


Abstract
Smad proteins transduce signals from transforming growth factor-β (TGF-β) receptors and regulate transcription of target genes. TGF-β is implicated in the regulation of the smooth muscle cell specific gene SM22α, but little is known about how Smads are involved in SM22α gene transcription. In this report, we demonstrate that TGF-β activation of the SM22α promoter is Smad dependent in C3H10T1/2 cells, BALB 3T3 cells and neural crest Monc-1 cells. We find that the promoter region from –162 to +41 is sufficient to up-regulate the reporter gene upon TGF-β induction. Smad3, Smad1 and Smad4 are found in TGF-β inducible complexes that bind to a region containing a Smad binding site (SBS) and a medea box. Both the SBS and medea box are necessary for complex formation and are functionally important. Smad4 is limiting for TGF-β induction, and Smad3, but not Smad1, significantly contributes to maximal activation. Time course luciferase assays and time course gel mobility shift assays reveal that the Smad3/4 complex is largely responsible for the immediate response of the SM22α promoter to TGF-β induction, and also contributes to the maximal promoter activity. We further demonstrate that AP-1 elements contribute to induction of the SM22α promoter by TGF-β.
INTRODUCTION
During development and growth of the vasculature, recruitment and differentiation of smooth muscle cells (SMCs) is a critical process. SMCs provide hemostatic control and protect new endothelium-lined vessels against rupture or regression. SMCs also assist endothelial cells in acquiring specialized functions in different vascular beds and maintain vascular tone and integrity in the adult (1). The mechanisms of SMC differentiation from various precursors remain obscure, but transforming growth factor-β (TGF-β) family ligands and downstream signaling intermediates have been shown to play roles in SMC differentiation in a variety of systems. TGF-β1, TGF-β receptors (including endoglin, TβR-II, TβR-I and Alk1) and signaling intermediates have all been identified by homologous recombination in the mouse as critical for embryonic angiogenesis. Loss of any of these components leads to defects in smooth muscle cell recruitment and/or differentiation during angiogenesis in the developing embryo and extraembryonic structures (2). In vitro studies have shown that TGF-β1 can induce differentiation of C3H10T1/2 cells to a smooth muscle phenotype directly (3). Likewise, TGF-β can induce neural crest stem cells to express SMC differentiation markers (4), suggesting that the role of TGF-β in regulating SMC differentiation is fairly broad.
To understand the molecular mechanism of SMC differentiation, the expression of SMC specific genes such as α-SM actin, SM myosin heavy chain and SM22α has been studied by several groups (5,6). Among these, SM22α is one of the earliest and most widely expressed SMC markers identified (7–10). SM22α is a 22-kDa calponin-related protein that interacts with other contraction-associated proteins such as tropomyosin and F-actin. SM22α expression is specific for smooth muscle, and the 5′ regulatory regions necessary for tissue specific expression have been well characterized. TGF-β can induce expression of SM22α, and this transcriptional regulation is under at least partial control of a TGF-β control element (TCE) in the 5′ regulatory region. Although the TCE had been shown to bind to gut-enriched Kruppel-like factor (GKLF), GKLF repressed the TGF-β-dependent increases of SM22α promoter activity, and it is unclear what factors bind the TCE under physiological conditions (11). Other elements in the promoter responsive to TGF-β, including CArG elements, have been identified (12), but the mechanism whereby TGF-β regulates these elements has not been fully elucidated.
TGF-β signaling is initiated by ligand binding to the transmembrane receptor serine-threonine kinases, TβR-I and TβR-II. Signaling from the receptors to the nucleus occurs predominantly by phosphorylation of receptor-associated Smad proteins (R-Smads) (13,14), which combine with the common Smad, Smad4, and translocate into the nucleus where they function as transcription factors alone or in association with other DNA binding factors. Much work clearly demonstrates, however, that other pathways including those activated by MAP and JNK kinases and ras also play a role (15). The aim of this study was to investigate the function of Smad proteins in TGF-β-induced SM22α gene transcription. Using reporter gene assays with the SM22α promoter and mutational analysis, we find that the SM22α promoter is Smad-dependent. We further demonstrate that Smad proteins bind to a bipartite site in the first exon and 5′ flanking sequence of the SM22α gene. We provide evidence that Smad proteins are responsible for the immediate SM22α gene activation upon TGF-β stimulation. We further show that AP-1 proteins may contribute to Smad-dependent activation of the SM22α promoter.
MATERIALS AND METHODS
Plasmids
Smad expression vectors have been described (16,17). SM22α promoter luciferase constructs (a generous gift from Dr Julian Solway, University of Chicago, Chicago, IL) have been described (8). Promoter mutants were generated using the Quikchange™ site-directed mutagenesis kit (Stratagene) according to the manufacturer’s recommendations. The Smad binding site (SBS) sequence GTCT (from nucleotide –157 to –154) was substituted to GAGA for p-162SM22m4, CAGAC (from nucleotide –95 to –91) to CACTG for p-162SM22m5, GTCT (from nucleotide +6 to +9) to CAGT for p-162SM22m6, CAGAC (from nucleotide +19 to +22) to CACTG for p-162SM22m7 and CAGA (from nucleotide –58 to –55) to CACT for p-162SM22m8. For TCE mutant construct p-162TCEm, the wild type sequence 5′-GGAGTGAGTGGGGCGGCCCG-3′ (from nucleotide –132 to –112) was substituted by 5′-GGAGTtAtTGttGCGGCCG-3′. Medea box mutant (p-162-MBm) was created by converting the sequence GCCGGCG (from nucleotide –11 to –5) to GCCttCG (18,19). Mutant plasmid p-162-SM-dm at both SBS and medea box were generated according to the individually mutated sequences. All plasmids were sequenced prior to use.
