

Abstract
Multidrug resistance mechanisms underlying the intractability of malignant melanomas remain largely unknown. In this study, we demonstrate that the development of multidrug resistance in melanomas involves subcellular sequestration of intracellular cytotoxic drugs such as cis-diaminedichloroplatinum II (cisplatin; CDDP). CDDP is initially sequestered in subcellular organelles such as melanosomes, which significantly reduces its nuclear localization when compared with nonmelanoma/KB-3-1 epidermoid carcinoma cells. The melanosomal accumulation of CDDP remarkably modulates melanogenesis through a pronounced increase in tyrosinase activity. The altered melanogenesis manifested an ≈8-fold increase in both intracellular pigmentation and extracellular transport of melanosomes containing CDDP. Thus, our experiments provide evidence that melanosomes contribute to the refractory properties of melanoma cells by sequestering cytotoxic drugs and increasing melanosome-mediated drug export. Preventing melanosomal sequestration of cytotoxic drugs by inhibiting the functions of melanosomes may have great potential as an approach to improving the chemosensitivity of melanoma cells.
Keywords: cancer, melanosomes, skin, tumor therapy, multidrug resistance
The incidence of melanomas, one of the most aggressive forms of cancer, has increased rapidly and now ranks fifth among the most common cancers in the United States (1). According to the American Cancer Society, there will be ≈62,000 new diagnoses of melanoma in the U.S. in 2006, and ≈7,900 people will die of this disease (see www.cancer.org). Melanomas are intrinsically resistant to radiation therapy and to many chemotherapeutic drugs that act on DNA, microtubules, and/or topoisomerases. Thus, researchers have recently begun to focus on the development of immunochemotherapy of melanomas, with some promising results (2, 3).
Unraveling the mechanisms of drug resistance in melanomas could help to improve current therapeutic approaches; however, the precise mechanisms underlying these refractory properties are still unknown (4–7). Previously, we reasoned that one potential mechanism for the intractability of melanomas to chemotherapy might be attributed to the presence of melanosomes in those tumors (7). Melanosomes are membrane-bound compartments that are unique subcellular organelles found in pigmented cells and that protect melanocytes against the harmful effects of toxic intermediates produced during melanin synthesis (8).
As we have recently proposed, one potential mechanism that might account for multidrug resistance (MDR) in melanomas is intracellular melanosomal drug trapping and active melanosome-mediated drug export (7). In this study, we further verified these hypotheses by directly comparing the melanosomal sequestration of cytotoxic drugs such as cis-diaminedichloroplatinum II (CDDP) in melanoma and nonmelanoma cells and by examining melanogenic changes that are related to extracellular melanosome export in melanoma cells treated with CDDP.
Results and Discussion
Cytoplasmic and Melanosomal Sequestration of Alexa Fluor-Labeled CDDP.
Melanoma cells are intrinsically resistant to many chemotherapeutic drugs that act on DNA (Fig. 1A and Fig. 5B, which is published as supporting information on the PNAS web site), microtubules, and/or topoisomerases (data not shown). To unravel the mechanism of drug resistance in melanomas, we examined whether cytotoxic drugs in melanoma cells could be trapped in subcellular organelles (such as melanosomes, lysosomes, the Golgi apparatus, and/or other vesicles), and we compared the intracellular retention of Alexa Fluor 546-labeled CDDP (AF-CP) (AF546CP; Invitrogen–Molecular Probes) both in MNT-1 melanotic melanoma (9) and in KB-3-1 epidermoid carcinoma (10–12) cell lines after incubation with the chemical for 2 h. As shown in Fig. 1B, intracellular accumulation of the drug was observed in the cytoplasm of MNT-1 cells but not in the nuclei of those cells (Fig. 1 B4 and B5). In contrast, the nonmelanoma cells (KB-3-1) accumulated significant amounts of AF-CP both in the cytoplasm and in the nuclei (Fig. 1 B1 and B2). These data suggest that the cytoplasmic trapping of AF-CP in melanoma cells prevents its further nuclear accumulation.
