

Abstract
Background:
Uracil DNA glycosylases (UDGs) are major repair enzymes that protect DNA from mutational damage caused by uracil incorporated as a result of a polymerase error or deamination of cytosine. Four distinct families of UDGs have been identified, which show very limited sequence similarity to each other, although two of them have been shown to possess the same structural fold. The structural and evolutionary relationships between the rest of the UDGs remain uncertain.
Results:
Using sequence profile searches, multiple alignment analysis and protein structure comparisons, we show here that all known UDGs possess the same fold and must have evolved from a common ancestor. Although all UDGs catalyze essentially the same reaction, significant changes in the configuration of the catalytic residues were detected within their common fold, which probably results in differences in the biochemistry of these enzymes. The extreme sequence divergence of the UDGs, which is unusual for enzymes with the same principal activity, is probably due to the major role of the uracil-flipping caused by the conformational strain enacted by the enzyme on uracil-containing DNA, as compared with the catalytic action of individual polar residues. We predict two previously undetected families of UDGs and delineate a hypothetical scenario for their evolution.
Conclusions:
UDGs form a single protein superfamily with a distinct structural fold and a common evolutionary origin. Differences in the catalytic mechanism of the different families combined with the construction of the catalytic pocket have, however, resulted in extreme sequence divergence of these enzymes.
Background
Mutagenic uracil appears in DNA opposite to guanine as a result of misincorporation or of deamination of cytosine. Similarly, the deamination process generates thymine opposite guanine in those organisms that undergo cytosine methylation [1,2]. DNA is safeguarded from the consequences of these events by the activity of uracil DNA glycosylases (UDGs), which remove uracil (and sometimes thymine) from the sugar backbone of DNA without breaking the phosphodiester bonds in the backbone. There are different types of these enzymes in the three superkingdoms of life. The best studied family of UDGs, typified by the Escherichia coli Ung protein, is largely specific for uracil and is present in a variety of bacteria, eukaryotes and large eukaryotic DNA viruses [1,3,4]. The mismatch-specific uracil DNA glycosylases (MUGs) have been identified in eukaryotes and several bacteria and, unlike the Ung-family enzymes, are additionally active on G:T mismatches [5,6]. Comparison of the crystal structures of these two enzymes has shown that they are structurally very similar, despite the low sequence similarity [5]. Subsequently, two other classes of UDGs have been characterized, one from thermophilic archaea and several bacteria [7,8] (hereinafter called AUDG) and the other from vertebrates (SMUG) [9]. The latter enzyme has a high specificity for uracil and for single-stranded substrates. The single-strand-specific UDGs (ssUDGs) are believed to be functionally similar to the UNGs and MUGs because they possess motifs similar to the catalytic motifs of the latter enzymes despite supposedly lacking significant sequence similarity to them [9]. In contrast, the structural and evolutionary affinities of the AUDGs are uncertain [8]. Thus, considerable structural diversity appears to exist among the UDGs, their generally similar catalytic activities notwithstanding.
Here, using sequence profile searches, multiple alignment analysis and structural comparisons, we unify all known UDGs into a single protein superfamily and predict a common α/β fold for them. We additionally detect several new probable UDGs that are distinct from the already characterized families, and explore the evolutionary scenarios that could have resulted in the observed phyletic distribution of these enzymes.
