

Abstract
Background:
Standard archival sequence databases have not been designed as tools for genome annotation and are far from being optimal for this purpose. We used the database of Clusters of Orthologous Groups of proteins (COGs) to reannotate the genomes of two archaea, Aeropyrum pernix, the first member of the Crenarchaea to be sequenced, and Pyrococcus abyssi.
Results:
A. pernix and P. abyssi proteins were assigned to COGs using the COGNITOR program; the results were verified on a case-by-case basis and augmented by additional database searches using the PSI-BLAST and TBLASTN programs. Functions were predicted for over 300 proteins from A. pernix, which could not be assigned a function using conventional methods with a conservative sequence similarity threshold, an approximately 50% increase compared to the original annotation. A. pernix shares most of the conserved core of proteins that were previously identified in the Euryarchaeota. Cluster analysis or distance matrix tree construction based on the co-occurrence of genomes in COGs showed that A. pernix forms a distinct group within the archaea, although grouping with the two species of Pyrococci, indicative of similar repertoires of conserved genes, was observed. No indication of a specific relationship between Crenarchaeota and eukaryotes was obtained in these analyses. Several proteins that are conserved in Euryarchaeota and most bacteria are unexpectedly missing in A. pernix, including the entire set of de novo purine biosynthesis enzymes, the GTPase FtsZ (a key component of the bacterial and euryarchaeal cell-division machinery), and the tRNA-specific pseudouridine synthase, previously considered universal. A. pernix is represented in 48 COGs that do not contain any euryarchaeal members. Many of these proteins are TCA cycle and electron transport chain enzymes, reflecting the aerobic lifestyle of A. pernix.
Conclusions:
Special-purpose databases organized on the basis of phylogenetic analysis and carefully curated with respect to known and predicted protein functions provide for a significant improvement in genome annotation. A differential genome display approach helps in a systematic investigation of common and distinct features of gene repertoires and in some cases reveals unexpected connections that may be indicative of functional similarities between phylogenetically distant organisms and of lateral gene exchange.
Background
Functional annotation of genomes is a critical aspect of the genomics enterprise. Without reliable assignment of gene function at the appropriate level of specificity, new genome sequences are plainly useless. The primary methodology used for genome annotation is the sequence database search, the results of which allow transfer of functional information from experimentally characterized genes (proteins) to their uncharacterized homologs in newly sequenced genomes [1,2,3]. However, general-purpose, archival sequence databases are not particularly suited for the purpose of genome annotation. The quality of the annotation of a new genome produced using a particular database critically depends on the reliability and completeness of the annotations in the database itself. As far as annotation is concerned, the purpose of primary sequence databases is to faithfully preserve the description attached to each sequence by its submitter. In their capacity as sequence archives, such databases include no detailed documentation in support of the functional annotations. Furthermore, primary sequence databases are not explicitly structured by either evolutionary or functional criteria. These features, which are inevitable in archival databases, seriously impede their utility as resources for genome annotation, particularly when an automated or semi-automated approach is attempted [4,5]. At its worst, this situation results in a notorious vicious circle of error amplification - an inadequately annotated database is used to produce an error-ridden and incomplete annotation of a new genome, which in turn makes the database even less useful [6,7,8].
One way out of this 'Catch-22' situation is to use a different type of database for genome annotation, namely databases in which sequence information is organized by structural, functional or phylogenetic criteria, or a combination thereof. For example, the KEGG [9] and WIT [10] databases are primarily function-oriented and organize protein sequences from completely and partially sequenced genomes according to their known or predicted roles in biochemical pathways, although WIT also provides a phylogenetic classification. In contrast, the SMART database [11] is organized on a structural principle and provides a searchable collection of common protein domains. All these databases share a fundamental common feature - they encapsulate carefully verified knowledge on protein structure, function and/or evolutionary relationships, and therefore, at least in principle, provide for a more robust mode of genome annotation than general-purpose databases and may serve as a stronger foundation for partially automated approaches to genome analysis.