Fig. 1.

Retention of AF-CP in MNT-1 and KB-3-1 cells. (A) The intrinsic MDR phenotype in MNT-1 cells is compared with nonmelanoma cells (KB-3-1). Cytotoxic curves were determined by 72-h MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide] assays. MNT-1 cells and several other melanoma cell lines included in the National Cancer Institute 60 cell line panel showed a similar level of CDDP (cisplatin) resistance compared with multidrug-sensitive MES-SA (human sarcoma) cells (Fig. 5B). A representative graph is shown. (B) Retention of AF-CP in MNT-1 and KB-3-1 cells. The cells were treated with 200 units/ml (1 unit is defined as the reagent solution required to label 25 ng of DNA in vitro) of AF-CP (red) for 2 h at room temperature, washed with cold PBS, and fixed and stained with DAPI (blue). Images were acquired with a Zeiss LSM 510 confocal microscope. B1, B2, B4, and B5 are representative confocal images of the double staining; B3 and B6 show controls not treated with AF-CP that were stained with DAPI alone to assess the vitality of cells and the integrity of the nuclei in MNT-1 cells and in KB-3-1 cells. (C and D) Colocalization of AF-CP with the melanosome stage II marker HMB45. Cells were treated with 10 units/ml AF-CP (C1 and D1) at 37°C for 60 min, fixed with 4% paraformaldehyde, and stained with the antibody against HMB45 (C2 and D2) and DAPI (C3). C4 and D4 are the overlaid images from C and D, respectively. D3 shows phase-contrast micrographs; cells are labeled a–g for identification.
To determine whether AF-CP was localized in melanosomes, we performed immunofluorescence confocal analysis of the distribution of AF-CP with the stage II melanosome marker HMB-45. Our results indicated that AF-CP was predominantly distributed in the cytoplasm and was sometimes concentrated in the region proximal to the plasma membrane, where it colocalized with HMB45 (Fig. 1C). Of note, the cytoplasmic accumulation of AF-CP was markedly reduced in more highly pigmented cells, an indication of the presence of later stages of melanosomes, such as stage III/IV or IV melanosomes (e.g., in the cells labeled c, d, and g of Fig. 1D3). These data are consistent with the absence of HMB-45 staining in mature melanosomes due to epitope masking by melanin (Fig. 1 D2 and D4). Stage II or II/III melanosomes seem to possess greater capacities to accumulate CDDP; however, the underlying mechanisms for this accumulation remain to be determined. We previously studied Alexa Fluor- and [14C]-labeled CDDP and found that they had similar biological behavior in terms of their drug uptake, efflux, and subcellular distribution in KB-3-1 cells and their CDDP-resistant derivatives (10, 12). Hence, AF-CP reflects at least some biological properties of unmodified CDDP.
X-Ray Mapping of CDDP in Melanosomes of Melanoma Cells.
It also has been noted that AF-CP forms monofunctional adducts on genomic DNAs and might have some differences compared with unmodified CDDP with respect to reactivity to its cellular target (S. J. Lippard, personal communication). To verify the results obtained from the experiments with AF-CP, we directly analyzed the intracellular retention of the unmodified CDDP both in MNT-1 and in KB-3-1 cells by using an x-ray microprobe (13) (Fig. 2). To validate the results presented in Fig. 1B, we compared the intracellular retention of CDDP in MNT-1 and KB-3-1 after a 2.5-h incubation with 104 μM CDDP. As shown in Fig. 2, the intracellular accumulation of CDDP (i.e., Pt) in MNT-1 cells (Fig. 2A5) was much less than was observed in KB-3-1 control cells (Fig. 2A2), which manifested enhanced nuclear accumulation of CDDP accompanied by a typical apoptotic morphology, including nuclear disruption and plasma membrane blebbing (Fig. 2A1), that was not seen in MNT-1 cells (Fig. 2A4). Clearly, these data reveal fundamental differences between melanoma and nonmelanoma cells in terms of their subcellular CDDP distributions, which could account for differences in chemosensitivity in in vitro cellular models (Fig. 1A) and in melanoma patients (2).