Results and discussion
Characterization of the UDG superfamily using iterative database searches
An iterative PSI-BLAST search [10] (cut-off for inclusion of sequences into the position-specific scoring matrix e < 0.01) initiated with the sequence of the TM0511 protein, the prototype member of the AUDG family, retrieved, with statistically significant e values, not only its orthologs and highly conserved paralogs from a variety of organisms, but also the classical MUGs and the Drosophila ssUDG. In addition, these searches resulted in the detection of uncharacterized UDG homologs from the bacteria Deinococcus radiodurans, Campylobacter jejuni and Neisseria meningitidis. The next round of iterative searches initiated with the sequences of the newly detected UDG homologs resulted in the retrieval of the Ung family of UDGs without any false positives. Thus, by using multiple profile searches, it was possible to connect all known UDGs, as well as several putative new ones through statistically significant sequence similarity. Clustering of the proteins of the emerging UDG superfamily using reciprocal retrieval in BLASTP searches as a criterion led to the identification of six distinct families. These are: UNG (orthologs of E. coli Ung); MUG (orthologs of E. coli Mug); AUDG; ssUDG; a previously undetected family that includes members from the genus Neisseria, Mycobacterium leprae, C. jejuni and Zymomonas mobilis (UDGX); and another new family including members from D. radiodurans and Rhodococcus erythropolis (DRUDG). Proteins from each of these families were aligned separately, and the regions corresponding to conserved secondary-structure elements were identified. The available three-dimensional structures of Ung and Mug were superimposed, and the resulting structural alignment was used to combine the multiple alignments of all six UDG families (Figure 1). Comparison of the multiple alignment with the available structures showed conservation of the principal structural elements (Figure 1), indicating that all proteins of the UDG superfamily adopt the same α/β fold as Ung and Mug. This predicted structural unity of the UDGs, along with the subtle but significant sequence similarity, suggests a common evolutionary origin for the entire superfamily.
Figure 1.
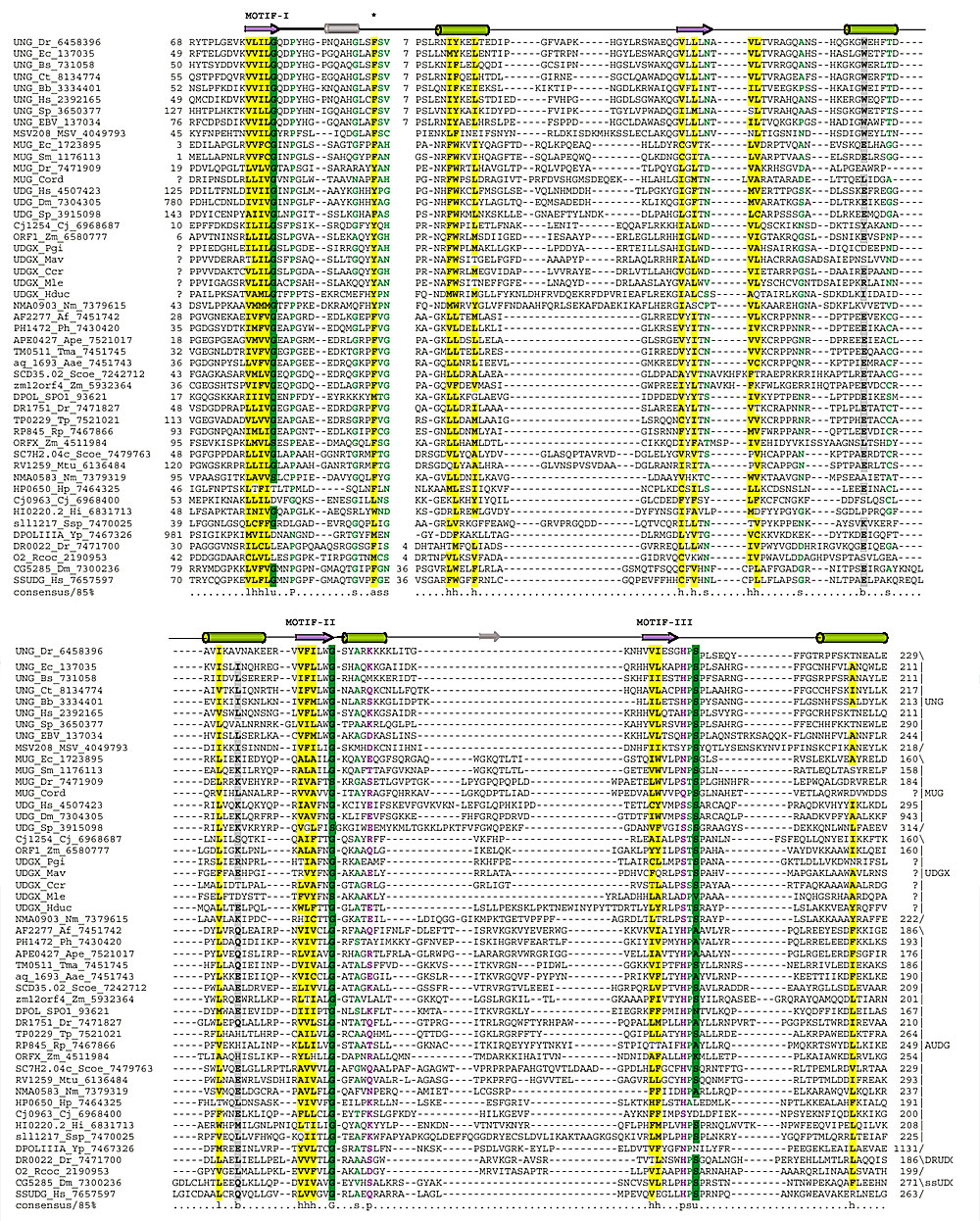
