor Odyssey infrared imaging/scanning system.
RNAi.
Spastin RNAi was achieved by using a duplex siRNA (antisense, 5′-rArUrArCrCrArUrUrCrCrArCrArGrCrUrUrGrCrUrCrCrUrUrCrUrG-3′; sense, 5phos-rGrArArGrGrArGrCrArArGrCrUrGrUrGrGrArArUrGrGrUAT). Cells were transfected with duplex siRNA at 20 nM duplex by using Silentfect (Bio-Rad) for 48 h. A scrambled siRNA was used at the same concentration as a control (sense, 5phos-rCrUrUrCrCrUrCrUrCrUrUrUrCrUrCrUrCrCrCrUrUrGrUGA; antisense, rUrCrArCrArArGrGrGrArGrArGrArArArGrArGrArGrGrArArGrGrA). Atlastin RNAi was achieved by using a short hairpin RNA (shRNA) that was previously shown to suppress atlastin (23). A control shRNA to alphafetoprotein (GGATCTGTGCCAAGCTCAGTTCAAGAGACTGAGCTTGGCACAGATCCTTTTTT) was cloned into pLentilox (24). Plasmids were use to transfect Cos-7 cells, and cells were examined 48–72 h after transfection.
Supplementary Material
Acknowledgments
We thank Dr. Craig Blackstone [National Institutes of Health (NIH), Bethesda] for advice and sharing reagents. This work was supported by NIH/National Institute of Neurological Disorders and Stroke Grants R01 NS043298 and K02 NS046472, the Spastic Paraplegia Foundation (B.L.), and NIH/Mental Health Grant T32 MH15174-28 (to K.E.).
Abbreviations
- MT
microtubule
- HSP
hereditary spastic paraplegia
- YFP
yellow fluorescent protein
- MIT
microtubule interacting and transport
- HA
hemagglutinin
- RNAi
RNA interference
- siRNA
short interfering RNA
- C-tail
cytoplasmic tail.
Footnotes
Conflict of interest statement: No conflicts declared.
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Fink J. K. Ann. Neurol. 2002;51:669–672. doi: 10.1002/ana.10258. [DOI] [PubMed] [Google Scholar]
- 2.Casari G., De Fusco M., Ciarmatori S., Zeviani M., Mora M., Fernandez P., De Michele G., Filla A., Cocozza S., Marconi R., et al. Cell. 1998;93:973–983. doi: 10.1016/s0092-8674(00)81203-9. [DOI] [PubMed] [Google Scholar]
- 3.Hansen J. J., Durr A., Cournu-Rebeix I., Georgopoulos C., Ang D., Nielsen M. N., Davoine C. S., Brice A., Fontaine B., Gregersen N., Bross P. Am. J. Hum. Genet. 2002;70:1328–1332. doi: 10.1086/339935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Reid E., Kloos M., Ashley-Koch A., Hughes L., Bevan S., Svenson I. K., Graham F. L., Gaskell P. C., Dearlove A., Pericak-Vance M. A., et al. Am. J. Hum. Genet. 2002;71:1189–1194. doi: 10.1086/344210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hazan J., Fonknechten N., Mavel D., Paternotte C., Samson D., Artiguenave F., Davoine C. S., Cruaud C., Durr A., Wincker P., et al. Nat. Genet. 1999;23:296–303. doi: 10.1038/15472. [DOI] [PubMed] [Google Scholar]
- 6.Evans K. J., Gomes E. R., Reisenweber S. M., Gundersen G. G., Lauring B. P. J. Cell Biol. 2005;168:599–606. doi: 10.1083/jcb.200409058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Roll-Mecak A., Vale R. D. Curr. Biol. 2005;15:650–655. doi: 10.1016/j.cub.2005.02.029. [DOI] [PubMed] [Google Scholar]
- 8.Patel H., Cross H., Proukakis C., Hershberger R., Bork P., Ciccarelli F. D., Patton M. A., McKusick V. A., Crosby A. H. Nat. Genet. 2002;31:347–348. doi: 10.1038/ng937. [DOI] [PubMed] [Google Scholar]