The database of Clusters of Orthologous Groups of proteins (COGs) is a phylogenetic classification of proteins encoded in completely sequenced genomes [12]. An attempt has been made to organize these proteins into groups of orthologs, direct evolutionary counterparts related by vertical descent [13,14]. Because of lineage-specific duplications, orthologous relationships in many cases exist between gene (protein) families, rather than between individual proteins, hence 'orthologous groups' (including only lineage-specific duplications in a COG is the principle of this analysis; in practice, because of insufficient resolution of sequence comparisons, certain COGs may include ancestral duplications). The principal phylogenetic classification in the COG database is overlaid with functional classification and annotation based on detailed sequence and structure analysis and published experimental data. The COG system has been designed as a platform for evolutionary analyses and for phylogenetic and functional annotation of genomes. The COGNITOR program associated with the COGs allows one to fit new proteins into existing COGs. The central tenet of this analysis is that, if it can be shown that the protein under analysis is an ortholog of functionally characterized proteins from other genomes, this functional information can be transferred to the analyzed protein with considerable confidence. In addition to COGNITOR, the COG system includes certain higher-level functionalities, such as analysis of phylogenetic patterns and co-occurrence of genomes in COGs. The current (as of 1 June, 2000) system consists of 2,112 COGs that encompass about 27,000 proteins from 21 completely sequenced genomes [15].
Here we describe the application of the COGs to the systematic annotation and evolutionary analysis of two recently sequenced archaeal genomes, those of the euryarchaeon Pyrococcus abyssi [16] and the crenarchaeon Aeropyrum pernix [17]. These genomes were selected to compare the utility of the COGs for the annotation of two types of genomes - one that is closely related to another genome already included in the system, as Pyrococcus abyssi is to P. horikoshii, and one that represents a group previously not covered by the COGs, the Crenarchaeota. We show here the relatively low error rate of the COG-assisted analysis and its contribution to a significant number of new functional predictions. Emphasis is on using the COG approach to identify features of the A. pernix genome that are shared among all Archaea and those that distinguish Crenarchaeota from Euryarchaeota. Thus this work had a dual focus: first, to explore the potential of the COG system for genome annotation; and second, to use the COG approach to reveal important trends in archaeal genome evolution. It should not be construed as a comprehensive analysis of any particular genome or a comprehensive comparative and evolutionary study; addressing each of these tasks would require the use of several additional methodologies.
Results and discussion
The protocol for genome annotation using the COG database
Figure 1 depicts the steps of the procedure used for the COG-based genome annotation. This protocol is not limited to straightforward COGNITOR analysis but also takes advantage of the phylogenetic information encapsulated in the COGs, primarily in the form of phylogenetic patterns, which can be used to guide the search for missing COG members (described in detail in [18]). Briefly, whenever one of the analyzed genomes was unexpectedly not represented in a COG, additional analysis was undertaken to identify possible diverged members by using an iterative database search with the PSI-BLAST program, or to detect members that could have been missed in the original genome annotation by using translating searches with the TBLASTN program. In the present analysis of two archaeal genomes, such unexpected absences involved COGs represented in all or most of the other species or in all other archaea. Conversely, unexpected occurrences of the analyzed genomes in COGs, for example the first archaeal member of a purely bacterial COG, was examined case by case to detect likely horizontal gene transfer events and novel functions in archaeal genomes.
Figure 1.
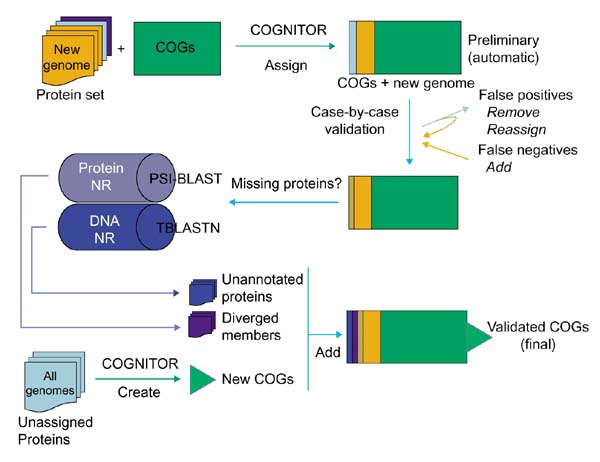
A flow chart of the genome annotation process using COGs. NR is the Non-Redundant sequence database at the National Center for Biotechnology Information.
