

Abstract
The pituitary glycoprotein hormones LH and FSH regulate the reproductive cycle and are sensitive to feedback by gonadal steroids. The common α-subunit shared by these hormones is transcriptionally repressed by androgen receptor (AR) in the presence of its ligand dihydrotestosterone. This identifies at least one mechanism that contributes to AR-dependent suppression of gonadotropin synthesis. Repression of α-subunit transcription by AR requires only the sequences within the first 480 bp of the promoter. While this region contains a high-affinity binding site for AR, this element does not mediate the suppressive effects of androgens. Instead, two other elements within the promoter-regulatory region (α-basal element and cAMP-regulatory element), which are important for expression of the α-subunit gene in gonadotropes, mediate the effects of AR. This suggests that AR inhibits activity of the α-subunit promoter by interfering with the transcriptional properties of the proteins that bind to α-basal element and the cAMP-regulatory elements. Furthermore, transfection analysis of various mutant ARs identified both the DNA-binding and ligand-binding domains of the receptor as critical for repression. Comparisons with the MMTV promoter revealed distinct structural requirements that underlie the trans-activation and transrepression properties of AR.
INTRODUCTION
All glycoprotein hormones share the same α-subunit (1). This subunit dimerizes with unique β-subunits to generate LH, FSH, TSH, and chorionic gonadotropin (CG) (1). This family of heterodimeric glycoproteins are critical regulators of reproduction and metabolism. Since the α-subunit is required for the production of all glycoprotein hormones, its expression must occur in each population of cells that produce these hormones. Thus, the α-subunit gene is expressed in the gonadotropes (LH and FSH) and thyrotropes (TSH) of the pituitary and in the trophoblast cells of the placenta (CG).
Regulation of this gene in gonadotropes involves input from both the hypothalamus and the gonads (2). The hypothalamic neuropeptide GnRH stimulates synthesis and secretion of gonadotropins by binding a specific G protein- coupled receptor on the surface of gonadotropes (2, 3). This increase in synthesis and secretion is accompanied by changes in transcription of the α-subunit gene (7, 8). This gene is also regulated through feedback signals from the gonads (2, 4–6). Typically, estrogens and androgens synthesized in response to gonadotropin stimulation repress transcription of the α- and gonadotropin β-subunit genes (4–6, 9–12). Mechanistically, negative feedback by gonadal steroids may reflect either direct regulatory effects occurring at the level of the pituitary or an indirect mechanism involving inhibition of GnRH synthesis and secretion from hypothalamic neurons (4–6). Several lines of evidence, however, suggest that direct transcriptional regulation of the α-subunit gene by androgens occurs within the pituitary and constitutes an important component of negative feedback (9, 11, 13–16).
Unlike negative regulation by glucocorticoid receptor, which predominantly exerts its negative regulatory effects by interacting with the transcription factor AP-1, the mechanism by which AR negatively influences transcription is less well understood. In one of the first reports on negative regulation by AR, we demonstrated that transcription of the α-subunit gene was repressed by androgens when promoter constructs, together with AR, were introduced into a gonadotrope cell line, αT3 (9). Repression was dependent on both the presence of AR and its ligand, dihydrotestosterone (DHT). This study also identified a high-affinity binding site for androgen receptor (ARE) located between the tandem CREs and the CCAAT box in the proximal promoter region of the α-subunit gene (Fig. 1) (9). This site mapped to a previously identified promoter element, the junctional response element or JRE, which is important for expression in the placenta and has significant sequence identity to an ARE/glucocorticoid response element consensus site (9). Based on these studies, we postulated that direct binding of activated AR to the JRE represses transcription of the α-subunit gene, accounting, at least partially, for the physiological regulation of the α-subunit by androgens. In the present study, however, we show instead that AR represses promoter activity via a mechanism that does not require a high-affinity binding site for the steroid receptor.
Fig. 1. Multiple Transcriptional Elements Contribute to Expression of the α-Subunit Gene in Gonadotropes.
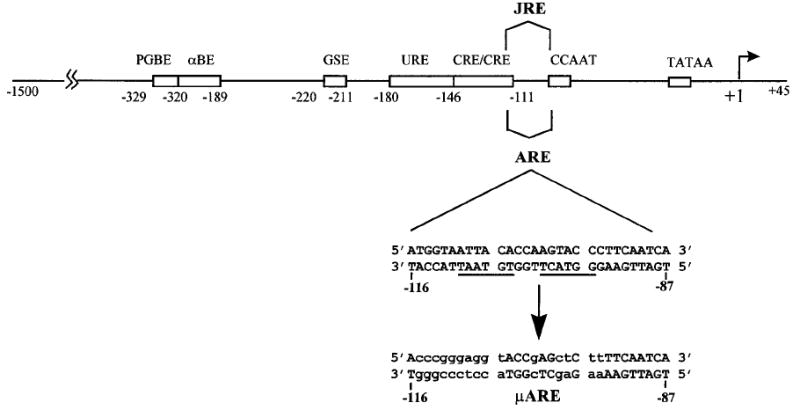
Regulatory elements required for expression of the human α-subunit gene in gonadotropes are depicted by boxes, and the positions of each are indicated by the bases relative to the transcriptional start site (arrow at +1). PGBE, αBE, GSE, URE, CRE, JRE, CCAAT (CCAATT box), and TATAA (TATAA box). ARE is the high-affinity binding site of AR. Sequences between −116 and −87 includes the ARE. μARE, sequence to which promoter was mutated to determine functional significance of the ARE.
The transcriptional mechanism required for expression of the α-subunit gene in gonadotropes is complex, involving an array of regulatory elements located between −330 and −95 bp of the 5′-flanking region (17–19). These include two tandem cAMP-regulatory elements (CREs), a GATA-binding site within the upstream regulatory element (URE), the gonadotrope-specific element (GSE), the pituitary glycoprotein hormone basal element (PGBE), and α-basal element (αBE) (Fig. 1) (17–19). A CCAAT box also contributes minimally to promoter activity (our unpublished data). Using mutations within these important elements, we report herein that repression occurs via a mechanism requiring the presence of two promoter elements, αBE and the tandem CREs. Mediation of androgen suppression by these two elements suggests that AR interferes with the transcriptional properties of the proteins that bind them. Furthermore, our studies identify regions within the DNA-binding domain (DBD) and the ligand-binding domain (LBD) of AR that are critical for repression and demonstrate that there are distinct receptor structural requirements for mechanisms of activation and repression by AR.