Multiple alignment of the UDG superfamily. The secondary-structure elements of the core UDG fold are shown in color above the multiple alignment. Some nonconserved elements in the MUG structure from E. coli are indicated in gray. The coloring of the alignment positions is according to the 85% consensus that includes the following categories of amino acid residues: h, hydrophobic, l, aliphatic, a, aromatic, shaded yellow (YFWLIVMA); s, small, individual letters colored green (SAGTVPNHD); p, polar, colored purple (STQNEDRKH); u, tiny, shaded green (GAS); and b, big, shaded gray (KREQWFYLMI). Af, Archaeoglobus fulgidus; Bb, Borrelia burgdorferi; Bs, Bacillus subtilis; Cj, Campylobacter jejuni; Ct, Chlamydia trachomatis; Dm, Drosophila melanogaster; Dr, Deinococcus radiodurans; Ec, Escherichia coli; Hi, Haemophilus influenzae; Hp, Helicobacter pylori; Hs, Homo sapiens; Mtu, Mycobacterium tuberculosis; Ph, Pyrococcus horikoshii; Rp, Rickettsia prowazekii; Sc, Saccharomyces cerevisiae; Scoel, Streptomyces coelicolor; Sp, Schizosaccharomyces pombe; Ssp, Synechocystis sp.; Tp, Treponema pallidum; Uu, Ureaplasma urealyticum; Yp, Yersinia pestis. The numbers at each end of each sequence are amino-acid positions and indicate the extent of the domain in each protein. The numbers within the alignment indicate inserts that have not been shown. The conserved motifs discussed in the text are designated I, II and III; the conserved aromatic (aliphatic) residue involved in the stacking interaction with uracil is indicated by an asterisk.
The sequence conservation in the UDG superfamily is concentrated primarily in three motifs, with the two motifs located near the amino and carboxyl termini corresponding to the substrate-binding pocket (Figures 1,2). The ancestral core fold of the UDG superfamily consists of a central parallel four-strand β sheet with a 2-1-3-4 topology, which is associated with four helices; the substrate-binding pocket is formed by the regions located after strand 1 and strand 4 (Figure 2). The central conserved motif corresponds to a sharp turn between strand 3 and the adjacent helix 3, which is one of the most characteristic structural features of the UDG super-family and is probably required to support the enzyme conformation needed to accommodate the DNA. In both structurally characterized members of this superfamily (Udg and Mug), a conserved aromatic residue located in the loop preceding the conserved helix 1 (Figures 1,2) mediates binding of the attacked uracil from the DNA double helix via stacking interactions [11]. This aromatic residue is replaced by an aliphatic residue in a small subset of the UDGs, and the loop may contain poorly conserved short helices in some of the UDGs, such as Mug. This position is highly conserved in the entire UDG superfamily (Figure 1), which suggests that a similar mechanism of uracil binding is universal in the UDGs.
Figure 2.