Fig. 2.

Subcellular localization of CDDP by a hard x-ray microprobe. (A) X-ray mapping of the Pt-containing compound (CDDP) in MNT-1 cells (A5) and in KB-3-1 cells (A2) treated with 104 μM CDDP. A1 and A4 are unstained electron micrographs corresponding to the orientation of the x-ray micrographs in A2 and A5/A6, respectively; A3 shows an MNT-1 cell without CDDP treatment (control); A6 is a Zn distribution image (which also serves as a morphological control of the MNT-1 cell shown in A5 treated with CDDP). The asterisk in A1 indicates the disruption of nuclear membrane in KB-3-1 cells. The red asterisks in A4–A6 outline the position of nuclear membranes in MNT-1 cells. (B) Melanosomal localization of CDDP by hard x-ray microprobe. B1 is an electron micrograph of an unstained MNT-1 cell treated with a sublethal dose (6.7 μM) of CDDP for 7 days; B2 and B3 depict x-ray mapping of CDDP and other elements such as Zn in the MNT-1 cell. (C) Typical emission spectrum from high-Pt areas of an MNT-1 melanoma cell treated with 6.7 μM CDDP, based on the average intensity of several melanosomes, the cytoplasm, and the extracellular space of the cell. a.u., arbitrary units; Nu, nucleus; EM, electron micrograph.
We next analyzed the intracellular retention of unmodified CDDP in MNT-1 cells that were treated with a sublethal concentration (2 μg/ml or 6.7 μM) of CDDP (Fig. 2B). In this case, the intracellular accumulation of CDDP was markedly reduced in the nuclei of the cells when compared with that in the cytoplasm (Fig. 2B2), consistent with the data in Fig. 1B. To determine whether CDDP was localized in melanosomes, we compared the pattern of the subcellular uptake of Zn with that of Pt. We found that the Pt-containing compounds colocalized well with Zn, which was predominantly colocalized with melanosomes in MNT-1 cells (Fig. 2 B2 and B3). The melanosomal localization of Zn and Pt was further confirmed by the emission spectrum of the elements, which was generated directly from melanosomes (only the Pt and Zn emission spectrums are shown in Fig. 2C). Thus, we could colocalize >50% of the Pt-containing compound (CDDP) within melanosomes. These data indicated that the Pt-containing compounds are indeed mainly trapped in subcellular organelles such as melanosomes.
Altered Melanogenesis and Accelerated Melanosome Export in CDDP-Treated Melanoma Cells.
Subsequently, we asked the question: What is the fate of melanosomes that accumulate cytotoxic drugs? We speculated that, in melanocytes and in melanomas, export of melanosomes might provide protection to the cells from the cytotoxic effects of melanin intermediates and anticancer agents, resulting in MDR in these cells. To further examine the role of melanogenesis in conferring MDR to melanomas, we treated MNT-1 cells with 6.7 μM CDDP for 2 weeks, which resulted in increased melanogenesis and increased numbers of melanosomes (Fig. 3A). These changes included a pronounced decrease in cellular proliferation concomitant with a 6- to 10-fold increase in the biogenesis of stage II and II/III melanosomes (Fig. 3 A1–A3) when compared with melanomas treated with high doses (104 μM or 400 μM) of CDDP (Figs. 2A4 and 3A2, respectively). We also found a marked (7.3-fold) increase in intracellular pigmentation (Fig. 3B2) and an 8-fold increase in the secretion of melanin-containing melanosomes (Fig. 3 B1, B3, B4, and C). The rate-limiting melanogenic enzymatic activity mediated by tyrosinase was increased 11-fold in CDDP-treated MNT-1 cells as compared with untreated controls (Fig. 3D), suggesting that enhanced melanogenesis in CDDP-treated cells may be due to enhanced tyrosinase activity. Our data suggest that various anticancer drugs that possess different modes of action (e.g., the antimicrotubule drug vinblastine) can also modulate melanogenesis in melanoma cells (Fig. 5, and Supporting Results and Discussion and Fig. 6, which are published as supporting information on the PNAS web site), suggesting that the increase in melanogenesis is a general stress-response mechanism in melanoma cells.