RESULTS
The High-Affinity Binding Site for Androgen Receptor (ARE) Is Not Required for Promoter Repression
Negative transcriptional regulation by AR was studied by using luciferase reporter vectors driven by wild type or mutant human α-subunit promoters. Promoter constructs were introduced into αT3 cells along with expression vectors for either AR (pCMVhAR) or GH (CMV-GH). Cotransfection of CMV-GH was included to control for changes in α-subunit promoter activity due to the co-transfection paradigm. Both AR and GH are expressed from the cytomegalovirus (CMV) promoter. Cells were then maintained in media containing 100 nm DHT. In the presence of pCMVhAR, activity of the wild type −1500 bp promoter (−1500 wt) was repressed approximately 2-fold when compared with the CMV-GH control (Fig. 2A). A mutation through the ARE (μARE, see Fig. 1) did not affect that ability of the receptor to repress promoter activity (Fig. 2A). Furthermore, deletion of promoter sequences upstream of −485 did not diminish AR regulation, demonstrating that the −485 bp promoter-proximal region mediates the suppressive effect of AR.
Fig. 2. Androgen Repression of the α-Subunit Gene Requires Only Sequences within the First 485 bp of the Promoter and Not Receptor Binding.
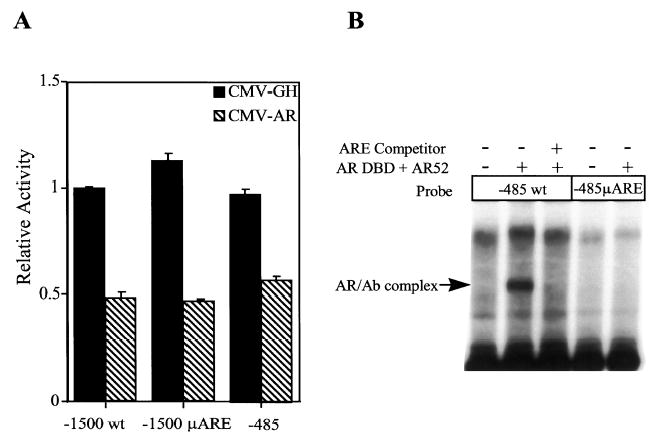
The luciferase gene driven by different promoter constructs was assayed for responsiveness to cotransfected AR. The promoter constructs studied are −1500 to +45 bp of the human α-subunit gene with an intact ARE (−1500 wt), −1500 to +45 bp of the human α-subunit gene with a block replacement mutation through the ARE (−1500 μARE), and −485 to +45 of the human α-subunit gene. A, αT3 cells were cotransfected with the luciferase constructs, RSV-βgalactosidase, and expression vectors for either GH (CMV-GH, black bars) or AR (pCMVhAR, hatched bars), and cells were maintained in the presence of 100 nm DHT. Luciferase activity of each construct was normalized to β-galactosidase activity to control for transfection efficiency. Data represent the luciferase/β-galactosidase activity of each construct normalized to the luciferase/β-galactosidase activity of the wild type construct (−1500wt) in the presence of CMV-GH. Transfections were done a minimum of three times and the error bars represent m. B, Electrophoretic mobility shift analysis. Radiolabeled promoter fragment (10 fmol) spanning from −485 to +45bp of either the wild type promoter (−485wt) or the promoter containing a mutation in the ARE (−485 μARE) was incubated with purified DBD of the rat AR (AR-DBD) and an antibody specific for AR (AR52). A double-stranded oligonucleotide containing the sequence for the α-subunit promoter ARE (5′-GTCATGGTAATTACACCAAGRACCCTTCAA-3′) was added to indicated reactions at a 200-fold concentration that of added probe.
Disruption of AR binding to the α-subunit promoter was confirmed by electrophoretic mobility shift analysis (Fig. 2B). A promoter fragment spanning −485 to +45bp was radiolabeled and incubated with purified DBD of the rat AR (AR-DBD) and an antibody specific for AR (AR52). In the presence of AR-DBD and AR52, a single major complex bound to the wild type promoter (−485 wt). This band was eliminated when excess unlabeled α-subunit ARE competitor was added to the reaction. No AR complex was formed on the promoter containing a mutation through the ARE (−485 μARE).
The above data demonstrate that androgen-dependent suppression occurs in the absence of a high-affinity binding site for AR. This suggested that AR interferes with the activity of a transcription factor(s) required for promoter activity either by binding directly to the factor or indirectly by competing for a common transcriptional adapter. Thus, to examine this possibility further, we elected to determine whether repression by AR mapped to other regulatory elements within the α-subunit promoter.
Repression by AR Requires Basal Promoter Elements Important for Expression of the α-Subunit in Gonadotropes
To identify regulatory elements required for AR repression, we took advantage of numerous studies from this laboratory and others that have elucidated a complex array of elements required for activity of the α-subunit promoter in gonadotropes (Fig. 1) (17–19). In short, we examined the effects of AR on a series of promoter constructs containing block replacement mutations through each of these elements. Details of the mutations are explained elsewhere (19). As shown in Fig. 3, promoter constructs containing mutations through the PGBE (ΔPGBE), GSE (ΔGSE), or the URE (ΔURE) were all repressed by AR, although to a slightly lesser degree than the wild type promoter. In contrast, promoters containing mutations through the αBE or the CREs were not significantly repressed by AR, demonstrating the importance of these sites in AR-mediated repression. It is important to note that the activity of each promoter mutant was typically more than 150-fold greater than a promoterless control (19). Thus, loss of repression is not the result of promoter constructs being compromised to such an extent that no further reduction in promoter activity is possible, but rather a result of the specific mutations in the αBE and CREs.
Fig. 3. Repression by AR Requires the αBE and CREs.
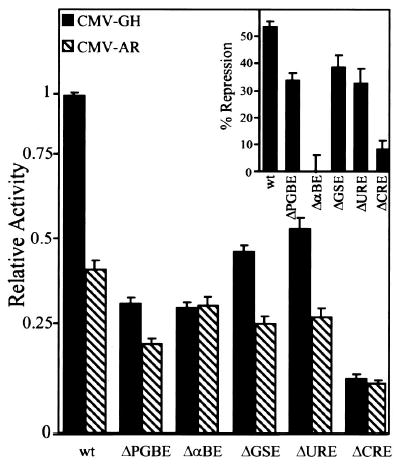
Promoter constructs carrying mutations through various response elements (see Fig. 1) were cotransfected with either CMV-GH (black bars) or pCMVhAR (hatched bars). Transfections were as described in the legend of Fig. 2 where relative activity represents the luciferase/β-galactosidase activity of each promoter made relative to the wild type promoter co-transfected with CMV-GH. The relative activity of each construct in the presence and absence of AR was used to calculate the percent repression (inset).