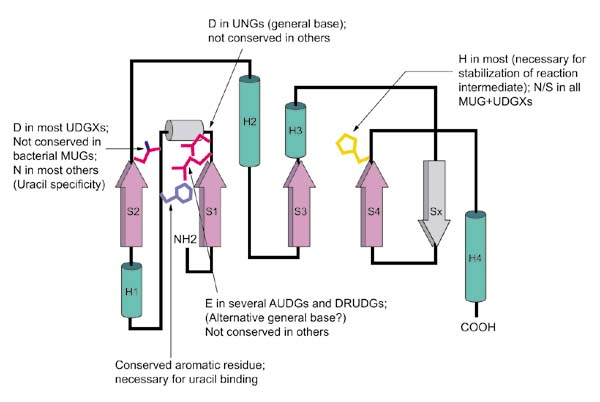
The topology of the UDG superfamily core fold, with the conserved and unique features of different families. The core secondary-structure elements of the UDG fold are colored as in Figure 1 and numbered according to their order in the sequence. The elements observed only in the MUGs are shown in gray. The conserved motif I occurs after strand 1 and motif II occurs after strand 4 and forms the active-site pocket in the three-dimensional structure.
Catalytic mechanism
The experimental determination of a similar catalytic activity in diverse members of the UDG superfamily and the conservation of the substrate-binding site suggest a generally conserved catalytic mechanism. However, several family-specific features predict interesting differences in the catalytic properties of the individual families. On the basis of studies on Ung-family enzymes such as those from herpesviruses, it has been suggested that protonation of the O2 of the flipped-out uracil is carried out by the conserved histidine in motif III, which acts as a general acid [3,12]. Studies on the E. coli Ung enzyme, however, have shown that this conserved histidine does not act as a general acid, but instead is neutral and acts as an electrophile [13,14]. On this basis, it has been proposed that the electrophilic interaction stabilizes the developing enolate on the uracil O2 in course of its excision [13,14]. This reaction is assisted by the conserved aspartate in motif I that acts as a general base and directs a water molecule for the nucleophilic attack [3,12,13]. The MUGs and the new family of bacterial UDGs (UDGX) identified here lack both the conserved electrophile (histidine) and the general base (aspartate) (Figure 1), which suggests that these are less efficient enzymes [5]. The remaining UDG families typically contain the electrophilic histidine, but not the general base aspartate in motif I (Figure 1). A subset of the AUDGs and the newly identified DRUDG family, however, contain a glutamate one position upstream of the aspartate present in motif I of the UNGs (Figure 1); this glutamate could act as an alternative general base for this subset of UDGs. Additionally, the loop formed by motif III also helps in clamping on the phosphate backbone to allow recognition of the target nucleotide by the active site [11]. The discrimination of uracil over cytosine in enzymes of the UNG family depends on the asparagine located near the end of the core strand 2 (Figures 1,2). An asparagine or aspartate is conserved in the majority of the UDG superfamily enzymes in this position, with the exception of some members of the MUG, AUDG and UDGX families. Both E. coli Mug and its human ortholog, TDG, have been shown to act on powerfully mutagenic alkylated bases such as etheno-cytosine [15]. Mutational replacement of asparagine by aspartate in the human uracil DNA glycosylase (UNG) results in its acquiring cytosine glycosylase activity [16]. This substitution, which occurs naturally in several UDGX family proteins, along with other replacements of asparagine in this position in different members of this superfamily (Figure 1), is probably an adaptation for removal of mutagenic alkylated bases such as etheno-cytosine.
Evolution
On the basis of the conservation of functionally important residues in the UDG superfamily, a parsimonious, although speculative, scheme for the evolution of these enzymes can be proposed (Figure 3). The ancestral uracil DNA glycosylase probably possessed the core fold with an asparagine at the end of strand 2 and a histidine at the end strand 4 and most closely resembed the AUDG and DRUDG families. From this ancestral form, the high-activity forms such as the UNG class could have evolved by acquiring the general base in motif I. The acquisition of the glutamate in motif I of the AUDGs and DRUDGs could represent independent evolution of the same type of high-activity enzyme. The lower-activity forms, such as the MUGs and the UDGXs, could have evolved by replacement of the ancestral electrophilic histidine that stabilized the reaction intermediate by another polar residue such as serine or asparagine. The localization of the active site formed by long loops on the same side of the UDG molecules probably resulted in relaxation of the selective constraints on their sequences beyond the maintenance of the general shape of the binding pocket. Moreover, even the charged residues in the binding pocket are not entirely constrained, because the enzyme mechanism seems to depend more critically on the steric strain caused by base-flipping than on the base or other residues that stabilize the intermediate. These features of the UDGs probably contributed to the evolution of a very high level of sequence divergence, without a single residue conserved throughout the superfamily, which is unusual for homologous enzymes that catalyze essentially the same reaction.