Assessment of computational assignment of proteins to COGs
Proteins were assigned to COGs by two rounds of automated comparison using COGNITOR, each followed by manual checking of the assignments. The first round attempts to assign proteins to existing COGs; typically, >90% of the assignments are made in this step. The second round serves two purposes: first, to assign paralogs that could have been missed in the first round to existing COGs; and second, to create new COGs from those proteins that remained unassigned. With the goal of determining the optimal level of automation for such tasks, we assessed the performance of the automated procedure for annotating the A. pernix genome, which belongs to a major taxon, Crenarchaeota, that so far has not been represented in the COG database. For comparative purposes, the performance of the automated procedure for annotating proteins from Pyrococcus abyssi was also evaluated. P. abyssi is a member of the Euryarchaeota and is closely related to P. horikoshii, which is currently represented in the COGs. The data are shown in Table 1. Three main classes of protein assignments are considered: true positives, false positives and false negatives.
Table 1.
Assignment of proteins to COGs
| Category | A. pernix | P. abyssi |
| Proteins assigned by COGNITOR | 1,123 | 1,421 |
| Proteins included in COGs* | 1,102 | 1,404 |
| True positives | 1,062 | 1,381 |
| Pre-existing COGs | 1,011 | 1,339 |
| New COGs | 27 | 3 |
| Divided† | 24 | 39 |
| False positives | 44 | 31 |
| Not accepted | 21 | 17 |
| Reassigned to a related COG | 21 | 14 |
| Reassigned to an unrelated COG | 2 | 0 |
| False negatives‡ | 17 | 9 |
*Includes true positives, reassigned false positives, and false negatives. †Not included in 'To preexisting COG' or 'To new COG' numbers. ‡Proteins added during manual checking.
True positives are proteins that were correctly assigned either to an existing COG or to a COG that was created as a result of adding the new species. After a detailed examination of the COGNITOR results, 95% of the automatically assigned A. pernix proteins and 97% of the automatically assigned P. abyssi proteins were classified as true positives. As expected, the number of COGs created as a result of adding each species significantly differed. P. abyssi, which belongs to a previously represented clade, contributed only three new COGs, each representing a conserved family missing in P. horikoshii. In contrast, 27 new COGs were created as a result of adding A. pernix proteins.
False positives are proteins that were incorrectly assigned to a COG, and these fall into two classes. The first class are those proteins that needed to be removed altogether (that is, not included in any COG). In such cases, although the criterion that the query protein had at least three genome-specific best hits to members of the given COG was formally met, a detailed examination showed that these hits most probably arose by chance (see the Materials and methods section). The second class are those proteins that were assigned to one COG by COGNITOR, but subsequently moved to another COG. Most often (21 of 23 cases for A. pernix, and all of the cases for P. abyssi), the proteins were moved to a related COG (for example, between two COGs that include distinct, but related families of ATPases).
False negatives are proteins that were not assigned to any COG by COGNITOR because they failed the three-best-hit criterion (see the Materials and methods section), but were included subsequently as a result of additional sequence comparisons initiated upon examination of unexpected phylogenetic patterns. Again, the occurrence of false negatives for P. abyssi was about half that of A. pernix (1% versus 2%). Of the 17 such omissions identified among A. pernix proteins, 11 occurred as a result of pre-processing the protein sequences for low-complexity regions using the SEG program. When COGNITOR was run without filtering, these proteins were automatically assigned to the COGs. The remainder, including all false negatives seen for P. abyssi, failed the three-best-hit criterion because they showed only weak similarity to the members of the respective COG. However, given that they were the best candidates for filling unexpected gaps in phylogenetic patterns, and also because they contained the typical sequence motifs of the respective families, these proteins were included in the COGs.