The above data imply that repression by AR involves a mechanism in which transcription factors binding αBE and the tandem CREs must be present. Recently, we reported that a number of transcription factors in the cAMP response element binding protein/activating transcription factor (ATF) family, including cAMP response element binding protein, cAMP response element modulator protein, ATF1, ATF2, and Jun, bind the CREs in the human α-subunit promoter (20). Additional studies showed that a natural CRE variant having a single point mutation in the core palindrome (TGACGTCA to TGATGCTA) is active in gonadotropes but binds only Jun and ATF2. Interestingly, in the presence of AR, a promoter containing the variant CRE(−1500 TGAT) is repressed to the same extent as a promoter containing the palindromic CRE (−1500 WT). This demonstrates that Jun and ATF2 are included in the subset of transcription factors required for promoter repression by AR (Fig. 4). Since the variant CRE is present in all species other than primates, the data suggest that AR-mediated repression of the α-subunit is not limited to primates.
Fig. 4. A Variant Form of the CRE Is Repressed by AR.
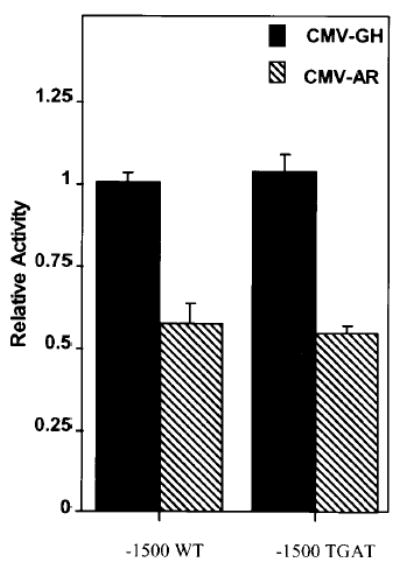
The CREs of the human α-subunit promoter were replaced with a variant form of the CRE that contains a C-to-T mutation in the core of each CRE (Var, TGACGTCA to TGATGTCA). Fifteen hundred base pairs of the human α-subunit promoter containing the variant CRE were used to drive expression of the luciferase reporter gene. The luciferase constructs were transfected into αT3 cells with RSV-β-galactosidase and either CMV-GH (black bars) or pCMVhAR (hatched bars). Transfections are as described in the legend of Fig. 2.
Repression by AR Requires Intact DBDs and LBDs
To more fully establish the mechanism by which AR represses transcription of the α-subunit gene, we addressed whether specific domains of AR are required for repression. Most structure-function studies of AR have focused on determining the regions of the receptor required for stimulation of transcription. Thus, by identifying the regions of the receptor required for repression, we not only learn about the mechanism of α-subunit gene regulation but also about the functional determinants required for the suppressive effects of AR.
To establish receptor requirements for repression, a variety of mutant ARs were cotransfected with the wild type −1500-bp luciferase reporter construct. ARs having deletions that span the amino-terminal domain (Δ14–150, Δ142–337, Δ338–499, Δ500–558) retained their ability to repress α-subunit promoter activity (Fig. 5A). Thus, repression does not require regions within the amino terminus of AR. In contrast, deletion of the DBD (Δ538–614) or the LBD (Δ660–919) eliminated AR-dependent repression, indicating that these two domains are critical. The requirement for the DBD was further substantiated using a point mutant that has a cysteine-to-alanine mutation within the first zinc finger (C576A). This mutation, which disrupts DNA-binding activity, eliminated repression by the receptor. A fourth mutation, which deletes the nuclear localization signal (Δ615–633), also prevents promoter repression. Since this mutant is poorly translocated into the nucleus, it is unclear whether this region is directly involved in repression (21). Together, these studies identify two distinct domains of AR that are required for suppression of the α-subunit promoter; namely, the DBD and the LBD.
Fig. 5. The DBD and LBD of the AR Are Required for Transcriptional Repression.
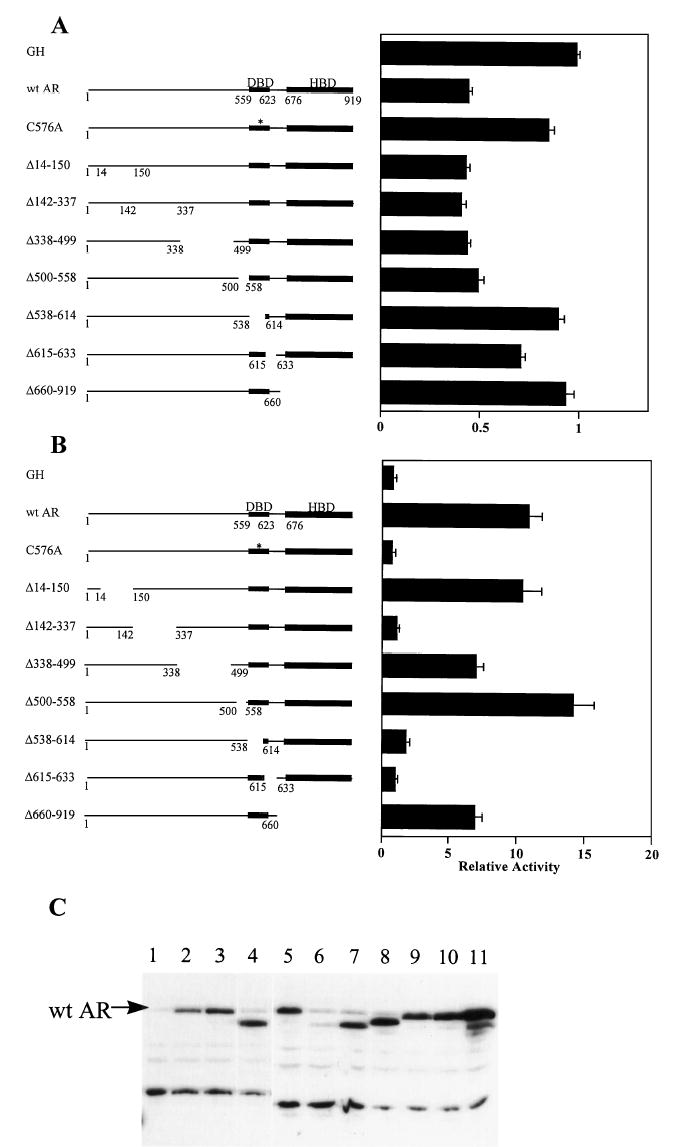
A, The wild type −1500-bp α-subunit promoter construct was cotransfected with expression vectors for either GH, AR, or various AR mutants as depicted to the left of the graph. Transfections were as described in the legend of Fig. 2, and the bar graph represents the luciferase/β-galactosidase activity of the wild type −1500-bp promoter in the presence of each expression vector relative to that cotransfected with CMV-GH. B, A MMTV promoter driving expression of luciferase was cotransfected with expression vectors for either GH, AR, or various AR mutants as depicted to the left of the graph. The bar graph represents the luciferase/β-galactosidase activity of the MMTV promoter in the presence of each expression vector relative to that cotransfected with CMV-GH. C, Western blot analysis of AR vectors expressed in transfected αT3 cells. CMV expression vectors of wild type AR and each AR mutant were transfected into αT3 cells and Western blot analysis performed using 40 μg of whole cell lysate. CMV-5 (empty vector, lane 1), pCMVhAR (lanes 2 and 5), C576A (lane 3), Δ660–919 (lane 4), Δ14–150 (lane 6), Δ142–337 (lane 7), Δ338–499 (lane 8), Δ500–558 (lane 9), Δ538–614 (lane 10), and Δ615–633 (lane 11).