Fig. 3.

Altered melanogenesis in CDDP-treated MNT-1 cells. (A) Shown is the effect of CDDP on the biogenesis of melanosomes at different stages (I–IV, stage I–IV melanosomes); melanosome numbers were counted from unstained micrographs. (B) Culture medium (B1) and cell pellets dissolved in the lysis buffer (B2) in MNT-1 cells in the presence (6.7 μM CDDP) or absence (control) of CDDP treatment for 2 weeks. B3 and B4 are conventionally stained electron micrographs of melanosomes secreted into the medium of MNT-1 cells treated with 6.7 μM CDDP. (C) Protein determination of secreted melanosome pellets in B1. (D) Tyrosinase activity from both secreted melanosomes (D1) and cell pellets (D2).
The consequences of enhanced melanogenesis within melanosomes would be an increase in the production of melanin and its intermediates, which are also cytotoxic to the cells. To prevent the cytoplasmic toxicity of these altered melanosomes (e.g., stage III/IV with an acidic compartment and cytotoxic drugs) in melanoma cells, the melanosomes must be exported from the cells by means of melanosome transfer (Fig. 3 B and C). We also confirmed the intracellular trapping of CDDP by using inductively coupled plasma MS in secreted melanosomes (see Supporting Methods, which is published as supporting information on the PNAS web site).
Obviously, an increase in the generation of stage II/III melanosomes may have a direct effect on increased melanosomal trapping (Fig. 1D). We also compared CDDP sensitivity between amelanotic SK-MEL-28 cells (containing stage I and II melanosomes) and SK-MEL-24 cells (having a few stage I melanosomes). Our results indicated that SK-MEL-28 cells have 10-fold higher levels of CDDP resistance than SK-MEL-24 cells in terms of the numbers of colonies formed after cytotoxic treatment (Fig. 7, which is published as supporting information on the PNAS web site). Thus, the components that regulate the dynamics of melanosomes (i.e., melanosome numbers, melanosomal trapping, and export) are likely involved in drug resistance.
Melanosomal membrane transporters, because of their direct roles in drug transport or sequestration, might play an important role in conferring MDR in melanomas. We have recently identified a cluster of ATP-binding cassette transporters (ABCA9, ABCB5, ABCC2, and ABCD1) that are overexpressed in melanoma cells (14, 15). In particular, ABCB5 was found to be preferentially expressed in pigment-producing cells, including melanotic melanoma and the majority of amelanotic melanoma cells (15). Experiments are needed to verify whether these ABC transporters are directly involved in melanosomal sequestration of cytotoxic drugs such as CDDP.
Melanosomal Enzymes, Melanin Content, and Drug Sensitivity.