Annotation of Aeropyrum pernix and Pyrococcus abyssii protein sets
Aeropyrum pernix has been reported to encode 2,694 putative proteins in a 1.67 megabase (Mb) genome [17]. Of these, 633 proteins were assigned a function or partial characterization in the original report, on the basis of sequence comparison with proteins in the GenBank, SWISS-PROT, EMBL, PIR and Owl databases. Each of these databases contains individually annotated proteins. In contrast, the COG database annotates protein families rather than individual proteins, and the method used for the construction of the COGs often allows distant relationships to be discerned. Thus, use of the COG database for the annotation of a newly sequenced genome would probably increase the number of functional assignments. Indeed, we have assigned 1,102 A. pernix proteins to COGs. Some of these proteins (154) are members of COGs belonging to the uncharacterized (S) group, about which little is known except that they form a conserved family [12]. Subtracting these, annotation has been added to 315 proteins - an increase of about 50% compared with the original annotation. These include, among others, the key glycolytic enzymes glucose-6-phosphate isomerase (APE0768, COG0166) and triosephosphate isomerase (APE1538, COG0149), and the pyrimidine biosynthetic enzymes orotidine-5'-phosphate decarboxylase (APE2348, COG0284), uridylate kinase (APE0401, COG0528), cytidylate kinase (APE0978, COG1102), and thymidylate kinase (APE2090, COG0125). Similarly, important functions in DNA replication and repair were confidently assigned to a significant number of A. pernix proteins that in the original annotation were described simply as a 'hypothetical protein'. Examples include the bacterial-type DNA primase (COG0358), the large subunit of the archaeal-eukaryotic-type primase (COG2219), a second ATP-dependent DNA ligase (COG1423), three paralogous photolyases (COG1533), and several helicases and nucleases of different specificities.
The case of the large subunit of the archaeal-eukaryotic primase illustrates well the contribution of different types of inference to genome annotation. COGNITOR failed to assign an A. pernix protein to this COG. Given the ubiquity of this subunit in euryarchaea and eukaryotes [19], however, and the presence of a readily detectable small primase subunit in A. pernix (COG1467), a more detailed analysis was undertaken by running PSI-BLAST searches against the NR database with all members of the original COG as starting queries. When the Archaeoglobus fulgidus primase sequence (AF0336) was used to initiate the search, the A. pernix counterpart (APE0667) was indeed detected at a statistically significant level.
An interesting case of reannotation of a protein with a critical function, which also resulted in more general conclusions, is the archaeal uracil DNA glycosylase (UDG; COG1573). The members of this COG are currently annotated either as a putative DNA polymerase (APE0427 from A. pernix and AF2277 from A. fulgidus) or as a hypothetical protein. However, UDG activity has been experimentally shown for the respective proteins from Thermotoga maritima [20] and A. fulgidus [21]. The reason for the erroneous annotation as a DNA polymerase is the independent fusion of the uracil DNA glycosylase with DNA polymerases in bacteriophage SPO1 and in Yersinia pestis. Although these fusions hampered the correct annotation in the original analysis of the archaeal genomes, they seem to be functionally informative, suggesting that this type of UDG functions in conjunction with the replicative DNA polymerase. This is consistent with a recent report that archaeal DNA polymerases stall in the presence of uracil before misincorporating adenine [22].
Additions, subtractions and changes to the A. pernix protein set
In all, 1,102 of the predicted 2,694 A. pernix proteins (41%) were included in the COGs, whereas 1,404 of the predicted 1,765 P. abyssi proteins (79%) were included. The percentage of A. pernix proteins included in the COGs was significantly less than the average (72%) for the other five archaeal protein sets currently included in the COG database (Table 2). It seems likely that this is due to an overestimate of the total number of ORFs in the A. pernix genome. Many of the ORFs with no similarity to proteins in sequence databases (1,538, or 57.1% [17]) overlap with ORFs from conserved families, including COG members. On the basis of the average representation of all genomes in the COGs (67%) and the average for the other archaea (72%), one could estimate the total number of A. pernix proteins to be between 1,550 and 1,700. This range is consistent with the size of the A. pernix genome (1.67 Mb) given the gene density of about one gene per kilobase that is typical of bacteria and archaea. Considering that A. pernix is the first crenarchaeon sequenced, and is also the only archaeal aerobe sequenced so far, one might expect that the representation of A. pernix proteins in COGs could be somewhat lower than the average for the Euryarchaeota. Taking 60% as a conservative estimate, this puts the upper limit of protein-coding genes in A. pernix at about 1,900. Complete reconstruction of the A. pernix proteome is beyond the scope of this work, but 849 ORFs, originally annotated as proteins, that significantly overlapped with COG members could be confidently excluded, which brings the number of genes to a maximum of 1,873.