Figure 3.
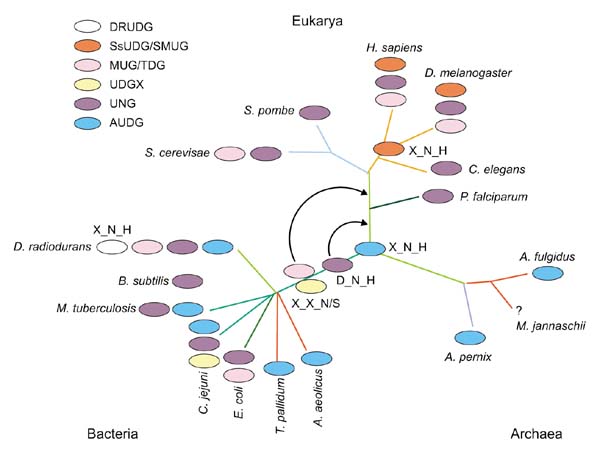
A hypothetical evolutionary scenario for the UDG superfamily. The different families are shown in different colors and potential order and lineage of derivation is indicated on the standard phylogenetic model for the three domains of life. The representation of the active-site pocket residues typical of that set is shown next to each class at the point of derivation. The first position is the general base represented by an aspartate in the UNGs, the second position is the uracil/cytosine discrimination site occurring after the core strand 2, and the third position is typically represented by a histidine that acts as an electrophile. The X at a given position denotes lack of conservation. In some of the AUDGs and the DRUDGs, a glutamate could function as alternative general base.
The phyletic distribution of the UDGs shows partial complementarity between different families, which suggests that they perform at least partially overlapping functions in different organisms (Table 1). Each completely sequenced genome, with the apparent exception of the archaea Methanococcus jannaschii and Methanobacterium thermoautotrophicum, encodes at least one member of the UDG superfamily, with a maximum of four members in the case of the radioresistant bacterium D. radiodurans (Table 1). Each of these families, with the exception of the ssUDGs, which so far are limited to animals, shows a patchy spread over a wide phylogenetic range, which suggests important roles for horizontal gene transfer and lineage-specific gene loss in the evolution of the UDGs. The presence of AUDG in at least one bacteriophage and of UNGs in large eukaryotic DNA viruses (Table 1) point to one possible type of vehicle for horizontal dissemination of these enzymes. The phyletic distribution of the UDGs suggests that the AUDGs could be the ancestral form, possibly inherited from the last common ancestor of all extant life forms. This seems to be compatible with the apparent ancestral layout of the active center of these enzymes (see above). The UNGs appear to be a primitive bacterial form, whereas the MUG-UDGX group could have been derived at a later stage of bacterial evolution. The separation between UNGs and MUG-UDGX could have been driven by selection for distinct functional niches, a uracil-specific enzyme in the case of the former and a G:U/T mismatch repair enzyme in the latter. The UDGX and MUG families show a closer relationship to each other than to other families of the superfamily, suggesting a relatively recent divergence and a similar mismatch repair function. The DRUDGs appear to be specialized derivatives that emerged within one bacterial lineage followed by limited dispersal, at least in the currently sampled bacterial taxa. Under this scenario, AUDGs have been displaced in some of the bacteria, and possibly in the ancestral eukaryotes, by the UNGs and MUGs.
Table 1.