Cell culture and transfections
C3H10T1/2 (10T1/2) and Balb 3T3 cells (American Type Culture Collection, Manassas, VA), and Smad3 knockout fibroblast cells [S3KO, a gift from Dr Ester Piek (National Cancer Institute, Bethesda, MD) (20)] were grown and maintained in Dulbecco’s modified Eagle’s medium (DMEM) (Mediatech, Herndon, VA) supplemented with 10% fetal bovine serum (FBS) and 2 mM glutamine (Invitrogen, Gaithersburg, MD). Prior to transfection, cells were plated at 6 × 104 cells/well in six-well plates and incubated until they reached 70% confluency. Monc-1 cells were a gift from Dr David Anderson (California Institute of Technology, Pasadena, CA) and were grown as described (21). Before transfection, Monc-1 cells were grown in DMEM containing 10% FBS for 2 days to induce smooth muscle differentiation. Cells were transiently transfected (in triplicate) with LipofectAMINE 2000 (Invitrogen) according to the manufacturer’s recommendations. Each well received luciferase reporter construct, Renilla luciferase and other expression plasmids as indicated. The total transfected DNA was adjusted with pBSK+ DNA and/or pCDNA3.0. After transfection, the medium was replaced with fresh medium and incubated for 24 h before being treated for 16 h or the indicated time with TGF-β (5 ng/ml) (R and D Systems, Minneapolis, MN) or vehicle alone in media containing 0.2% FBS. As indicated in some experiments, a constitutively active TGF-β receptor construct was used instead of TGF-β. Luciferase activity of cell lysates was determined using the Dual-Luciferase Assay System from Promega (Madison, WI). The relative luciferase activity values were normalized to Renilla activity. All measurements represent the mean of duplicate corrected luciferase measurements from triplicate wells ± SEM. Results were analyzed using ANOVA with pairwise comparisons between relevant groups. All experiments were performed at least twice with representative results shown.
Preparation of nuclear extracts and electrophoretic mobility shift assays (EMSAs)
10T1/2 cells were plated at a density of 3 × 106/15 cm dish, grown to 70% confluency as above, and then starved in 0.2% serum medium overnight and treated with or without 5 ng/ml TGF-β for the times indicated. Nuclear extracts were prepared as described (22). Protein concentrations were measured using BCA Protein Assay Reagent (Pierce). Oligonucleotide probes were labelled and EMSAs were performed as described (22). In competitive or antibody supershift assays, competitor oligonucleotides or Smad antibodies (Dr Peter ten Dijke, The Netherlands Cancer Institute, Amsterdam, The Netherlands) were added simultaneously to the reaction buffer with labeled probes and incubated on ice for 45 min.
Reverse transcriptase–polymerase chain reaction (RT–PCR)
Cells were treated with TGF-β for the time indicated in DMEM containing 0.2% FBS. Total RNA was extracted using Trizol (Invitrogen) following the manufacturer’s instructions. RT–PCR was performed using the Superscript First Strand Synthesis System for RT–PCR (Invitrogen) and primers specific to SM22α or cyclophilin (as a loading control). Primer sequences are available upon request.
RESULTS
SM22α gene expression is Smad dependent
To determine the role of Smad proteins in regulating TGF-β-induced SM22α gene transcription, we first identified the minimum promoter region necessary for TGF-β induction in 10T1/2 cells. The SM22α promoter region from –441 to +41 bp has been shown to correctly direct arterial expression of SM22α during development (23). We found that either the regions from –162 to +41 bp or from –441 to +41 bp gave identical fold-activation to TGF-β induction in 10T1/2 cells (data not shown). The –162 to +41 bp construct was used in all subsequent studies. To determine if TGF-β induction of this construct was dependent on Smad proteins, we used a well-characterized dominant interfering Smad4 construct (Smad4ΔM4) (24). Smad4ΔM4 was cotransfected with the reporter plasmid into 10T1/2 cells and luciferase activity determined. Smad4ΔM4 significantly reduced the level of TGF-β induced luciferase activity (Fig. 1A), which demonstrated that Smad proteins play an essential role in full activation of the SM22α gene by TGF-β.
Figure 1.

The SM22α promoter is dependent on Smads. (A) Effect of a Smad4 mutant on SM22α promoter activity. p-162SM22luc plasmid was transfected with pcDNA3.0 or Smad4 mutant expression vector (Smad4ΔM4) into 10T1/2 cells and luciferase activity determined with or without TGF-β. (B) Activity of the SM22α promoter with mutations at potential Smad binding sites. The wild type or mutant plasmids were transfected into 10T1/2 cells and luciferase activity determined as described following treatment as indicated. (C) The sequence of SM22α promoter region from –162 to +41 bp. Numbers correspond to the transcription start site. The potential Smad binding, TCE and CArG sites are underlined and named as indicated. *P < 0.01 for comparison to TGF-β treated control.
Smads bind to the consensus sequences CAGA or GTCT (Smad binding sites or SBS) which confer TGF-β responsiveness (25). Sequence analysis of the SM22α 5′ regulatory region identified five potential Smad binding sites located at –157, –95, –58, +6 and +19 (Fig. 1C). To determine whether these sites function in TGF-β induction, each SBS was individually mutated and the mutant reporters used in transient transfection experiments. The SBS mutation, –162m6, decreased TGF-β induction by >50% compared to the wild type (Fig. 1B). However, the other SBS mutations –162m4, –162m5, –162m7 or –162m8 only had a slight effect on promoter response to TGF-β. These results suggest that Smad proteins may selectively activate SM22α transcription in response to TGF-β through the SBS at position +6.
Both Smad binding site and the upstream medea box are necessary for formation of a TGF-β inducible complex
Examination of the sequence surrounding the SBS at +6 uncovered an upstream GCCGGCG sequence (Fig. 2A). This GC-rich stretch matches the binding sequence for the Drosophila Smad4 homolog, medea (26) and resembles Smad responsive elements in the Id1 (27), Smad6 (28) and Col7A (29) promoters. To determine whether the SBS and medea box could bind Smad proteins following TGF-β stimulation, EMSAs were performed with nuclear extracts from 10T1/2 cells and DNA probes containing both the SBS and medea box (SM-AB) or individual sites alone (SBS or medea box). As shown in Figure 2B, the TGF-β induced complex bound to the DNA sequence containing both the SBS and medea box. The medea box alone also formed an inducible complex of similar size but lower intensity with the nuclear extracts from TGF-β treated cells, while SBS alone was not sufficient for the formation of an inducible complex. We also saw a complex of about the same size as the induced complex that was present in extracts from uninduced cells that disappeared following TGF-β treatment (Fig. 2B, lane 5). This complex did not appear to contain Smad proteins and loss of binding had no effect on promoter activity (data not shown).
Figure 2.