Table 2.
Comparison of proteins in COGs for archaeal species
| Species* COGs | Genome size (Mb) | ORFs | Percentage of ORFs in COGs | |
| Total | in COGs | |||
| Af | 2.18 | 2,411 | 1,755 | 73 |
| Mj | 1.74 | 1,747 | 1,252 | 72 |
| Mth | 1.75 | 1,871 | 1,339 | 72 |
| Ph | 1.74 | 2,072 | 1,333 | 64 |
| Pa | 1.77 | 1,765 | 1,404 | 79 |
| Ap | 1.67 | 2,694 | 1,102 | 41 |
| Ap† | 1.67 | 1,873 | 1,129 | 60 |
*For abbreviations see the Materials and methods section. †After adjusting for new ORFs and removal of likely false ORFs. This adjusted number of genes is the upper estimate of the actual total number of genes in A. pernix.
Despite the apparent over-representation of ORFs in A. pernix, we nonetheless added 28 previously unidentified ORFs that represent conserved protein families, including such functionally indispensable proteins as chorismate mutase (APE0563a, COG1605), translation initiation factor IF-1 (APE_IF-1, COG0361), and seven ribosomal proteins (APE_rpl21E, COG2139; APE_rps14, COG0199; APE_rpl29, COG0255; APE_rplX, COG2157; APE_rpl39E, COG2167; APE_rpl34E, COG2174; APE_rps27AE, COG1998). These missed genes were identified by searching the genome sequence translated in all six frames for possible members of COGs with unexpected phylogenetic patterns. For example, the translation initiation factor IF-1 COG0361 contained exactly one protein from each of the species represented in COGs, except for A. pernix. Considering the importance of this protein in translation and its conservation across all species in the COGs, it seemed unlikely that it would be missing from A. pernix, and, indeed, a highly conserved IF-1 ortholog was readily identified in translating searches with the respective COG members as queries. Not unexpectedly, all newly identified A. pernix genes encode small proteins.
Conservation of the core of archaeal COGs shows that A. pernix is a typical archaeon
Because A. pernix is the first crenarchaeal genome to be completely sequenced, it was important to investigate whether or not the conserved core of archaeal genes previously identified by comparative analysis of euaryarchaeal genomes [19] is shared by the crenarchaea. The data in Table 3 indicate that this is indeed the case - in all functional categories of COGs, the majority of COGs that contain representatives from all five euryarchaeal species also include A. pernix. A very similar conclusion has been independently reached in a recent cluster analysis of archaeal proteins [23]. The fraction of the A. pernix gene set that belongs to this conserved core, approximately 30%, is very similar to those in each of the euryarchaea if the number of predicted A. pernix genes is adjusted as described above (Figure 2). Furthermore, the breakdown pattern of the proteins into members of COGs including all archaeal species, those in COGs with a subset of archaeal species, those in COGs with no other archaeal species, and those not included COGs, appeared to be conserved in A. pernix and each of the Euryarchaeota, indicating common evolutionary trends (Figure 2).
Table 3.
Phyletic distribution of the archaeal COGs
| Functional category | Number of COGs including all five euryarchaeal species | Number of COGs including all five euryarchaeal species and A. pernix | Number of COGs including a subset of euryarchaeal species and A. pernix | Number of COGs including A. pernix but none of the euryarchaeal species |
| Translation and ribosome biogenesis | 113 | 109 | 17 | 0 |
| Transcription | 26 | 25 | 11 | 0 |
| Replication, recombination, repair | 38 | 27 | 15 | 2 |
| Cell division and chromosome partitioning | 3 | 1 | 0 | 0 |
| Post-translational modification, | 19 | 15 | 4 | 2 |
| protein turnover, chaperones | ||||
| Cell envelope biogenesis, outer membrane | 8 | 7 | 8 | 0 |
| Cell motility and secretion | 8 | 8 | 6 | 0 |
| Inorganic ion transport and metabolism | 13 | 9 | 27 | 2 |
| Signal transduction | 4 | 4 | 5 | 1 |
| Carbohydrate transport and metabolism | 20 | 18 | 13 | 2 |
| Energy production and conversion | 36 | 22 | 33 | 18 |
| Amino acid transport and metabolism | 34 | 29 | 58 | 5 |
| Nucleotide transport and metabolism | 34 | 25 | 6 | 3 |
| Coenzyme metabolism | 25 | 21 | 30 | 2 |
| Lipid metabolism | 8 | 6 | 15 | 2 |
| General functional prediction only | 75 | 59 | 60 | 8 |
| Uncharacterized | 67 | 45 | 82 | 4 |
| Total | 514 | 416 | 273 | 50 |
Figure 2.