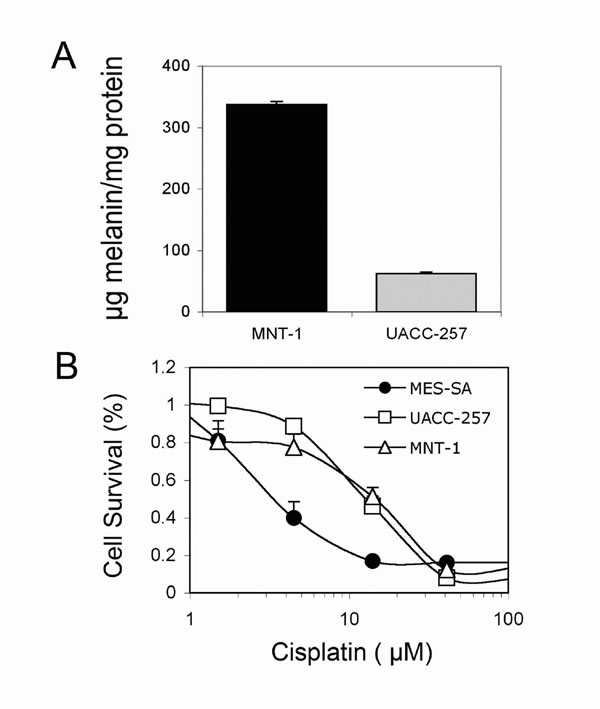
It has not been determined whether tyrosinase and melanin are directly involved in drug resistance. There is evidence in the literature that, in vitro, melanin inhibits the cytotoxic effects of both doxorubicin and daunorubicin, but not CDDP, in MOLT 4 cells, because CDDP does not bind to melanin (16), suggesting that melanin might play a role in conferring drug resistance in vivo. Thus, we also compared the drug-resistant phenotypes of MNT-1 and UACC-257 melanoma cells, which have large differences in melanin content (Fig. 5A), and found that there is no difference in CDDP sensitivity between these two cell lines (Fig. 5B). Furthermore, Pak et al. (17) initially reported that tyrosinase-related protein 2 (TYRP2) confers CDDP resistance that is independent of tyrosinase activity, tyrosinase-related protein 1 (TYRP1), and melanin content, suggesting that tyrosinase activity and melanin are not absolute requirements for the development of drug resistance. Other melanosomal enzymes, such as TYRP2, may possess the ability to confer drug resistance.
In summary, the development of MDR in melanomas involves the subcellular sequestration of intracellular CDDP (Fig. 4). CDDP is initially trapped in subcellular organelles such as melanosomes, which significantly prevents its further nuclear accumulation when compared with nonmelanoma cancer cells (Figs. 1 and 2). In the second phase, melanosomal uptake of CDDP remarkably alters the melanogenic pathway, which manifests as a pronounced increase in tyrosinase activity and intracellular pigmentation (Fig. 3). Finally, the altered melanogenesis accelerates extracellular transport of melanosomes that contain CDDP.
Fig. 4.

Schema of CDDP resistance mechanisms in melanomas. Melanoma cells become resistant to CDDP anticancer drugs by several sequentially occurring mechanisms (7). In an initial phase, drug resistance could occur as a result of reduced drug influx (e.g., reduced endocytosis, reduced activity of an importer, or increased energy-dependent efflux). Once they enter cells, drugs could be ensnared in subcellular organelles such as melanosomes (Figs. 1 and 2) or other vesicles and exported from cells by enhanced melanosome transfer mechanisms that are conserved in melanocytes (Fig. 3). I–IV, stage I–IV melanosomes.
Our experimental data suggest that CDDP resistance in melanomas is associated with melanosome-mediated trapping and export of CDDP. Thus, many therapeutic implications could be derived from this study. In principle, the components of the entire melanogenic pathway could be molecular targets for the therapy of melanomas. Targeting of the rate-limiting steps of the melanogenic pathway may provide novel approaches to modulating MDR in melanomas. For example, inhibition of melanogenesis by down-regulation of melanosome biogenesis would be an effective way to dramatically reduce the intracellular trapping of cytotoxic drugs and enhance genomic cytotoxicity.
Materials and Methods
Cell Culture and Cellular Drug Sensitivity Assays.
Both MNT-1 and KB-3-1 cells have been described in refs. 9 and 10. Cellular drug sensitivity was evaluated by the MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide] assay in triplicate or quadruplicate, as described in ref. 18.
Accumulation of AF-CP and Immunofluorescence Confocal Microscopy.
Confocal microscopic analysis of intracellular CDDP accumulation was assessed by staining cells with AF-CP, as described in ref. 12. Cells were seeded in Lab-Tek chamber slides (Nalge Nunc International, Rochester, NY) and incubated with AF-CP at a final concentration of 10–200 units/ml (1 unit is defined as the reagent solution required to label 25 ng of DNA in vitro) in the presence or absence of CDDP for the indicated times. The slides were fixed in 4% paraformaldehyde and stained with the monoclonal antibody HMB-45 against Pmel17 (DAKO). Confocal microscopic analysis was performed as described in ref. 19. Image analysis was performed with the LSM 510 confocal microscope and its associated colocalization software (Zeiss).