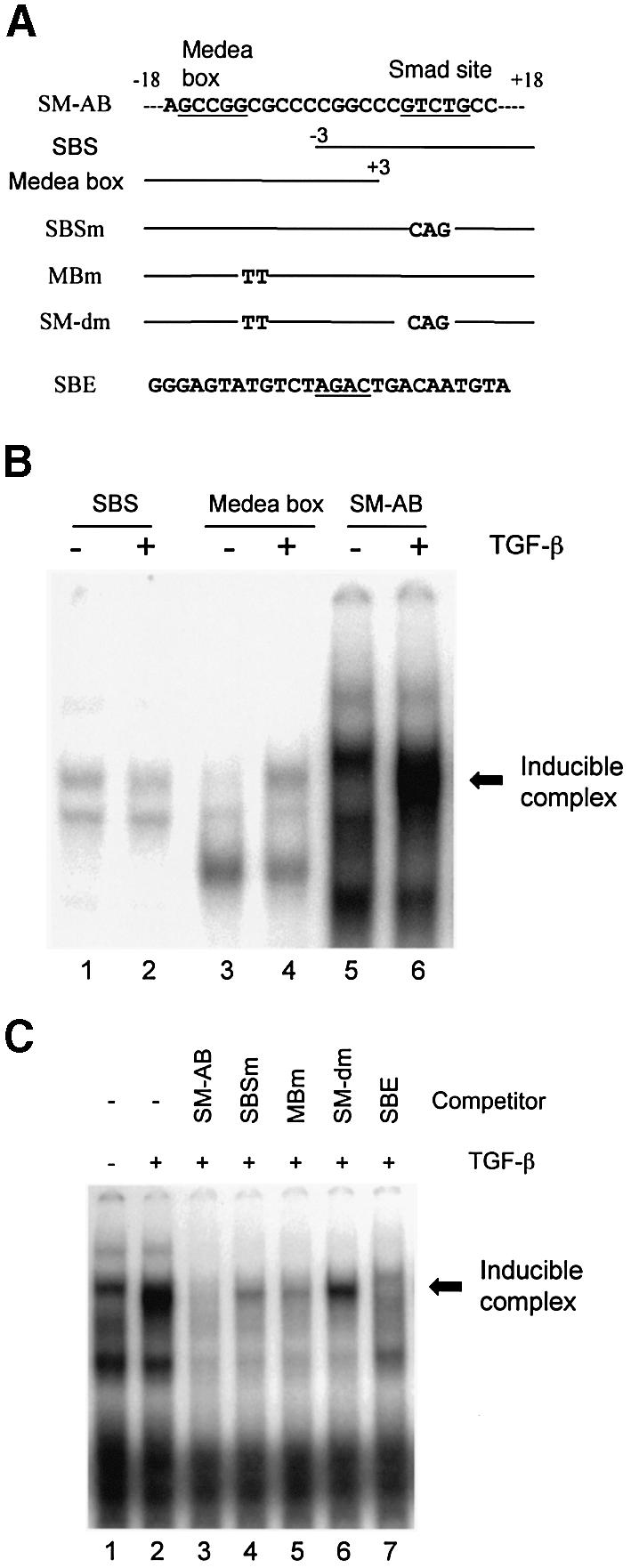
Smad proteins interact with the SM22α promoter sequence. (A) Sequence scheme of DNA fragments used in the EMSAs. Numbers correspond to distance from the transcription start site of the SM22α gene. The SBS and medea box are underlined. Substituted nucleotides for the SBS and medea box are shown. (B) EMSA with SBS and medea box. Nuclear extracts from 10T1/2 cells treated with vehicle (–) or TGF-β1 (5 ng/ml) (+) for 30 min were incubated with 32P-labeled probes for SBS, medea box, or both sites (SM-AB) and resultant complexes resolved as described. The TGF-β induced complexes in lanes 4 and 6 are indicated by arrows. (C) EMSAs with the competitive oligos were performed using labeled SM-AB and indicated cold competitor oligonucleotides.
To determine further the role of the medea box and SBS in binding TGF-β inducible proteins, the SBS and medea box were mutated separately and jointly as shown in Figure 2A. Competitive EMSA (Fig. 2C) showed that although the wild type sequence could completely compete for binding, the inducible interaction was reduced, although not completely, by the individual mutations at either SBS or medea box (lanes 4 and 5). The double mutation was unable to significantly compete for complex formation (lane 6). Together these data suggest that both SBS and medea box are necessary for the formation of the TGF-β-inducible complex. Additionally, the previously characterized Smad binding element (SBE) (30) was able to efficiently compete the formation of an inducible complex (lane 7), further demonstrating the involvement of Smad protein(s) in forming a TGF-β inducible complex.
Both the SBS and medea box contribute to SM22α gene activation
In order to determine the functional importance of the medea box and SBS, individual and joint mutations were created at the SBS and medea box in the p-162SM22luc, and luciferase activity determined following transient transfection. As shown in Figure 3A, the individual mutation at either the SBS or medea box significantly reduced promoter activity. The double mutation, however, did not further decrease reporter gene expression, suggesting that the SBS and medea box do not have a synergistic effect in response to TGF-β induction. In order to determine whether the effects we observed were unique to 10T1/2 cells, we performed similar transfection experiments in two other cell lines. In Balb 3T3 fibroblasts, introduction of the double mutation had a similar effect, lowering the induction of luciferase activity (Fig. 3B), but not eliminating it completely. As a second model of smooth muscle differentiation, we used the neural crest stem cell line Monc-1. In Monc-1 cells, TGF-β induced an ∼2-fold activation of SM22α promoter activity, and in contrast to 10T1/2 and Balb 3T3 cells, this induction was completely abolished by combined mutations in the Smad and medea sites (Fig. 3C). Similar experiments performed using the longer –441 promoter demonstrated that it, too, was inhibited to the same degree by the double mutations (data not shown). This demonstrates that Smad sites play critical roles in regulating SM22α promoter activity in multiple cell types, and are absolutely required in at least one.
Figure 3.

(A) Effect of mutations at SBS and medea box on SM22α promoter activity. Wild type (-162) or plasmids with mutation at SBS (-162-Sm), medea box (-162-MBm) or both sites (-162-SM-dm) were transfected into 10T1/2 cells and luciferase activity determined. (B and C) Wild type or mutant reporter plasmids as indicated were transfected into Balb 3T3 (B) or Monc-1 (C) cells and luciferase activity determined. *P < 0.01 for comparison to TGF-β treated -162 construct.