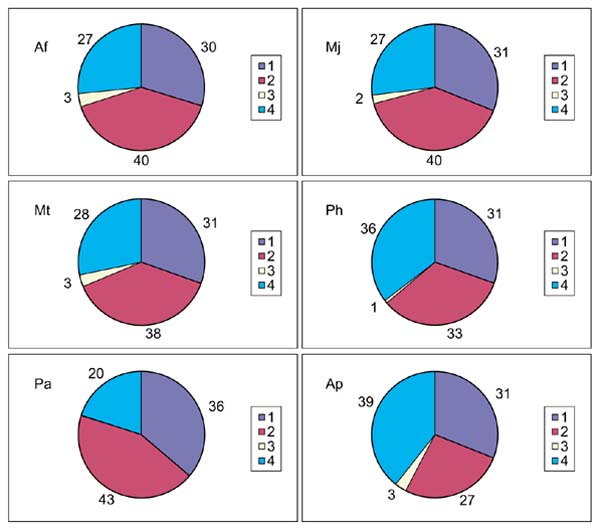
The main phylogenetic patterns for the predicted proteins encoded in six archaeal genomes. Af, Archaeoglobus fulgidus; Mt, Methanobacterium thermoautotrophicum, Pa, Pyrococcus abyssi; Mj, Methanococcus jannaschii; Ph, Pyrococcus horikoshii; Ap, Aeropyrum pernix. 1, members of COGs including all archaeal species; 2, members of COGs including a subset of archaeal species; 3, members of COGs that include no archaeal species other then the analyzed one; 4, not in COGs. The percentage of proteins in each category is indicated.
The matrix of co-occurrence of genomes in the COGs (Table 4) shows that the gene repertoire of A. pernix overlaps to a much greater extent with those of Euryarchaeota than with those of bacteria or yeast. Typically, there are fewer common COGs between A. pernix and euryarchaeal species than there are among the latter, although some preferential co-occurrence of A. pernix with the two species of Pyrococcus is notable (Table 4). To further assess the relationships between genomes, we used the co-occurrence data to construct a distance matrix (see the Materials and methods section), which in turn was used for generating a cluster dendrogram and neighbor-joining and least-square trees (Figure 3). This analysis not only unequivocally placed A. pernix within the archaeal domain, but even grouped it with the Pyrococcus species in the cluster dendrogram (Figure 3a) and the neighbor-joining tree (Figure 3b), although not in the least-square tree where it was positioned at the base of the archaeal branch and outside of the Euryarchaeota (data not shown). The outcome of this type of analysis, which is conceptually similar to recent attempts at constructing 'gene content' evolutionary trees [24,25,26], is a mixed reflection of phylogenetic relationships and similarities or differences in gene repertoires related to the lifestyles of the respective organisms. The contribution of the latter non-phylogenetic factors is well illustrated by the clustering of parasitic bacteria such as, for example, Haemophilus influenzae and Helicobacter pylori, which contradicts the obvious phylogenetic affinity of the former with Escherichia coli, and the deep branching of the mycoplasmas, the most degraded bacterial parasites, instead of the phylogenetically justified grouping with Bacillus subtilis (Figure 3). By the same token, it appears most likely that clustering of A. pernix with the pyrococci primarily reflects some common aspects of their metabolism which remain to be identified. A contribution of preferential lateral gene exchange to this grouping also seems possible. Some genes shared by A. pernix and the pyrococci, to the exclusion of the rest of the Euryarchaeota, are discussed below. A. pernix did not show any closer relationship to yeast than did the euryarchaea (Table 4 and Figure 3a,b). Thus, at least at the level of co-occurrence in COGs, or in other words, the fraction of shared orthologs, we see no support for the hypothesis of the origin of eukaryotes from crenarchaea [27,28].