The same receptor constructs were also analyzed for their ability to stimulate activity of the mouse mammary tumor virus promoter, which contains androgen-responsive hormone-response elements. Unlike repression, transcriptional stimulation requires a domain within the amino terminus (Δ142–337, Fig. 5B). An intact DBD is also essential. Minor roles in activation by a second region within the amino terminus [amino acids (aa) 338–499] and the LBD are suggested by the less than optimal activation response when these regions are deleted. Importantly, Western blot analysis confirmed that each of these mutant receptors are expressed in transfected αT3 cells (Fig. 5C). These findings are consistent with previously reported studies on activation of transcription by AR (21, 22).
To further validate the involvement of the DBD and LBD in repression of α-subunit promoter activity, we cotransfected the wild type −1500-bp reporter construct with two additional mutants, one containing only the DBD (aa 507–660) and the other the DBD plus the LBD (aa 507–919). Both constructs were fully capable of promoter repression (Fig. 6A). In contrast, they were incapable of stimulating mouse mammary tumor virus (MMTV) promoter activity (Fig. 6B). On the basis of these data, it is tempting to conclude that the DBD alone contains the necessary information required for suppression of α-subunit promoter activity. It is important to recognize, however, that this is only true when the amino-terminal domain of AR is absent, as a construct missing only the LBD fails to repress (Δ660–919; Fig. 5B). Thus, in the context of the entire receptor, both the DBD and LBD are required for suppression of transcription.
Fig. 6. The DBD of AR Is Sufficient for Repression of the α-Subunit Promoter.
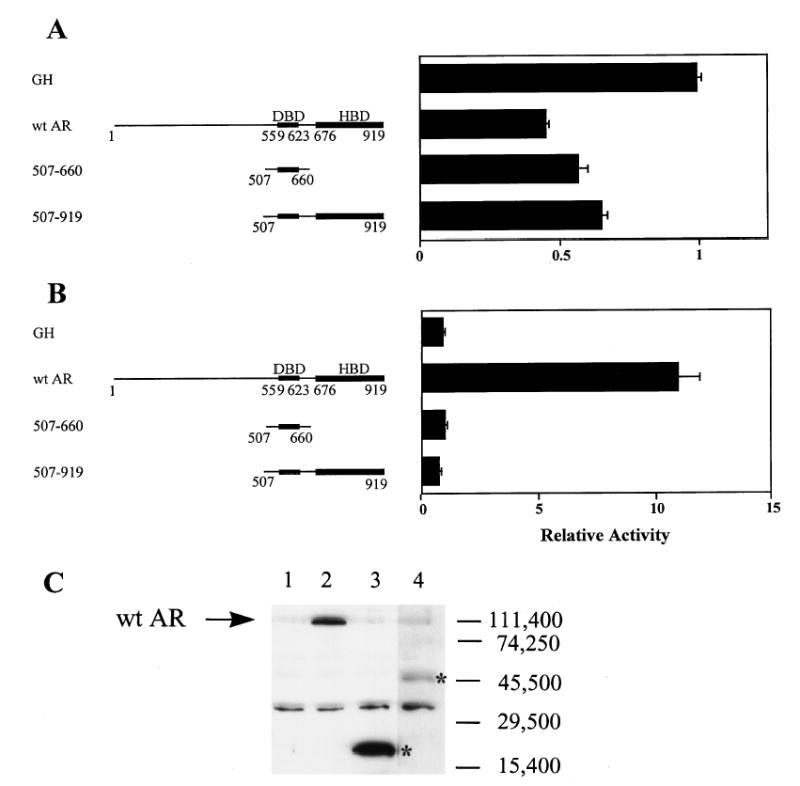
A, The wild type −1500-bp α-subunit promoter construct was cotransfected with expression vectors for either GH, AR, or two AR mutants containing only the DBD (aa 507–660) or the DBD plus the LBD (aa 507–919) as depicted to the left of the graph. Transfections were as described in the legend of Fig. 2 and the bar graph represents the luciferase/β-galactosidase activity of the wild type −1500-bp promoter in the presence of each expression vector relative to that cotransfected with CMV-GH. B, The MMTV promoter driving expression of luciferase was cotransfected with expression vectors for either GH, AR, or two AR mutants containing only the DBD (aa 507–660) or the DBD plus the LBD (aa 507–919). The bar graph represents the luciferase/β-galactosidase activity of the MMTV promoter in the presence of each expression vector relative to that cotransfected with CMV-GH. C, Western blot analysis of AR vectors expressed in transfected αT3 cells. CMV expression vectors of wild type AR and each AR mutant were transfected into αT3 cells and Western blot analysis performed using 40 μg of whole cell lysate. CMV-5 (empty vector, lane 1), pCMVhAR (lane 2), 507–660 (lane 3), and 507–919 (lane 4). Migrations of the AR mutants 507–660 and 507–919 are marked with an asterisk.
DISCUSSION
AR represses transcription of several genes (9, 23–28). Nevertheless, the mechanism(s) responsible for this suppression have only recently begun to emerge (29–31). This report is among the first to examine how AR attenuates activity of a promoter within its natural context. More importantly, there are several features that distinguish the α-subunit promoter from other promoters negatively regulated by AR, suggesting that transcriptional repression by AR can occur through multiple mechanisms that depend on promoter context. As far as we know, this is the first example of steroid receptor-dependent negative regulation being mediated by a paired combination of regulatory elements that bind other specific transcription factors.