Phyletic distributions of the six families of UNGs
| Species/family | UNG* | AUDG* | MUG | SsUDG* DRUDG* |
| + UDGX* | ||||
| Bacteria | ||||
| Escherichia coli | 1 | 1 (MUG) | ||
| Haemophilus influenzae | 1 | 1 | ||
| Neisseria meningitidis | 1 | 1 | 1 (UDGX) | |
| Rickettsia prowazekii | 1 | |||
| Campylobacter jejuni | 1 | 1 | 1 (UDGX) | |
| Helicobacter pylori | 1 | 1 | ||
| Bacillus subtilis | 1 | |||
| Mycoplasma genitalium | 1 | |||
| Mycoplasma pneumoniae | 1 | |||
| Ureaplasma urealyticum | 1 | |||
| Deinococcus radiodurans | 1 | 1 | 1 (MUG) | 1 |
| Mycobacterium tuberculosis 1 | 1 | |||
| Streptomyces coelicolor | 1 | 2 | ||
| Synechocystis sp. | 1 | |||
| Chlamydia trachomatis | 1 | |||
| Chlamydophila pneumoniae 1 | ||||
| Treponema pallidum | 1 | |||
| Borrelia burgdorferi | 1 | 1(d)† | ||
| Aquifex aeolicus | 1 | |||
| Thermotoga maritima | 1 | |||
| Archaea | ||||
| Aeropyrum pernix | 1 | |||
| Archaeoglobus fulgidus | 1 | |||
| Pyrococcus horikoshii | 1 | |||
| Methanobacterium | ||||
| thermoautotrophicum | ||||
| Methanococcus jannaschii | ||||
| Eukaryota | ||||
| Saccharomyces cerevisiae | 1 | (r)‡ | ||
| Schizosaccharomyces pombe1 | 1(MUG) | |||
| Caenorhabditis elegans | 1 | |||
| Drosophila melanogaster | (?)§ | 1(MUG) | 1 | |
| Homo sapiens | 1 | 1(MUG) | 1 | |
| Large DNA viruses | ||||
| Poxviruses | 1 | |||
| Herpesviruses | 1 | |||
| Bacteriophages SPO1 | 1 |
*The number of detected representatives of each family is indicated for each species. Note that duplication is uncharacteristic of the UNGs. †(d) indicates a possibly disrupted version in which the amino-terminal conserved motifs are not detectable; ‡(r) indicates an apparent recent loss in S. cerevisiae, as the gene is retained in the related yeast Candida albicans; §(?) indicates the unusual lack of a detectable UNG in both the genome and EST sequences.
The AUDGs are fused to two distinct DNA polymerases - a DNA polymerase IIIα subunit in Yersinia pestis and a polymerase of the A family (homolog of bacterial Pol I) in bacteriophage SP01. This fusion is the cause of many erroneous annotations of AUDG family members as 'putative phage-type DNA polymerases' that are found in current sequence databases. The fusion with the polymerases suggests that the functioning of the AUDGs, and possibly other UDGs, could be tightly coupled to that of the DNA replication apparatus. This may be particularly important in the archaea, whose polymerases stall at uridines in the template strand [17]. Given this possible function of AUDGs in replication and the fundamental role of UDGs in repair, the apparent absence of these enzymes in two archaeal methanogens is unexpected. Although these archaeal genomes could encode extremely divergent members of the UDG superfamily that escaped detection even in the present detailed analysis, it seems more likely that in these archaea the UDGs have been displaced by unrelated enzymes of the α-helical MutY super-family [18].
Eukaryotes encode UNG- and MUG-family enzymes that are not found in archaea and are closely related to their bacterial orthologs. This strongly suggests acquisition from bacterial endosymbionts (including mitochondria), followed by displacement of the UDG inherited from the common ancestor with archaea (probably AUDG). The MUG-family enzymes from animals have low-complexity segments on either side of the DNA glycosylase domain. In the case of Drosophila these are particularly expanded and are associated with two minor groove DNA-binding motifs, the AT hooks [19]. This motif is found in many chromosomal proteins and could help in the translocation of the enzyme to specific sites in chromatin, such as matrix attachment regions. Interestingly, different eukaryotic lineages show notable differences in their repertoires of UDGs, with only an UNG-family enzyme so far detected in the nematode Caenorhabditis elegans. The phylogenetic affinity of the ssUDGs, which have been detected up to now only in coelomates, is hard to discern, because they have evolved distinct structural features, such as long inserts, that are not seen in the other members of the UDG superfamily. The presence of a histidine in motif III suggests that the ssUDGs could have evolved from a UNG-like enzyme by rapid divergence. The evolutionary divergence and the origin of acquisition of a distinct DNA glycosylase may correlate with the need for an enzyme that can meet the particular DNA repair needs of multicellular animals, such as the repair of frequently transcribed DNA.