Smad3, Smad4 and Smad1 are present in the nuclear complexes that bind to SM22α promoter CAGA box
In the above gel shift experiments, we observed that TGF-β induced complexes were formed with both SM-AB, which contains both SBS and medea box, and medea box alone probes. The formation of either of these two complexes was completely eliminated by a consensus Smad binding element (compare with Fig. 2C; data for medea box alone is not shown). To determine which Smads are present in these TGF-β-inducible complexes, the nuclear extracts were incubated with well-characterized antibodies specific to Smads 1, 2, 3, 4 or 5 (31). We detected an obvious supershift of TGF-β dependent binding complex with anti-Smad3 and anti-Smad4 IgG using the SM-AB probe (Fig. 4A, lanes 5 and 7). A faint supershift band could also be observed with anti-Smad1 IgG (Fig. 4A, lane 3), and a slight reduction in complex intensity was observed with anti-Smad2 IgG. As demonstrated in panel B, anti-Smad1 IgG could supershift more than half of the complex formed by medea box (Fig. 4B, lane 3). Smad4 antibody showed the same degree of supershift to the complex formed by medea box alone as that by the larger SM-AB fragment containing both medea box and SBS (Fig. 4B, lane 6). Similar to the SM-AB probe, a small decrease in band intensity was seen in the complex formed with the medea box probe and anti-Smad2 IgG. Unlike SM-AB, however, anti-Smad5 IgG also caused a decrease in band intensity. We conclude that the TGF-β-inducible complex that binds SM-AB contains predominantly Smad3 and Smad4, with a small amount of Smad1, while the complex that binds the medea box contains predominantly Smad1 and Smad4 with a small contribution of Smad5 and Smad3. The presence of Smad2 in either complex cannot be completely ruled out. It is unlikely that the differences in binding seen are due to differences in expression of the various Smads, as expression levels as measured by semi-quantitative RT–PCR were roughly equal (data not shown).
Figure 4.
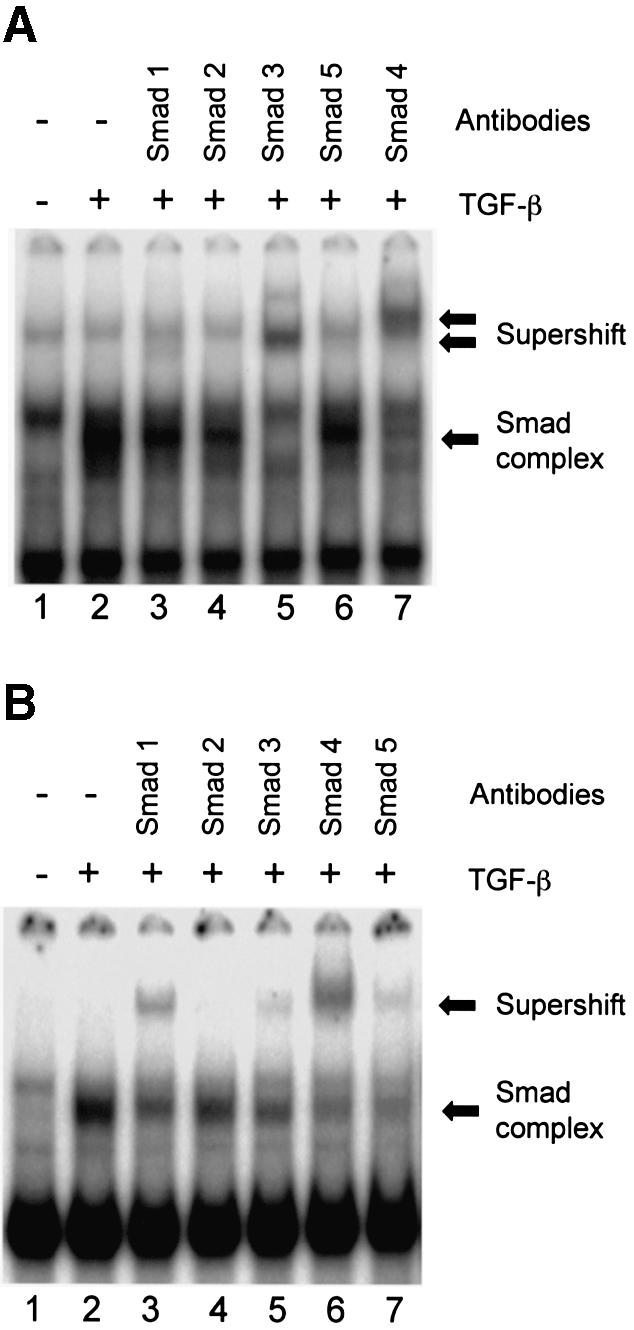
Supershift assays of proteins binding to the SBS and medea box. Nuclear extracts from 10T1/2 cells treated with vehicle (–) or TGF-β1 (+) for 30 min were incubated with radiolabeled SM-AB (A) or medea box (B). Anti-Smad antibodies were simultaneously added to the reaction. Smad complexes and antibody supershifted complexes are indicated.
Smad3 is critical to the TGF-β induction of SM22α gene promoter activity
In order to determine which Smads that bind to the SM22α cis elements contribute to promoter activation, we performed co-expression studies in 10T1/2 cells using Smad expression plasmids and the p-162SM22luc reporter. Smad1, Smad3 or Smad4 expression plasmids were co-transfected individually or in combination with p-162SM22luc plasmid and luciferase activities assayed. Preliminary experiments indicated that co-transfection of Smad1 or Smad3 in the absence of additional Smad4 had little effect on promoter activity (data not shown), suggesting that Smad4 is limiting in this assay. Co-transfection of Smad4 had a small but significant effect on promoter activity, and promoter activity was further increased by the addition of Smad3 and Smad4 together (Fig. 5A). Co-expression of Smad1 and Smad4 had no effect above that of Smad4 alone on p162SM22luc activity. In separate experiments in 293 cells, immunoblot analysis showed that all three constructs were expressed at roughly equivalent levels following transient transfection (data not shown). Similarly, Smad2, which did not convincingly demonstrate significant DNA binding in the supershift assay (Fig. 4) did not have any effect on promoter activity (data not shown). The co-transfection of Smad3, Smad1 and Smad4 did not significantly alter promoter activity beyond that obtained in the presence of Smad3 and Smad4 together. These results suggest that Smad3 is the major R-Smad contributing to activation of the SM22α promoter induced by TGF-β. To confirm the importance of Smad3 in regulating SM22α promoter activity, we performed a dose response experiment using increasing amounts of transfected Smad3. As shown in Figure 5B, the effect of Smad3 was dose dependent, further suggesting an important role for Smad3 in the positive regulation of SM22α activity. To determine whether Smad3 was absolutely required for SM22α promoter activity, we examined p-162SM22luc reporter activity in Smad3 null fibroblast cells. We used an activated TGF-β receptor construct to induce reporter expression since these late passage fibroblasts appeared to lose responsiveness to exogenous ligand (data not shown), perhaps as a result of down regulation or loss of cell surface receptors. As seen in Figure 5C, constitutively active TβR-I did not have any effect on promoter activity in Smad3 knockout cells. When co-transfected with Smad3, however, the promoter response was recovered and showed approximately a 4-fold increase in reporter activity. Taken together, these data demonstrate that Smad3 plays a significant role in regulating SM22α promoter activity, and that the effect is specific. Although Smad1 can apparently bind to the involved promoter elements, it does not appear to contribute to transcriptional activation of the promoter, nor does overexpression of Smad1 inhibit promoter activity.