Multiple examples exist in the literature whereby nuclear receptors repress promoter activity by interfering with other transcription factors. In most cases, members of the Fos/Jun/ATF family, the Oct family, and NF-κB appear to be targets for transcriptional interference (32, 33). With the glucocorticoid receptor (GR), in vitro data suggest that repression is the result of direct interaction between components of the AP-1 complex, Fos and Jun, and GR (32, 34, 35). However, this mechanism has not been confirmed in vivo.
Even though the α-subunit promoter contains a high-affinity binding site for AR (9), our present study clearly establishes that this site does not mediate the negative transcriptional effect of androgens. Instead, two other regulatory elements (αBE and the tandem CREs) appear to be the targets for androgen-dependent negative regulation of the α-subunit promoter. The involvement of the CREs in AR repression suggests that AR may directly interact with members binding this element. Repression of the variant CRE, which binds only Jun and ATF2, further implicates these proteins in interactions with AR. Such a mechanism is supported by earlier finding of Kallio et al., who showed that AR inhibited c-Jun binding to an AP-1 site in vector DNA sequences (31). This information, together with the data on interactions between GR and c-jun, implicates c-jun as a common target for members of the steroid receptor family.
Although interaction between AR and c-jun may be important for the CRE involvement associated with repression of the α-subunit promoter, it clearly cannot fully explain the mechanism, since an additional element (αBE) is also required for AR-dependent suppression. The mutation that established the involvement of αBE in AR repression spans from −319 to −291 of the α-subunit promoter. This region was previously shown to contain two response elements important for promoter activity, αBE1 and αBE2 (19). Analysis of smaller mutations within each of these elements revealed that full response to AR requires that both elements be present (data not shown). Furthermore, earlier studies on the α-subunit promoter in gonadotropes revealed that complex interactions occur between proteins binding the αBE and CREs. This suggested that a shared coactivator may be involved in transducing the transcriptional signal from the proteins that bind αBE and the CREs (19, 20). Such a mechanism may help explain the requirement for two response elements in androgen repression by invoking this intermediate or coactivator as a target for AR.
Several recent reports indicate that the LBDs of multiple nuclear receptors interact with the CREB-binding protein, CBP, a coactivator required for activation of both CREB and AP-1 (36–38). This suggested that repression of AP-1 activity by steroid receptors results from a competition for limiting amounts of CBP. In this regard, participation of the CREs in transcriptional repression of the α-subunit gene also implicates CBP, or other coactivator that interacts with proteins binding the αBE and CREs, as a possible target for the observed promoter effects.
The co-requirement for αBE in AR-dependent suppression of α-subunit promoter activity is of interest in light of an even more recent report indicating that AR negatively regulates matrix metalloproteinase-I expression not through AP-1 but through a family of Ets-related transcription factors that are also required for positive regulation (29). This study revealed a direct protein-protein interaction between an Ets protein and AR. Interestingly, within αBE there is an Ets protein-binding site. Despite our earlier findings that the Ets site is not required for basal promoter activity (19), it remains possible that interactions with an Ets protein at this site may be a prerequisite for promoter repression. Thus, the need for two distinct elements may be explained through a mechanism in which AR is coupled to the promoter via the Ets protein and brought into proximity of the CRE and thereby facilitates its interference of the transcription factors bound to that element.
Another important feature of our study is the demonstration that repression of the α-subunit promoter requires two distinct domains of AR: the DBD and the LBD. Although both domains are required in the context of the full-length receptor, the DBD alone can function as an efficient repressor when isolated from both the amino-terminal domain and the LBD. This suggests that, in the presence of ligand, the LBD may prevent the amino-terminal half of the receptor from masking the suppressive activity of AR that resides within the DBD. The requirement for ligand in AR-dependent repression of the α-subunit promoter was previously established (9). Two other reports have identified regions of AR required for repression and differ from our study in that repression domains within the amino terminus of AR were identified (29, 31). In one study, the amino terminus (aa 38 and 296), the LBD, and the DBD (point mutation C562G) were all implicated as important for transcriptional repression by AR (31). A more recent report concluded that repression required multiple redundant domains in the amino terminus (29). One such domain, encompassing aa 510–536, was deemed sufficient for repression. All of our constructs with repressive activity contained this domain or other potential redundant sequences. Our experiments, therefore, did not directly test whether a redundant amino-terminal domain of AR is required for suppression of α-subunit promoter activity.
Although both activation and repression require an AR with an intact DBD and LBD, there are some important distinctions in the use of these domains. In activation, the DBD functions to bind the receptor to a response element, differing from its function in α-subunit repression where direct binding to a promoter-regulatory element is not required. The LBD also plays a role in both activation and repression. In our studies, removal of the LBD eliminates repression by the receptor. In contrast, with activation, the receptor is still capable of constitutive activity but to a lesser extent than the wild type. Importantly, there is a discrete domain within the amino-terminal region of AR that is required for activation that does not participate in repression, further underscoring that there are domain-specific differences in the stimulatory and repressive properties of AR. Our goal now is to further elucidate the roles of the DBD and LBD in repression of the α-subunit gene.
MATERIALS AND METHODS
Materials
DHT was purchased from Sigma (St. Louis, MO). Radionuclides were purchased from DuPont-New England Nuclear (Boston, MA). DNA-modifying enzymes and restriction enzymes were purchased from either Boehringer Mannheim (Indianapolis, IN) or GIBCO/BRL (Gaithersburg, MD).
DNA
All plasmid DNAs were prepared from overnight bacterial cultures using Qiagen DNA plasmid columns according to the supplier’s protocol (Qiagen, Chatsworth, CA). Oligonucleotides were purchased from Midland Scientific (Midland, Texas).
Purified AR and Antibody
Bacterially expressed and purified rat AR DBD (aa residues 460–704, rAR-DBD) was prepared as previously described (39). The antibody (AR52) used in the electrophoretic mobility shift assays is directed against an epitope (aa residues 527–541) that maps to exon B of the rat AR (41).
Clones
Generation of the human α-subunit promoter constructs was described previously (19, 20). Clones containing mutations in the PGBE, αBE, GSE, URE, CRE, and ARE were described previously as μ7, μ8, μ11, μ13, μ14, and μ15, respectively (19). The −1500 TGAT promoter construct is described elsewhere (20). Wild type human AR expression vector consists of the full-length AR cDNA fused to the CMV promoter (39). All AR mutants and GH are expressed from the CMV promoter. CMV-GH (43), AR mutants 507–660, 507–919, 1–660, (22), hAR Δ142–337 (44), Δ338–499 (45) and hAR(C576A), Δ14–150, Δ615–633, Δ538–614 (21) are described. Δ500–558 was constructed similarly to those described elsewhere (22). MMTV-Luc was generated by isolating a 2710 NheI/BamHI fragment containing the luciferase gene and SV40 sequences from pGL2-Basic (Promega, Madison, WI) and subcloning them into the NheI/BamHI sites downstream of the MMTV long terminal repeat in pMAMneo (CLONTECH, Palo Alto, CA). This resulted in the removal of a 3566-bp fragment from the pMAMneo vector containing the Neor cassette and most of the SV40 sequences.