Conclusions
Using sequence profile searches, multiple alignment analysis and protein structure comparisons, we have shown that all known UDGs form a single protein superfamily with a distinct structural fold and a common evolutionary origin. The extreme sequence divergence of different families of UDGs is probably due to differences in their biochemistry, with only the general shape of the protein molecule and the binding pocket being essential for the DNA glycosylase reaction per se. Although the UDG superfamily is nearly ubiquitous among cellular life forms, the individual families show limited and distinct phyletic distributions. The emerging evolutionary scenario for the UDGs involves multiple events of lateral gene transfer and lineage-specific gene loss. In addition, we predict two previously undetected families of UDGs; the experimental investigation of their functions is expected to broaden the current perspective on these critical repair enzymes.
Materials and methods
The databases used in this study were the Non-redundant Nucleotide and Protein and the Expressed Sequence Tags (EST) databases (National Center for Biotechnology Information) and the individual protein sequence databases of completely and partially sequenced genomes [20]. Local alignment searches were performed using the gapped version of the BLAST programs (BLASTPGP for proteins and TBLASTNGP for translating searches of nucleotide databases) [10]. Sequence profile searches were performed using the PSI-BLAST program [10] or using the HMMSEARCH program, with input hidden Markov models generated from multiple alignments using the HMMBUILD program [21]. The multiple alignments were generated using a combination of PSI-BLAST and CLUSTALW [22]. The statistically significant motifs were detected using the Gibbs sampling option of the MACAW program [23,24]. The three-dimensional structure visualization, alignment and modeling were carried out using the SWISS-PDB-Viewer program [25].
References
- Krokan HE, Standal R, Slupphaug G. DNA glycosylases in the base excision repair of DNA. Biochem J. 1997;325:1–16. doi: 10.1042/bj3250001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedberg EC, Walker GC, Siede W. DNA Repair and Mutagenesis. . Washington, DC: American Society for Microbiology; 1995.
- Savva R, McAuley-Hecht K, Brown T, Pearl L. The structural basis of specific base-excision repair by uracil-DNA glycosylase. Nature. 1995;373:487–493. doi: 10.1038/373487a0. [DOI] [PubMed] [Google Scholar]
- Mol CD, Arvai AS, Slupphaug G, Kavli B, Alseth I, Krokan HE, Tainer JA. Crystal structure and mutational analysis of human uracil-DNA glycosylase: structural basis for specificity and catalysis. Cell . 1995;80:869–878. doi: 10.1016/0092-8674(95)90290-2. [DOI] [PubMed] [Google Scholar]
- Barrett TE, Savva R, Panayotou G, Barlow T, Brown T, Jiricny J, Pearl LH. Crystal structure of a G:T/U mismatch-specific DNA glycosylase: mismatch recognition by complementary-strand interactions. Cell . 1998;92:117–129. doi: 10.1016/s0092-8674(00)80904-6. [DOI] [PubMed] [Google Scholar]
- Gallinari P, Jiricny J. A new class of uracil-DNA glycosylases related to human thymine-DNA glycosylase. Nature. 1996;383:735–738. doi: 10.1038/383735a0. [DOI] [PubMed] [Google Scholar]
- Sandigursky M, Franklin WA. Uracil-DNA glycosylase in the extreme thermophile Archaeoglobus fulgidus. . J Biol Chem. 2000;275:19146–19149. doi: 10.1074/jbc.M001995200. [DOI] [PubMed] [Google Scholar]
- Sandigursky M, Franklin WA. Thermostable uracil-DNA glycosylase from Thermotoga maritima, a member of a novel class of DNA repair enzymes. Curr Biol. 1999;9:531–534. doi: 10.1016/s0960-9822(99)80237-1. [DOI] [PubMed] [Google Scholar]