- 9.Ciccarelli F. D., Proukakis C., Patel H., Cross H., Azam S., Patton M. A., Bork P., Crosby A. H. Genomics. 2003;81:437–441. doi: 10.1016/s0888-7543(03)00011-9. [DOI] [PubMed] [Google Scholar]
- 10.Babst M., Wendland B., Estepa E. J., Emr S. D. EMBO J. 1998;17:2982–2993. doi: 10.1093/emboj/17.11.2982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Reid E., Connell J., Edwards T. L., Duley S., Brown S. E., Sanderson C. M. Hum. Mol. Genet. 2005;14:19–38. doi: 10.1093/hmg/ddi003. [DOI] [PubMed] [Google Scholar]
- 12.Zhao X., Alvarado D., Rainier S., Lemons R., Hedera P., Weber C. H., Tukel T., Apak M., Heiman-Patterson T., Ming L., et al. Nat. Genet. 2001;29:326–331. doi: 10.1038/ng758. [DOI] [PubMed] [Google Scholar]
- 13.Zhu P. P., Patterson A., Lavoie B., Stadler J., Shoeb M., Patel R., Blackstone C. J. Biol. Chem. 2003;278:49063–49071. doi: 10.1074/jbc.M306702200. [DOI] [PubMed] [Google Scholar]
- 14.Topp J. D., Gray N. W., Gerard R. D., Horazdovsky B. F. J. Biol. Chem. 2004;279:24612–24623. doi: 10.1074/jbc.M313504200. [DOI] [PubMed] [Google Scholar]
- 15.Eymard-Pierre E., Lesca G., Dollet S., Santorelli F. M., di Capua M., Bertini E., Boespflug-Tanguy O. Am. J. Hum. Genet. 2002;71:518–527. doi: 10.1086/342359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wharton S. B., McDermott C. J., Grierson A. J., Wood J. D., Gelsthorpe C., Ince P. G., Shaw P. J. J. Neuropathol. Exp. Neurol. 2003;62:1166–1177. doi: 10.1093/jnen/62.11.1166. [DOI] [PubMed] [Google Scholar]
- 17.Charvin D., Cifuentes-Diaz C., Fonknechten N., Joshi V., Hazan J., Melki J., Betuing S. Hum. Mol. Genet. 2003;12:71–78. doi: 10.1093/hmg/ddg004. [DOI] [PubMed] [Google Scholar]
- 18.Svenson I. K., Kloos M. T., Gaskell P. C., Nance M. A., Garbern J. Y., Hisanaga S., Pericak-Vance M. A., Ashley-Koch A. E., Marchuk D. A. Neurogenetics. 2004;5:157–164. doi: 10.1007/s10048-004-0186-z. [DOI] [PubMed] [Google Scholar]
- 19.Meyer T., Schwan A., Dullinger J. S., Brocke J., Hoffmann K. T., Nolte C. H., Hopt A., Kopp U., Andersen P., Epplen J. T., Linke P. Neurology. 2005;65:141–143. doi: 10.1212/01.wnl.0000167130.31618.0a. [DOI] [PubMed] [Google Scholar]
- 20.Scarano V., Mancini P., Criscuolo C., De Michele G., Rinaldi C., Tucci T., Tessa A., Santorelli F. M., Perretti A., Santoro L., Filla A. J. Neurol. 2005;252:901–903. doi: 10.1007/s00415-005-0768-1. [DOI] [PubMed] [Google Scholar]
- 21.Tessa A., Casali C., Damiano M., Bruno C., Fortini D., Patrono C., Cricchi F., Valoppi M., Nappi G., Amabile G. A., et al. Neurology. 2002;59:2002–2005. doi: 10.1212/01.wnl.0000036902.21438.98. [DOI] [PubMed] [Google Scholar]
- 22.Sanderson C. M., Connell J. W., Edwards T. L., Bright N. A., Duley S., Thompson A., Luzio J. P., Reid E. Hum. Mol. Genet. 2006;15:307–318. doi: 10.1093/hmg/ddi447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhu P. P., Soderblom C., Tao-Cheng J. H., Stadler J., Blackstone C. Hum. Mol. Genet. 2006;15:1343–1353. doi: 10.1093/hmg/ddl054. [DOI] [PubMed] [Google Scholar]
- 24.Rubinson D. A., Dillon C. P., Kwiatkowski A. V., Sievers C., Yang L., Kopinja J., Rooney D. L., Ihrig M. M., McManus M. T., Gertler F. B., et al. Nat. Genet. 2003;33:401–406. doi: 10.1038/ng1117. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}