Figure 5.

SM22α promoter activity is Smad3 dependent. (A) p-162SM22luc plasmid was transfected into 10T1/2 cells with indicated expression constructs and luciferase activity determined following TGF-β treatment. *P < 0.01 for comparison to TGF-β treated S4 or S4+S1. **P < 0.01 for comparison to TGF-β treated –162. (B) Smad3 activates SM22α promoter activity in a dose dependent manner. Increasing amounts of Smad3 expression construct were co-transfected with Smad4 and -162luc and luciferase activity determined following TGF-β stimulation. (C) Smad3 restores the SM22α promoter activity in Smad3 null fibroblasts. p-162SM22luc plasmid was transfected alone (-162) or with activated TβR-I (-162+Alk5*) or activated TβR-I plus Smad3 (-162+Alk5*+S3) into Smad3 null fibroblasts and luciferase activity determined. *P < 0.01 for the comparison-162+Alk5*+S3 to -162 or -162+Alk5*.
Smad proteins are critical in the early response of SM22α promoter to TGF-β induction
Many studies have indicated that Smad proteins accumulate in the nucleus immediately following the stimulation of TGF-β, but reporter gene analyses are typically performed after 16–48 h of induction with ligand. To analyze the dynamic regulation of the SM22α gene promoter upon TGF-β induction, we transfected the p-162SM22luc plasmid into 10T1/2 cells and performed luciferase assays at 2, 4, 6, 8 and 16 h after TGF-β treatment. As shown in Figure 6A, luciferase activity was significantly increased as early as 4 h after TGF-β addition. The maximal activity was achieved at 8 h of induction. After 16 h of stimulation, the promoter activity retained only 30% of the maximum. Similar results were obtained by examining expression of induction of endogenous transcripts, although there was a more sustained expression of SM22α mRNA as measured by RT–PCR in 10T1/2 cells (Fig. 6B).
Figure 6.

Smad proteins are responsible for the immediate response of SM22α promoter activity to TGF-β induction. (A) Time course of SM22α promoter activity. 10T1/2 cells were transfected with p-162SM22luc plasmid and treated with vehicle or TGF-β1 (5 ng/ml) for the indicated times and luciferase activity determined. P < 0.01 for comparison of untreated to TGF-β-treated for all time points except 2 h. (B) RT–PCR demonstrating SM22α mRNA expression. 10T1/2 cells were treated with TGF-β at the concentration and time indicated or left untreated. Total RNA was extracted and subjected to RT–PCR using primers specific to SM22α or cyclophilin (as a loading control) as indicated. (C and D) Time course EMSA of Smads and TCE complex. Nuclear extracts untreated or treated with TGF-β for the indicated times were incubated with SM-AB (SBS and medea box) (C) or TCE DNA oligo (D) and resolved as described. Complexes are indicated by arrows. (E) The effect of Smad binding on the early induction of SM22α promoter activity. Wild type (-162) and mutant plasmid (-162-SM-dm) were transfected into 10T1/2 cells and luciferase activity assayed at the indicated times. P < 0.01 for the difference in percent activation from 4 to 6 h.
In order to determine what proteins are responsible for the early response of the SM22α promoter to TGF-β, EMSAs were performed at various times following TGF-β treatment to detect the dynamic interactions of Smads and TCE binding proteins with sequences in the SM22α promoter. TCE binding proteins have been reported to be important to TGF-β induced SM22α promoter activity, but this activity was observed at 48 h of induction and the TGF-β inducible TCE complex was only detected at 4 h after TGF-β induction (11). As shown in Figure 6C, the Smad complex was formed as early as 15 min after TGF-β treatment, reached the highest level at 30 min, and started to decrease after 2 h. The TCE complex, however, was not present until 4 h of induction (Fig. 6D). This suggests that de novo protein synthesis is not necessary for Smad activity and that the pre-existing Smads are rapidly translocated into the nucleus. Given the time course of the binding, however, the inducible protein or proteins that bind to the TCE, likely requires synthesis after TGF-β induction.
To determine further the role of Smad proteins in the early response of SM22α promoter to TGF-β, plasmid p-162SM22luc or the plasmid with double mutations at the SBS and medea box were transfected into 10T1/2 cells and luciferase activity determined after 4 or 6 h of TGF-β induction (Fig. 6E). We found that the double mutations, which eliminated the binding of Smads to the SM22α promoter, decreased the TGF-β induced activity to only 36% of control at 4 h. After 6 h of treatment, promoter activity was reduced to 51% of control values, suggesting that the Smad complex is responsible for the initial response of SM22α promoter to stimulation by TGF-β.
AP-1 cis-elements contribute to SM22α promoter activation by TGF-β
To determine the additional factor involved in TGF-β induced SM22α promoter activity, we examined the promoter sequence and found that a consensus AP-1 site is located ∼40 bp upstream of the Smad binding sites (Fig. 7A). AP-1 components have been shown to interact with Smads (32), and can contribute to responsiveness of transcriptional induction by TGF-β (33). In order to determine if the potential AP-1 binding site in the SM22α promoter contributes to induction by TGF-β, we mutated this site and performed reporter assays as for the Smad mutants. As shown in Figure 7B, mutation of the AP-1 consensus sequence caused a significant inhibition of activation by TGF-β. When mutations in the AP-1 site were combined with the Smad site, promoter activity was completely lost, suggesting a synergistic interaction between these two sites in regulating SM22α promoter activity.
Figure 7.