Table 4.
Co-occurrence of genomes in COGs: A. pernix groups within the archaeal domain
| Ap | Mj | Mth | Af | Ph | Pa | Tm | Ec | Bs | Ssp | Sc | |
| Ap | - | 275 | 273 | 151 | 203 | 168 | 424 | 321 | 349 | 386 | 387 |
| 836 | 561 | 563 | 685 | 634 | 677 | 412 | 515 | 487 | 450 | 449 | |
| - | 408 | 424 | 411 | 299 | 297 | 647 | 955 | 878 | 752 | 402 | |
| Mj | - | 157 | 164 | 252 | 258 | 507 | 460 | 496 | 481 | 524 | |
| 969 | 812 | 805 | 717 | 722 | 462 | 509 | 473 | 488 | 445 | ||
| - | 175 | 291 | 249 | 252 | 597 | 961 | 892 | 714 | 406 | ||
| Mth | - | 177 | 325 | 296 | 509 | 441 | 478 | 465 | 515 | ||
| 987 | 810 | 662 | 700 | 478 | 546 | 509 | 522 | 472 | |||
| - | 286 | 271 | 274 | 581 | 924 | 856 | 680 | 379 | |||
| Af | - | 355 | 330 | 569 | 483 | 526 | 540 | 593 | |||
| 1096 | 741 | 780 | 527 | 613 | 570 | 556 | 503 | ||||
| - | 192 | 194 | 532 | 857 | 795 | 646 | 348 | ||||
| Ph | - | 46 | 460 | 436 | 453 | 495 | 487 | ||||
| 933 | 894 | 473 | 497 | 480 | 438 | 446 | |||||
| - | 80 | 586 | 973 | 885 | 764 | 405 | |||||
| Pa | - | 470 | 431 | 451 | 501 | 505 | |||||
| 974 | 504 | 543 | 523 | 473 | 469 | ||||||
| - | 577 | 964 | 898 | 742 | 396 | ||||||
| Tm | - | 223 | 216 | 325 | 592 | ||||||
| 1059 | 836 | 843 | 734 | 467 | |||||||
| - | 634 | 522 | 468 | 384 | |||||||
| Ec | - | 339 | 480 | 813 | |||||||
| 1470 | 1331 | 990 | 657 | ||||||||
| - | 234 | 212 | 194 | ||||||||
| Bs | - | 464 | 750 | ||||||||
| 1365 | 901 | 615 | |||||||||
| - | 301 | 236 | |||||||||
| Ssp | - | 632 | |||||||||
| 1202 | 570 | ||||||||||
| - | 281 | ||||||||||
| Sc | - | ||||||||||
| 851 | |||||||||||
| - |
*In each cell, the middle line is the number of COGs in which the given two species co-occur; the top line is the number of COGs in which the genome in the corresponding row, but not the one in the corresponding column, is represented; conversely, the bottom line is the number of COGs in which the genome in the corresponding column, but not the one in the corresponding row, is represented. The diagonal cells show the total number of COGs that include representatives from the given genome. The cells that show the co-occurrence data among archaea show the numbers of COGs in red, and the cells that show the co-occurrence data for archaea and yeast show the numbers of COGs in blue. For abbreviations, see the Materials and methods section.
Figure 3.

Classification of genomes by co-occurrence in the COGs. (a) A cluster dendrogram. (b) A neighbor-joining unrooted tree. For abbreviations, see the Materials and methods section.
Within the conserved archaeal core, there is a considerable number of genes that either comprise unique archaeal COGs (examples of these are shown in Table 5) or are sporadically present in certain bacterial species, but not in eukaryotes. These COGs are of particular interest from the viewpoint of the 'standard model' of evolution of the three domains of life, which applies primarily to the information-processing systems of the cell and places the tree root between bacteria and archaea-eukaryotes [29,30], because they can be considered synapomorphies defining the archaeal state (the sporadic representation of some such COGs in bacteria is most likely explained by horizontal gene transfer). Such characteristic archaeal proteins include, for example, the archaeal-type Holliday junction resolvase, the ATPase subunit of the archaeal-specific TopoVI (Table 5) and the archaeal DnaG-like primase with its unique domain organization (COG0358 [19]). The finding that these proteins are shared by Euryarchaeota and Crenarchaeota is important because it suggests that the respective genes were most probably present in the common ancestor of archaea and eukaryotes, but have been displaced in the eukaryotic lineage.