Cell Culture and Transfections
αT3 cells were grown in DMEM supplemented with 5% FBS, 5% horse serum, penicillin, and streptomycin (GIBCO, Grand Island, NY) (46). Transfection and enzyme assays were performed as described previously (19). Briefly, each luciferase construct (1.25 μg) was cotransfected with 60 ng of either CMV-GH or pCMVhAR expression vectors and 0.42 μg RSV-β-galactosidase using 5 μl lipofectamine. The lipofectamine/DNA solution was replaced with complete medium containing 100 nm DHT after 12–16 h. Cells were harvested 48 h later using 150 μl of lysis buffer (Promega). Luciferase activity was quantified by luminescence using 10–20 μl lysate and 100 μl luciferase assay reagent (Promega). β-Galactosidase activity was quantified also by luminescence using the Galacto-light assay system (Tropix, Bedford, MA). For each sample, the luciferase/β-galactosidase activity of each construct was normalized to the luciferase/β-galactosidase activity of the wild type promoter in the presence of CMV-GH. The values were then averaged over a minimum of three independent experiments.
Electrophoretic Mobility Shift Analysis and Supershift Analysis
Preparation of nuclear extracts (47) and electrophoretic mobility shift analysis were done as previously described (9, 19). Briefly, rAR-DBD (100 ng) was preincubated at 4 C for 15 min in binding buffer (12.5 mm HEPES, pH 7.9, 25 mm KCl, 10 mm MgCl2, 10% glycerol, 0.5% Triton X-100, 0.5 mM dithiothreitol) with 1 μg of poly(deoxyinosinic-deoxycytidylic)acid and 200 ng each of Escherichia coli DNA and salmon sperm DNA. Approximately 10 fmol of probe were added to the reaction, and binding was allowed to proceed for 10 min at 4 C. Subsequently, 1 μg of anti-AR DBD antibody, AR-52, was added to the appropriate binding reactions, and incubations were continued for an additional 20 min at room temperature. An oligonucleotide containing the sequence for the α-subunit promoter ARE (5′-GTCATGGTAATTACACCAAGRACCCTTCAA-3′) was added to indicated reactions at a concentration 200× that of added probe. Competitor was added to the reaction before the addition of probe. After an additional incubation for 20 min at 4 C, samples were subjected to electrophoresis in 4% polyacrylamide gels (0.5× Tris-borate-EDTA) at 4 C for 4–5 h. Gels were dried and visualized by autoradiography.
Western Blot Analysis
Expression plasmids (15 μg) were transfected into αT3 cells seeded onto 100-mm plates at a density of 1.5 × 106 cells per plate as described above. After 48 h, cells were harvested by washing two times in PBS, adding 1 ml of TEN buffer (40 mm Tris, 7.5, 1 mm EDTA, 150 mm NaCl) and incubating at room temperature for 5 min. Cells were removed from the plates, pelleted, and resuspended in 150 μl 0.25 m Tris-HCl (7.8). Cells were lysed by a series of three freeze-thaw cycles, the debris was removed by centrifugation, and the supernatant was assayed for protein. Lysate (40 μg) was resolved on an SDS-PAGE gel using a 4% stacking gel and a 10% resolving gel as described elsewhere (19). Proteins were then transferred to a nitrocellulose membrane (Schleicher & Schuell, Keene, NH) and rinsed in a solution of PBS-Tween (0.1% Tween 20 in PBS). The membrane was blocked using blotto (0.1% Tween, 5% Carnation nonfat dry milk, 0.02% sodium azide in PBS) at room temperature for 8 h and then incubated with the AR antibody AR52 at a dilution of 1:2000 in blotto overnight at room temperature. The membrane was rinsed two times for 15 min each at room temperature in PBS-Tween and then incubated with the secondary antibody (anti-rabbit horseradish peroxidase 1:7500, Santa Cruz Bio-technology, Santa Cruz, CA) for 2 h at room temperature in blotto. Membranes were washed in PBS-Tween twice for 5 min, once for 25 min, and twice for 10 min followed by one quick rinse in PBS alone. Proteins were visualized by chemiluminescence using the enhanced chemiluminescence Western blotting detection reagents by Amersham (Arlington Heights, IL).
Footnotes
This work was supported by NIH Grants DK-28559 and HD-34032 (to J.H.N.) and DK-08975–02 (to L.L.H.).