Isolation of Secreted Melanosomes and Electron Microscopy (EM).
MNT-1 cells were treated in the presence or absence of 6.7 μM CDDP for 2 weeks. The cell culture medium was collected in the presence of 1 mM PMSF and 1× protease inhibitor cocktails (Sigma–Aldrich) and centrifuged at 503 × g to remove cell debris. The supernatants were collected and ultracentrifuged at 157,882 × g for 1 h at 4°C. The melanosome-containing pellets were then resuspended in PBS and stored at −80°C before use. The protein concentrations were determined by the bicinchoninic acid method (BCA; Pierce). The identity of secreted melanosomes was confirmed by conventional EM (Fig. 3 B3 and B4).
Measurement of Melanin Content and Melanogenic Assays.
Melanin content was determined as described in ref. 20. Melanogenic assays were performed by examining tyrosinase activity, as described in ref. 21.
Direct Mapping of Metal Complex Uptake by a Hard X-Ray Microprobe.
MNT-1 melanoma cells were treated with a series of CDDP concentrations (0, 6.7, 104, and 400 μM) at 37°C at different time points. The cells were then trypsinized and prepared for EM analysis by high-pressure freezing, freeze substitution in acetone/glutaraldehyde, and embedding in LR White or Lowicryl HM20 resin (Electron Microscopy Sciences, Hatfield, PA). Gold specimen carriers were used instead of brass. The sections were nominally cut to a thickness of 300 nm and were mounted on indexed gold grids. The unstained sections were imaged by a 300-kV transmission electron microscope to provide correlative ultrastructural data. The unstained images were examined and photographed. The specimens were further analyzed by using the 2-ID-D fluorescence x-ray microprobe at the Advanced Photon Source (Argonne National Laboratory) (13).
Supplementary Material
Acknowledgments
We thank S. J. Lippard for comments on AF-CP, D. W. Shen and S. Ambudkar for discussion and suggestions, and G. Leiman for assistance in preparing the manuscript. This work was supported by the Intramural Research Program of the National Institutes of Health, National Cancer Institute, Center for Cancer Research. Use of the Advanced Photon Source at the Argonne National Laboratory was supported by the U.S. Department of Energy, Office of Science, Basic Energy Sciences, under Contract W-31-109-Eng-38.
Abbreviations
- mdr
multidrug resistance
- cisplatin or cddp
cis:-diaminedichloroplatinum II
- AF-CP
Alexa Fluor 546-labeled CDDP.
Footnotes
Conflict of interest statement: No conflicts declared.
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Jemal A., Murray T., Ward E., Samuels A., Tiwari R. C., Ghafoor A., Feuer E. J., Thun M. J. CA Cancer J. Clin. 2005;55:10–30. doi: 10.3322/canjclin.55.1.10. [DOI] [PubMed] [Google Scholar]
- 2.Atkins M. B., Buzaid A. C., Houghton A. N., Jr. In: Cutaneous Melanoma. 4th Ed. Balch C., Houghton A., Sober A., Soong S., editors. St. Louis: Quality Medical Publishing; 2003. pp. 589–604. [Google Scholar]
- 3.Dudley M. E., Wunderlich J. R., Yang J. C., Sherry R. M., Topalian S. L., Restifo N. P., Royal R. E., Kammula U., White D. E., Mavroukakis S. A., et al. J. Clin. Oncol. 2005;23:2346–2357. doi: 10.1200/JCO.2005.00.240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Grossman D., Altieri D. C. Cancer Metastasis Rev. 2001;20:3–11. doi: 10.1023/a:1013123532723. [DOI] [PubMed] [Google Scholar]