- Haushalter KA, Todd Stukenberg MW, Kirschner MW, Verdine GL. Identification of a new uracil-DNA glycosylase family by expression cloning using synthetic inhibitors. Curr Biol. 1999;9:174–185. doi: 10.1016/s0960-9822(99)80087-6. [DOI] [PubMed] [Google Scholar]
- Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parikh SS, Mol CD, Slupphaug G, Bharati S, Krokan HE, Tainer JA. Base excision repair initiation revealed by crystal structures and binding kinetics of human uracil-DNA glycosylase with DNA. EMBO J . 1998;17:5214–5226. doi: 10.1093/emboj/17.17.5214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodson ML, Michaels ML, Lloyd RS. Unified catalytic mechanism for DNA glycosylases. J Biol Chem. 1994;269:32709–32712. [PubMed] [Google Scholar]
- Drohat AC, Xiao G, Tordova M, Jagadeesh J, Pankiewicz KW, Watanabe KA, Gilliland GL, Stivers JT. Heteronuclear NMR and crystallographic studies of wild-type and H187Q Escherichia coli uracil DNA glycosylase: electrophilic catalysis of uracil expulsion by a neutral histidine 187. Biochemistry. 1999;38:11876–11886. doi: 10.1021/bi9910880. [DOI] [PubMed] [Google Scholar]
- Drohat AC, Jagadeesh J, Ferguson E, Stivers JT. Role of electrophilic and general base catalysis in the mechanism of Escherichia coli uracil DNA glycosylase. . Biochemistry. 1999;38:11866–11875. doi: 10.1021/bi9910878. [DOI] [PubMed] [Google Scholar]
- Saparbaev M, Laval J. 3,N4-ethenocytosine, a highly mutagenic adduct, is a primary substrate for Escherichia coli double-stranded uracil-DNA glycosylase and human mismatch-specific thymine-DNA glycosylase. Proc Natl Acad Sci USA. 1998;95:8508–8513. doi: 10.1073/pnas.95.15.8508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kavli B, Slupphaug G, Mol CD, Arvai AS, Peterson SB, Tainer JA, Krokan HE. Excision of cytosine and thymine from DNA by mutants of human uracil-DNA glycosylase. EMBO J. 1996;15:3442–3447. [PMC free article] [PubMed] [Google Scholar]
- Greagg MA, Fogg MJ, Panayotou G, Evans SJ, Connolly BA, Pearl LH. A read-ahead function in archaeal DNA polymerases detects promutagenic template-strand uracil. Proc Natl Acad Sci USA. 1999;96:9045–9050. doi: 10.1073/pnas.96.16.9045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aravind L, Walker DR, Koonin EV. Conserved domains in DNA repair proteins and evolution of repair systems. Nucleic Acids Res. 1999;27:1223–1242. doi: 10.1093/nar/27.5.1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aravind L, Landsman D. AT-hook motifs identified in a wide variety of DNA-binding proteins. Nucleic Acids Res. 1998;26:4413–4421. doi: 10.1093/nar/26.19.4413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Microbial Genomes Blast Databases. http://www.ncbi.nlm.nih.gov/Microb_blast/unfinishedgenome.html
- Eddy SR. Profile hidden Markov models. Bioinformatics . 1998;14:755–763. doi: 10.1093/bioinformatics/14.9.755. [DOI] [PubMed] [Google Scholar]
- Thompson JD, Higgins DG, Gibson TJ. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. . Nucleic Acids Res. 1994;22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuler GD, Altschul SF, Lipman DJ. A workbench for multiple alignment construction and analysis. Proteins. 1991;9:180–190. doi: 10.1002/prot.340090304. [DOI] [PubMed] [Google Scholar]
- Neuwald AF, Liu JS, Lawrence CE. Gibbs motif sampling: detection of bacterial outer membrane protein repeats. Protein Sci. 1995;4:1618–1632. doi: 10.1002/pro.5560040820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guex N, Peitsch MC. SWISS-MODEL and the Swiss-Pdb-Viewer: an environment for comparative protein modeling. Electrophoresis . 1997;18:2714–2723. doi: 10.1002/elps.1150181505. [DOI] [PubMed] [Google Scholar]