Effect of AP1 site mutation on SM22α promoter activity. (A) Partial nucleotide sequence from SM22α promoter. AP1 site and Smad sites are underlined and mutant sequences of the various constructs are shown below the wild type sequence. (B) Loss of activity with AP1 site mutation. p-162SM22luc wild type (-162) and plasmids with mutations at Smad site (-162-Sm), AP1 site (-162-AP1m), or both (-162-SmAP1m) were transfected into 10T1/2 cells. Luciferase activity was assayed 16 h after TGF-β treatment.
DISCUSSION
SM22α is one of several SMC markers and serves as an excellent model for studies of developmentally regulated, lineage-specific gene expression in SMCs. Determining the mechanism of the transcriptional regulation of these marker genes is of obvious importance in understanding the molecular mechanism of SMC differentiation. TGF-β has been shown to up-regulate the expression of SMC differentiation markers through a TCE, serum response factor (SRF) box and other DNA elements (11,12,34,35). To date, the direct involvement of Smad proteins, the primary signaling intermediates downstream of the TGF-β receptors, has not been demonstrated.
In the present study, we have found that Smads play an essential role in TGF-β induced SM22α promoter activity. Four lines of evidence support our conclusion. First, we found that SM22α promoter activity is dependent on Smad signaling. Co-transfection of a dominant inhibitory Smad4 significantly reduced promoter activity induced by TGF-β. Second, disruption of one of the five potential Smad binding sites in the SM22α promoter significantly reduced TGF-β-induced promoter activity. Third, Smad proteins physically interact with SM22α promoter sequences. Smad3, Smad1 and Smad4 form a complex or complexes (perhaps with additional factors) that binds to a region containing a Smad binding site and an upstream medea box. Finally, Smad3 is necessary for SM22α promoter activity since the promoter is non-functional in Smad3 null fibroblast cells and co-transfection of Smad3 restores reporter gene expression. Taken together, these data demonstrate that Smad proteins are essential for full activation of the SM22α gene following TGF-β stimulation.
It is also clear from our data that in some cells, Smad proteins do not act alone to regulate SM22α expression. Although mutation of the SBS or medea box significantly reduces SM22α activity, it does not eliminate it. Mutation of the upstream AP-1 site completely abolishes SM22α promoter activity at early time points, and greatly diminishes it at later times (data not shown), while combined mutations at both Smad and AP-1 sites completely abrogates promoter activity. Interestingly, treatment of the cells with bone morphogenic protein 2 (BMP2) did not induce promoter activity (data not shown) even though Smad1 can bind to sites within the promoter. These results suggest that both Smads and AP-1 are important for SM22α promoter regulation, and that the interaction may be specific for Smad3. The extent of Smad interaction with proteins that can bind to AP-1 consensus sites has been well demonstrated. Although Smad proteins can clearly bind to fos and jun family members (32,36,37), the exact location and role of this binding remains unclear. In some systems AP-1 and Smads can act synergistically to activate transcription (32,36,38), while in others such interactions are clearly inhibitory (18,37,39,40). Similarly, it is not clear whether Smad and AP-1 proteins physically interact on or off DNA (18). Current data suggests that both interactions are possible, and may depend in part on the nature of the DNA sequences and AP-1 family members involved (18). One explanation for our results is that Smads can still interact with AP-1 in the absence of direct Smad–DNA binding, which would allow partial activation of the promoter in the Smad/medea mutants, and that AP-1 is an essential component of the activation complex and must bind to DNA or cooperatively facilitates Smad binding. This would explain the loss of activity of the AP-1 mutant promoter constructs.
Our results further suggest that Smad proteins are important for the early activation of SM22α promoter activity in response to TGF-β. Interestingly, the Smad site is located in the first exon, just downstream from the transcriptional start site. Regulatory elements located in the 5′ untranslated region (UTR) are thought to be important in early transcriptional activation (41,42). Smad proteins have been shown to bind in the 5′UTR of the c-jun promoter, which is rapidly induced following TGF-β stimulation (33). SM22α promoter activity can be detected at 4 h of TGF-β induction and reached a maximal level at 8 h of TGF-β stimulation. Smad proteins mediate at least part of this early induction since the Smad complex was observed as early as 15 min after TGF-β addition, and reached a maximal level at 30 min, with a decrease after 1–2 h of induction. Functional significance of this finding is demonstrated when mutation of the Smad binding region of SM22α promoter eliminates most of the early response to TGF-β induction. We propose a model whereby Smad activation and DNA binding is responsible for immediate induction of SM22α promoter activity. Continued expression relies on induction and maintenance by other cellular factors. In our hands AP-1 sites make an important contribution to this early effect, as evidenced by the incomplete loss of signaling in 10T1/2 and Balb 3T3 cells. In some cell types, for example in Monc-1 neural crest cells and Smad3 knockout mouse embryonic fibroblasts (KO MEFs), however, loss of Smad activity completely eliminated transcriptional activation. Other factors such as SRF and YY1 have been shown to regulate SM22α gene expression, and these may contribute to sustained activity (11,34,43). The molecular basis for these differences awaits further study.
Examination of the promoter sequences of other SMC-specific genes, including SM α-actin, SM myosin heavy chain and calponin, identified several potential Smad binding sites present in the 5′-flanking regions (data not shown). Similar to the SM22α gene, one or two CAGA boxes are located in the 5′UTR (35,44,45). It is therefore possible that other genes involved in SMC differentiation share a similar mechanism of Smad initiation followed by maintenance by other factors during TGF-β induced transcription.
Acknowledgments
ACKNOWLEDGEMENTS
The authors thank A. Roberts, J. Yoo and A. Symes for critical reading of the manuscript, and members of the Symes and Lechleider labs for helpful suggestions and encouragement. This work was supported by NIH grant HL65681 and a grant from the Lymphangioleiomyomatosis (LAM) Foundation to R.J.L.