- 5.Helmbach H., Rossmann E., Kern M. A., Schadendorf D. Recent Res. Cancer Res. 2003;161:93–110. doi: 10.1007/978-3-642-19022-3_9. [DOI] [PubMed] [Google Scholar]
- 6.Soengas M. S., Lowe S. W. Oncogene. 2003;22:3138–3151. doi: 10.1038/sj.onc.1206454. [DOI] [PubMed] [Google Scholar]
- 7.Chen K. G., Gottesman M. M. In: From Melanocytes to Malignant Melanoma. Hearing V. J., Leong S. P. L., editors. Totowa, NJ: Humana; 2005. pp. 593–606. [Google Scholar]
- 8.Hearing V. J. J. Dermatol. Sci. 2005;37:3–14. doi: 10.1016/j.jdermsci.2004.08.014. [DOI] [PubMed] [Google Scholar]
- 9.Kushimoto T., Basrur V., Valencia J., Matsunaga J., Vieira W. D., Ferrans V. J., Muller J., Appella E., Hearing V. J. Proc. Natl. Acad. Sci. USA. 2001;98:10698–10703. doi: 10.1073/pnas.191184798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shen D. W., Goldenberg S., Pastan I., Gottesman M. M. J. Cell. Physiol. 2000;183:108–116. doi: 10.1002/(SICI)1097-4652(200004)183:1<108::AID-JCP13>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 11.Chauhan S. S., Liang X. J., Su A. W., Pai-Panandiker A., Shen D. W., Hanover J. A., Gottesman M. M. Br. J. Cancer. 2003;88:1327–1334. doi: 10.1038/sj.bjc.6600861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liang X. J., Shen D. W., Chen K. G., Wincovitch S. M., Garfield S. H., Gottesman M. M. J. Cell. Physiol. 2005;202:635–641. doi: 10.1002/jcp.20253. [DOI] [PubMed] [Google Scholar]
- 13.Ilinski P., Lai B., Cai Z., Yun W., Legnini D., Talarico T., Cholewa M., Webster L. K., Deacon G. B., Rainone S., et al. Cancer Res. 2003;63:1776–1779. [PubMed] [Google Scholar]
- 14.Szakacs G., Annereau J. P., Lababidi S., Shankavaram U., Arciello A., Bussey K. J., Reinhold W., Guo Y., Kruh G. D., Reimers M., et al. Cancer Cell. 2004;6:129–137. doi: 10.1016/j.ccr.2004.06.026. [DOI] [PubMed] [Google Scholar]
- 15.Chen K. G., Szakacs G., Annereau J. P., Rouzaud F., Liang X. J., Valencia J. C., Nagineni C. N., Hooks J. J., Hearing V. J., Gottesman M. M. Pigm. Cell Res. 2005;18:102–112. doi: 10.1111/j.1600-0749.2005.00214.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Svensson S. P., Lindgren S., Powell W., Green H. Pigm. Cell Res. 2003;16:351–354. doi: 10.1034/j.1600-0749.2003.00030.x. [DOI] [PubMed] [Google Scholar]
- 17.Pak B. J., Li Q., Kerbel R. S., Ben-David Y. Melanoma Res. 2000;10:499–505. doi: 10.1097/00008390-200010000-00013. [DOI] [PubMed] [Google Scholar]
- 18.Chen G., Jaffrezou J. P., Fleming W. H., Duran G. E., Sikic B. I. Cancer Res. 1994;54:4980–4987. [PubMed] [Google Scholar]
- 19.Valencia J. C., Matsui K., Bondy C., Zhou J., Rasmussen A., Cullen K., Yu Z. X., Moss J., Ferrans V. J. J. Invest. Med. 2001;49:421–433. doi: 10.2310/6650.2001.33787. [DOI] [PubMed] [Google Scholar]
- 20.Virador V. M., Kobayashi N., Matsunaga J., Hearing V. J. Anal. Biochem. 1999;270:207–219. doi: 10.1006/abio.1999.4090. [DOI] [PubMed] [Google Scholar]
- 21.Hearing V. J., Ekel T. M. Biochem. J. 1976;157:549–557. doi: 10.1042/bj1570549. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}