References
- 1.Fiddes JC, Talmadge K. Structure, expression, and evolution of the genes encoding the human glycoprotein hormones. Recent Prog Horm Res. 1984;40:43–78. doi: 10.1016/b978-0-12-571140-1.50006-2. [DOI] [PubMed] [Google Scholar]
- 2.Haisenleder DJ, Dalkin AC, Marshall JC 1994 Regulation of gonadotropin gene expression; In: Knobil E, Neill JD (eds) The Physiology of Reproduction, ed 2. Raven Press, New York, pp 1793–813
- 3.Conn PM 1994 The molecular mechanism of gonadotropin-releasing hormone action in the pituitary. In: Knobil E, Neill JD (eds) The Physiology of Reproduction, ed 2. Raven Press, New York, pp 1815–1832
- 4.Brinkley HJ. Endocrine signalling and female reproduction. Biol Reprod. 1981;24:22–43. doi: 10.1095/biolreprod24.1.22. [DOI] [PubMed] [Google Scholar]
- 5.Desjardins C. Endocrine signalling and male reproduction. Biol Reprod. 1981;24:1–21. doi: 10.1095/biolreprod24.1.1. [DOI] [PubMed] [Google Scholar]
- 6.Gharib SD, Wierman ME, Shupnik MA, Chin WW. Molecular biology of the pituitary gonadotropins. Endocr Rev. 1990;11:177–199. doi: 10.1210/edrv-11-1-177. [DOI] [PubMed] [Google Scholar]
- 7.Chedrese PJ, Kay TWH, Jameson JL. Gonadotropin-releasing hormone stimulates glycoprotein hormone α-subunit messenger ribonucleic acid (mRNA) levels in αT3 cells by increasing transcription and mRNA stability. Endocrinology. 1994;134:2475–2481. doi: 10.1210/endo.134.6.7515001. [DOI] [PubMed] [Google Scholar]
- 8.Roberson MS, Misra-Press A, Laurance ME, Stork PJS, Maurer RA. A role for mitogen-activated protein kinase in mediating activation of the glycoprotein hormone α-subunit promoter by gonadotropin-releasing hormone. Mol Cell Biol. 1995;15:3531–3539. doi: 10.1128/mcb.15.7.3531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Clay CM, Keri RA, Finicle AB, Heckert LL, Hamernik DL, Marschke KM, Wilson EM, French FS, Nilson JH. Transcriptional repression of the glycoprotein hormone alpha subunit gene by androgen may involve direct binding of androgen receptor to the proximal promoter. J Biol Chem. 1993;268:13556–13564. [PubMed] [Google Scholar]
- 10.Keri RA, Andersen B, Kennedy GC, Hamernik DL, Clay CM, Brace AD, Nett TM, Notides AC, Nilson JH. Estradiol inhibits transcription of the human glycoprotein hormone α-subunit gene despite the absence of a high affinity binding site for estrogen receptor. Mol Endocrinol. 1991;5:725–733. doi: 10.1210/mend-5-5-725. [DOI] [PubMed] [Google Scholar]
- 11.Winters SJ, Ishizaka K, Kitahara S, Troen P, Attardi B. Effects of testosterone on gonadotropin subunit messenger ribonucleic acids in the presence or absence of gonadotropin-releasing hormone. Endocrinology. 1992;130:726–734. doi: 10.1210/endo.130.2.1370794. [DOI] [PubMed] [Google Scholar]
- 12.Keri RA, Wolfe MW, Saunders TL, Anderson I, Kendall S, Wagner T, Yeung J, Gorski J, Nett TM, Camper SA, Nilson JH. The proximal promoter of the bovine luteinizing hormone β-subunit gene confers gonadotrope-specific expression and regulation by gonadotropin-releasing hormone, testosterone, and 17β-estradiol in transgenic mice. Mol Endocrinol. 1994;8:1807–16. doi: 10.1210/mend.8.12.7708066. [DOI] [PubMed] [Google Scholar]
- 13.Naess O, Attramadal A, Aakvaag A. Androgen binding proteins in he anterior pituitary, hypothalamus, pre-optics area and brain cortex of the rat. Endocrinology. 1975;96:1–9. doi: 10.1210/endo-96-1-1. [DOI] [PubMed] [Google Scholar]
- 14.Thieulant M, Pelletier J. Evidence for androgen and estrogen receptors in castrated ram pituitary cytosol; in castrated ram pituitary cytosol: influence of time after castration. J Steroid Biochem. 1979;10:677–87. doi: 10.1016/0022-4731(79)90521-1. [DOI] [PubMed] [Google Scholar]
- 15.Bonsall RW, Rees HD, Michael RP. The distribution, nuclear uptake, and metabolism of [3H]dihydrotestoster-one in the brain, pituitary gland and genital tract of the rhesus monkey. J Steroid Biochem. 1985;23:389–98. doi: 10.1016/0022-4731(85)90184-0. [DOI] [PubMed] [Google Scholar]
- 16.Wierman ME, Wang C. Androgen selectively stimulates follicle-stimulating hormone β mRNA levels after gonadotropin-releasing hormone antagonist administration. Biol Reprod. 1990;42:563–71. doi: 10.1095/biolreprod42.3.563. [DOI] [PubMed] [Google Scholar]
- 17.Schoderbek WE, Kim KW, Ridgway EC, Mellon PL, Maurer RA. Analysis of DNA sequences required for pituitary-specific expression of the glycoprotein hormone alpha-subunit gene. Mol Endocrinol. 1992;6:893–903. doi: 10.1210/mend.6.6.1379672. [DOI] [PubMed] [Google Scholar]
- 18.Horn F, Windle JJ, Barnhart KM, Mellon PL. Tissue-specific gene expression in the pituitary: The glycoprotein hormone α-subunit gene is regulated by a gonadotrope-specific protein. Mol Cell Biol. 1992;12:2143–53. doi: 10.1128/mcb.12.5.2143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Heckert LL, Schultz K, Nilson JH. Different composite regulatory elements direct expression of the human α subunit gene to pituitary and placenta. J Biol Chem. 1995;270:26497–504. doi: 10.1074/jbc.270.44.26497. [DOI] [PubMed] [Google Scholar]
- 20.Heckert LL, Schultz K, Nilson JH. The cAMP response elements of the human alpha subunit gene bind similar proteins in trophoblasts and gonadotropes but have distinct functional sequence requirements. J Biol Chem. 1996;271:31650–31656. doi: 10.1074/jbc.271.49.31650. [DOI] [PubMed] [Google Scholar]
- 21.Zhou ZX, Sar M, Simental JA, Lane MV, Wilson EM. A ligand-dependent bipartite nuclear targeting signal in the human androgen receptor. J Biol Chem. 1994;269:13115–13123. [PubMed] [Google Scholar]
- 22.Simental JA, Sar M, Lane MV, French FS, Wilson EM. Transcriptional activation and nuclear targeting signals of the human androgen receptor. J Biol Chem. 1991;266:510–518. [PubMed] [Google Scholar]