REFERENCES
- 1.Carmeliet P. (2000) Mechanisms of angiogenesis and arteriogenesis. Nature Med., 6, 389–395. [DOI] [PubMed] [Google Scholar]
- 2.Goumans M.J. and Mummery,C. (2000) Functional analysis of the TGF-β receptor/Smad pathway through gene ablation in mice. Int. J. Dev. Biol., 44, 253–265. [PubMed] [Google Scholar]
- 3.Hirschi K.K., Rohovsky,S.A. and D’Amore,P.A. (1998) PDGF, TGF-β and heterotypic cell-cell interactions mediate endothelial cell-induced recruitment of 10T1/2 cells and their differentiation to a smooth muscle fate. J. Cell Biol., 141, 805–814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shah N.M., Groves,A.K. and Anderson,D.J. (1996) Alternative neural crest cell fates are instructively promoted by TGF-β superfamily members. Cell, 85, 331–343. [DOI] [PubMed] [Google Scholar]
- 5.Owens G.K. (1998) Molecular control of vascular smooth muscle cell differentiation. Acta Physiol. Scand., 164, 623–635. [DOI] [PubMed] [Google Scholar]
- 6.Hungerford J.E. and Little,C.D. (1999) Developmental biology of the vascular smooth muscle cell: building a multilayered vessel wall. J. Vasc. Res., 36, 2–27. [DOI] [PubMed] [Google Scholar]
- 7.Li L., Miano,J.M., Cserjesi,P. and Olson,E.N. (1996) SM22α, a marker of adult smooth muscle, is expressed in multiple myogenic lineages during embryogenesis. Circ. Res., 78, 188–195. [DOI] [PubMed] [Google Scholar]
- 8.Solway J., Seltzer,J., Samaha,F.F., Kim,S., Alger,L.E., Niu,Q., Morrisey,E.E., Ip,H.S. and Parmacek,M.S. (1995) Structure and expression of a smooth muscle cell-specific gene, SM22α. J. Biol. Chem., 270, 13460–13469. [DOI] [PubMed] [Google Scholar]
- 9.Zhang J.C., Kim,S., Helmke,B.P., Yu,W.W., Du,K.L., Lu,M.M., Strobeck,M., Yu,Q. and Parmacek,M.S. (2001) Analysis of SM22α-deficient mice reveals unanticipated insights into smooth muscle cell differentiation and function. Mol. Cell. Biol., 21, 1336–1344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Duband J.L., Gimona,M., Scatena,M., Sartore,S. and Small,J.V. (1993) Calponin and SM 22 as differentiation markers of smooth muscle: spatiotemporal distribution during avian embryonic development. Differentiation, 55, 1–11. [DOI] [PubMed] [Google Scholar]
- 11.Adam P.J., Regan,C.P., Hautmann,M.B. and Owens,G.K. (2000) Positive- and negative-acting Kruppel-like transcription factors bind a transforming growth factor β control element required for expression of the smooth muscle cell differentiation marker SM22α in vivo. J. Biol. Chem., 275, 37798–37806. [DOI] [PubMed] [Google Scholar]
- 12.Hautmann M.B., Madsen,C.S. and Owens,G.K. (1997) A transforming growth factor beta (TGF-β) control element drives TGF-β-induced stimulation of smooth muscle alpha-actin gene expression in concert with two CArG elements. J. Biol. Chem., 272, 10948–10956. [DOI] [PubMed] [Google Scholar]
- 13.Piek E., Heldin,C.H. and ten Dijke,P. (1999) Specificity, diversity and regulation in TGF-β superfamily signaling. FASEB J., 13, 2105–2124. [PubMed] [Google Scholar]
- 14.Massague J. and Wotton,D. (2000) Transcriptional control by the TGF-β/Smad signaling system. EMBO J., 19, 1745–1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mulder K.M. (2000) Role of Ras and Mapks in TGF-β signaling. Cytokine Growth Factor Rev., 11, 23–35. [DOI] [PubMed] [Google Scholar]
- 16.de Caestecker M.P., Parks,W.T., Frank,C.J., Castagnino,P., Bottaro,D.P., Roberts,A.B. and Lechleider,R.J. (1998) Smad2 transduces common signals from receptor serine-threonine and tyrosine kinases. Genes Dev., 12, 1587–1592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.de Caestecker M.P., Yahata,T., Wang,D., Parks,W.T., Huang,S., Hill,C.S., Shioda,T., Roberts,A.B. and Lechleider,R.J. (2000) The Smad4 activation domain (SAD) is a proline-rich, p300-dependent transcriptional activation domain. J. Biol. Chem., 275, 2115–2122. [DOI] [PubMed] [Google Scholar]
- 18.Verrecchia F., Vindevoghel,L., Lechleider,R.J., Uitto,J., Roberts,A.B. and Mauviel,A. (2001) Smad3/AP-1 interactions control transcriptional responses to TGF-β in a promoter-specific manner. Oncogene, 20, 3332–3340. [DOI] [PubMed] [Google Scholar]
- 19.Kim J., Johnson,K., Chen,H.J., Carroll,S. and Laughon,A. (1997) Drosophila Mad binds to DNA and directly mediates activation of vestigial by Decapentaplegic. Nature, 388, 304–308. [DOI] [PubMed] [Google Scholar]
- 20.Piek E., Ju,W.J., Heyer,J., Escalante-Alcalde,D., Stewart,C.L., Weinstein,M., Deng,C., Kucherlapati,R., Bottinger,E.P. and Roberts,A.B. (2001) Functional characterization of transforming growth factor β signaling in Smad2- and Smad3-deficient fibroblasts. J. Biol. Chem., 276, 19945–19953. [DOI] [PubMed] [Google Scholar]
- 21.Rao M.S. and Anderson,D.J. (1997) Immortalization and controlled in vitro differentiation of murine multipotent neural crest stem cells. J. Neurobiol., 32, 722–746. [DOI] [PubMed] [Google Scholar]
- 22.Pitts R.L., Wang,S., Jones,E.A. and Symes,A.J. (2001) Transforming growth factor-β and ciliary neurotrophic factor synergistically induce vasoactive intestinal peptide gene expression through the cooperation of Smad, STAT and AP-1 sites. J. Biol. Chem., 276, 19966–19973. [DOI] [PubMed] [Google Scholar]
- 23.Moessler H., Mericskay,M., Li,Z., Nagl,S., Paulin,D. and Small,J.V. (1996) The SM 22 promoter directs tissue-specific expression in arterial but not in venous or visceral smooth muscle cells in transgenic mice. Development, 122, 2415–2425. [DOI] [PubMed] [Google Scholar]
- 24.deCaestecker M.P., Hemmati,P., Larisch-Bloch,S., Ajmera,R., Roberts,A.B. and Lechleider,R.J. (1997) Characterization of functional domains within Smad4/DPC4. J. Biol. Chem., 272, 13690–13696. [DOI] [PubMed] [Google Scholar]
- 25.Yingling J.M., Datto,M.B., Wong,C., Frederick,J.P., Liberati,N.T. and Wang,X.F. (1997) Tumor suppressor Smad4 is a transforming growth factor β-inducible DNA binding protein. Mol. Cell. Biol., 17, 7019–7028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Xu X., Yin,Z., Hudson,J.B., Ferguson,E.L. and Frasch,M. (1998) Smad proteins act in combination with synergistic and antagonistic regulators to target Dpp responses to the Drosophila mesoderm. Genes Dev., 12, 2354–2370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Korchynskyi O. and ten Dijke,P. (2002) Identification and functional characterization of distinct critically important bone morphogenetic protein-specific response elements in the Id1 promoter. J. Biol. Chem., 277, 4883–4891. [DOI] [PubMed] [Google Scholar]
- 28.Ishida W., Hamamoto,T., Kusanagi,K., Yagi,K., Kawabata,M., Takehara,K., Sampath,T.K., Kato,M. and Miyazono,K. (2000) Smad6 is a Smad1/5-induced smad inhibitor. Characterization of bone morphogenetic protein-responsive element in the mouse Smad6 promoter. J. Biol. Chem., 275, 6075–6079. [DOI] [PubMed] [Google Scholar]
- 29.Vindevoghel L., Kon,A., Lechleider,R.J., Uitto,J., Roberts,A.B. and Mauviel,A. (1998) Smad-dependent transcriptional activation of human type VII collagen gene (COL7A1) promoter by transforming growth factor-β. J. Biol. Chem., 273, 13053–13057. [DOI] [PubMed] [Google Scholar]
- 30.Zawel L., Dai,J.L., Buckhaults,P., Zhou,S., Kinzler,K.W., Vogelstein,B. and Kern,S.E. (1998) Human Smad3 and Smad4 are sequence-specific transcription activators. Mol. Cell, 1, 611–617. [DOI] [PubMed] [Google Scholar]
- 31.Dennler S., Itoh,S., Vivien,D., ten Dijke,P., Huet,S. and Gauthier,J.M. (1998) Direct binding of Smad3 and Smad4 to critical TGF-β-inducible elements in the promoter of human plasminogen activator inhibitor-type 1 gene. EMBO J., 17, 3091–3100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhang Y., Feng,X.H. and Derynck,R. (1998) Smad3 and Smad4 cooperate with c-Jun/c-Fos to mediate TGF-β-induced transcription. Nature, 394, 909–913. [DOI] [PubMed] [Google Scholar]
- 33.Wong C., Rougier-Chapman,E.M., Frederick,J.P., Datto,M.B., Liberati,N.T., Li,J.M. and Wang,X.F. (1999) Smad3-Smad4 and AP-1 complexes synergize in transcriptional activation of the c-Jun promoter by transforming growth factor β. Mol. Cell. Biol., 19, 1821–1830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Strobeck M., Kim,S., Zhang,J.C., Clendenin,C., Du,K.L. and Parmacek,M.S. (2001) Binding of serum response factor to CArG box sequences is necessary but not sufficient to restrict gene expression to arterial smooth muscle cells. J. Biol. Chem., 276, 16418–16424. [DOI] [PubMed] [Google Scholar]
- 35.White S.L. and Low,R.B. (1996) Identification of promoter elements involved in cell-specific regulation of rat smooth muscle myosin heavy chain gene transcription. J. Biol. Chem., 271, 15008–15017. [DOI] [PubMed] [Google Scholar]
- 36.Liberati N.T., Datto,M.B., Frederick,J.P., Shen,X., Wong,C., Rougier-Chapman,E.M. and Wang,X.F. (1999) Smads bind directly to the Jun family of AP-1 transcription factors. Proc. Natl Acad. Sci. USA, 96, 4844–4849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Verrecchia F., Tacheau,C., Schorpp-Kistner,M., Angel,P. and Mauviel,A. (2001) Induction of the AP-1 members c-Jun and JunB by TGF-β/Smad suppresses early Smad-driven gene activation. Oncogene, 20, 2205–2211. [DOI] [PubMed] [Google Scholar]
- 38.Qing J., Zhang,Y. and Derynck,R. (2000) Structural and functional characterization of the transforming growth factor-β-induced Smad3/c-Jun transcriptional cooperativity. J. Biol. Chem., 275, 38802–38812. [DOI] [PubMed] [Google Scholar]
- 39.Verrecchia F., Pessah,M., Atfi,A. and Mauviel,A. (2000) Tumor necrosis factor-α inhibits transforming growth factor-β/Smad signaling in human dermal fibroblasts via AP-1 activation. J. Biol. Chem., 275, 30226–30231. [DOI] [PubMed] [Google Scholar]
- 40.Dennler S., Prunier,C., Ferrand,N., Gauthier,J.M. and Atfi,A. (2000) c-Jun inhibits transforming growth factor β-mediated transcription by repressing Smad3 transcriptional activity. J. Biol. Chem., 275, 28858–28865. [DOI] [PubMed] [Google Scholar]
- 41.Damert A., Leibiger,B. and Leibiger,I.B. (1996) Dual function of the intron of the rat insulin I gene in regulation of gene expression. Diabetologia, 39, 1165–1172. [DOI] [PubMed] [Google Scholar]
- 42.McCarthy T.L., Thomas,M.J., Centrella,M. and Rotwein,P. (1995) Regulation of insulin-like growth factor I transcription by cyclic adenosine 3′,5′-monophosphate (cAMP) in fetal rat bone cells through an element within exon 1: protein kinase A-dependent control without a consensus AMP response element. Endocrinology, 136, 3901–3908. [DOI] [PubMed] [Google Scholar]
- 43.Kim S., Ip,H.S., Lu,M.M., Clendenin,C. and Parmacek,M.S. (1997) A serum response factor-dependent transcriptional regulatory program identifies distinct smooth muscle cell sublineages. Mol. Cell. Biol., 17, 2266–2278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Takahashi K., Tazunoki,T., Okada,T., Ohgami,K., Miwa,T., Miki,A. and Shibata,N. (1996) The 5′-flanking region of the human smooth muscle cell calponin gene contains a cis-acting domain for interaction with a methylated DNA-binding transcription repressor. J. Biochem. (Tokyo), 120, 18–21. [DOI] [PubMed] [Google Scholar]
- 45.Min B.H., Foster,D.N. and Strauch,A.R. (1990) The 5′-flanking region of the mouse vascular smooth muscle alpha-actin gene contains evolutionarily conserved sequence motifs within a functional promoter. J. Biol. Chem., 265, 16667–16675. [PubMed] [Google Scholar]