- 23.Bellido T, Jilka RL, Boyce BF, Girasole G, Broxmeyer H, Dalrymple SA, Murray R, Manolagas SC. Regulation of interleukin-6, osteoclastogenesis, and bone mass by androgens. The role of the androgen receptor. J Clin Invest. 1995;95:2886–2895. doi: 10.1172/JCI117995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Leppa S, Mali M, Miettinen HM, Jalkanen M. Syndecan expression regulates cell morphology and growth of mouse mammary epithelial tumor cells. Proc Natl Acad Sci USA. 1992;89:932–936. doi: 10.1073/pnas.89.3.932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Metsis M, Timmusk T, Allikmets R, Saarma M, Persson H. Regulatory elements and transcriptional regulation by testosterone and retinoic acid of the rat nerve growth factor receptor promoter. Gene. 1992;121:247–254. doi: 10.1016/0378-1119(92)90128-c. [DOI] [PubMed] [Google Scholar]
- 26.Persson H, Lievre CA-L, Soder O, Villar MJ, Metsis M, Olson L, Ritzen M, Hokfelt T. Expression of beta-nerve growth factor receptor mRNA in Sertoli cells down-regulated by testosterone. Science. 1990;247:704–707. doi: 10.1126/science.2154035. [DOI] [PubMed] [Google Scholar]
- 27.Henttu P, Liao SS, Vihko P. Androgens up-regulate the human prostate-specific antigen messenger ribonucleic acid (mRNA), but down-regulate the prostatic acid phosphatase mRNA in the LNCaP cell line. Endocrinology. 1992;130:766–772. doi: 10.1210/endo.130.2.1370795. [DOI] [PubMed] [Google Scholar]
- 28.Chatterjee B, Majumdar D, Ozbilen O, Murty CV, Roy AK. Molecular cloning and characterization of cDNA for androgen-repressible rat liver protein, SMP-2. J Biol Chem. 1987;262:822–825. [PubMed] [Google Scholar]
- 29.Schneikert J, Peterziel H, Defossez P-A, Klocker H, de Laumoit Y, Cato ACB. Androgen receptorets protein interaction is a novel mechanism for steroid hormone-mediated down-modulation of matrix metalloproteinase expression. J Biol Chem. 1996;271:23907–23913. doi: 10.1074/jbc.271.39.23907. [DOI] [PubMed] [Google Scholar]
- 30.Palvimo JJ, Kallio PJ, Ikonen T, Mehto M, Janne OA. Dominant negative regulation of trans-activation by the rat androgen receptor: roles of the N-terminal domain and heterodimer formation. Mol Endocrinol. 1993;7:1399–1407. doi: 10.1210/mend.7.11.8114755. [DOI] [PubMed] [Google Scholar]
- 31.Kallio PJ, Poukka H, Moilanen A, Janne OA, Palvimo JJ. Androgen receptor-mediated transcriptional regulation in the absence of direct interaction with a specific DNA element. Mol Endocrinol. 1995;9:1017–1028. doi: 10.1210/mend.9.8.7476976. [DOI] [PubMed] [Google Scholar]
- 32.Herrlich P, Ponta H. Mutual cross-modulation of steroid/retinoic acid receptor and AP-1 transcription factor activities. Trends Endocrinol Metab. 1994;5:341–6. doi: 10.1016/1043-2760(94)90164-3. [DOI] [PubMed] [Google Scholar]
- 33.Beato M, Herrlich P, Schutz G. Steroid hormone receptors: many actors in search of a plot. Cell. 1995;83:851–7. doi: 10.1016/0092-8674(95)90201-5. [DOI] [PubMed] [Google Scholar]
- 34.Yamamoto KR, Pearce D, Thomas J, Miner JN 1992 Combinatorial regulation at a mammalian composite response element. In: McKnight SL, Yamamoto KR (eds) Transcriptional Regulation. Cold Spring Harbor Laboratory Press, Plainview, NY, pp 1169–1192
- 35.Schule R, Rangarajan P, Kliewer S, Ransone LJ, Bolado J, Yang N, Verma IM, Evans RM. Functional antagonism between oncoprotein c-Jun and the glucocorticoid receptor. Cell. 1990;62:1217–1226. doi: 10.1016/0092-8674(90)90397-w. [DOI] [PubMed] [Google Scholar]
- 36.Kwok RP, Lundblad JR, Chrivia JC, Richards JP, Bachinger HP, Brennan RG, Roberts SG, Green MR, Goodman RH. Nuclear protein CBP is a coactivator for the transcription factor CREB. Nature. 1994;370:223–226. doi: 10.1038/370223a0. [DOI] [PubMed] [Google Scholar]
- 37.Arias J, Alberts AS, Brindle P, Claret FX, Smeal T, Karin M, Feramisco J, Montminy M. Activation of cAMP and mitogen responsive genes relies on a common nuclear factor. Nature. 1994;370:226–229. doi: 10.1038/370226a0. [DOI] [PubMed] [Google Scholar]
- 38.Kamei Y, Xu L, Hainzel T, Torchia J, Kurokawa R, Gloss B, Lin S, Heyman R, Rose DW, Glass CK, Rosenfeld RG. A CBP integrator complex mediates transcriptional activation and AP-1 inhibition by nuclear receptors. Cell. 1996;85:403–414. doi: 10.1016/s0092-8674(00)81118-6. [DOI] [PubMed] [Google Scholar]
- 39.Tan J, Marschke KB, Ho KC, Perry ST, Wilson EM, French FS. Response elements of the androgen regulated C3 gene. J Biol Chem. 1992;267:4456–4466. [PubMed] [Google Scholar]
- 40.Quarmby VE, Kemppainen JA, Sar M, Lubahn DB, French FS, Wilson EM. Expression of recombinant androgen receptor in cultured mammalian cells. Mol Endocrinol. 1990;4:1399–1407. doi: 10.1210/mend-4-9-1399. [DOI] [PubMed] [Google Scholar]
- 41.Tan JA, Joseph DR, Quarmby VE, Lubahn DB, Sar M, French FS, Wilson EM. The rat androgen receptor: primary structure, autoregulation of its messenger RNA and immunocytochemical localization of the receptor protein. Mol Endocrinol. 1988;2:1276–1285. doi: 10.1210/mend-2-12-1276. [DOI] [PubMed] [Google Scholar]
- 42.Sar M, Lubahn DB, French FS, Wilson EM. Immunohistochemical localization of the androgen receptor in rat and human tissues. Endocrinology. 1990;127:3180–3286. doi: 10.1210/endo-127-6-3180. [DOI] [PubMed] [Google Scholar]
- 43.Keri RA, Nilson JH. A steroidogenic factor-1 binding site is required for activity of the luteinizing hormone B subunit promoter in gonadotropes of transgenic mice. J Biol Chem. 1996;271:10782–10785. doi: 10.1074/jbc.271.18.10782. [DOI] [PubMed] [Google Scholar]
- 44.Zhou Z, Sar M, French FS, Wilson EM 1993 Molecular biological aspects of the human androgen receptor relating to disease. In: Moudgil V (ed) Steroid Hormone Receptors: Basic and Clinical Aspects. Birkhauser, New York, pp 407–426
- 45.Langley E, Zhou ZX, Wilson EM. Evidence for an anti-parallel orientation of the ligand activated human androgen receptor. J Biol Chem. 1995;270:29983–29990. doi: 10.1074/jbc.270.50.29983. [DOI] [PubMed] [Google Scholar]
- 46.Windle JJ, Weiner RI, Mellon PL. Cell lines of the pituitary gonadotrope lineage derived by targeted oncogenesis in transgenic mice. Mol Endocrinol. 1990;4:597–603. doi: 10.1210/mend-4-4-597. [DOI] [PubMed] [Google Scholar]
- 47.Abmayr SM, Workman JL 1994 Preparation of nuclear and cytoplasmic extracts from mammalian cells. In: Ausubel FM, Brent R, Kingston RE, Moore DD, Seidman JG, Smith JA, Struhl K (eds) Current Protocols in Molecular Biology. Greene and Wiley Inter-Science, New York, pp 12.1.1–9