Table 5.
COGs represented in all archaea but not in other species: probable archaeal synapomorphies
| COG number | (Predicted) function | Comments |
| 2511 | Glu-tRNAGln amidotransferase B subunit | This protein is homologous to bacterial B subunits and an archaeal paralog but contains a synapomorphic insert, the so-called GAD domain, shared with bacterial aspartyl-tRNA synthetases [49] |
| 2016 | Predicted RNA-binding protein, contains PUA domain | PUA domain is most common in archaea and is found also in pseudouridine synthases, archaeosine synthases and glutamate kinases [50] |
| 1370 | Predicted RNA-binding protein, contains PUA domain | This form of the PUA domain is present as a stand-alone protein in A. pernix and A. fulgidus but is fused with the archaeosine synthase in the other euryarchaea [50] |
| 1746 | tRNA nucleotidyltransferase (CCA-adding enzyme) | Archaeal CCA-adding enzyme is only very distantly related to other members of the Polβ superfamily of nucleotidyltransferases [51] |
| 1395 | Predicted transcription regulators | The proteins of this family do not share similarity with other proteins beyond the DNA-binding helix-turn-helix domain [32] |
| 1389 | DNA topoisomerase VI, subunit B | These proteins contain an ATPase domain of the TopoII/MutL/HSP90/histidine kinase fold, but do not show a specific relationships to any other proteins of this class |
| 1591 | Holliday junction resolvase, archaeal-type | Distant homologs seen in some bacteria (L.A., K.S. Makarova and E.V.K., unpublished.observations) |
| 1571 | Predicted DNA-binding proteins, possibly nucleotidyl transferase or nuclease | These proteins consist of two distinct, predicted DNA-binding domains (OB-fold and Zn-ribbon) and an uncharacterized, probably enzymatic domain that is unique for archaea (see text) |
| 1491 | Predicted DNA-binding protein | These proteins contain the helix-hairpin-helix module, but otherwise, do not show significant similarity to any other proteins |
| 1938 | Predicted ATP-grasp-domain-containing enzymes | Only distantly related to other ATP-grasp proteins; predicted to possess ATP-dependent carboligase or similar activity [19] |
| 1407 | Predicted calcineurin-type phosphoesterase | Only distantly related to other phosphohydrolases of the calcineurin fold [37] |
| 1782 | Predicted metal-dependent RNase of the metallo-β-lactamase fold | In spite of significant similarity to other families of metallo-β-lactamases, this family shows a clear synapomorphy, the presence of the RNA-binding KH domain [52] |
| 1608 | Predicted kinase related to acetylglutamate kinase | Only distantly related to other kinases of the same fold |
| 1829 | Predicted kinase of the actin/HSP70/sugar kinase fold | Only distantly related to other kinases of the same fold |
| 1907 | Predicted kinase of the actin/HSP70/sugar kinase fold | Only distantly related to other kinases of the same fold |
| 1831 | Predicted metal-dependent hydrolase of the urease superfamily | Only distantly related to other hydrolases of the same superfamily [53] |
| 1571 | Predicted DNA-binding protein containing the Zn-ribbon module | |
| 2034 | Conserved membrane protein | |
| 2064 | Conserved membrane protein | |
| 1339, 2090, 1581, | Uncharacterized proteins unique to archaea | |
| 1460, 1786, 1701, 1931, 1909, 1888, 1382, 1849, 1630, 1303, 1325, 1679 |
Detailed sequence analysis of the core archaeal genes, which included comparison of the protein sequences from the respective COGs to pre-computed profiles for specific protein domains, resulted in the prediction of previously uncharacterized potential roles in conserved systems for some of them. For example, proteins in COG1571 contain two previously recognized domains, namely a nucleic-acid-binding OB-fold domain similar to those found in the ssDNA-binding protein RPA and in DNA polymerase subunits from archaea and bacteria [31] and a metal-binding Zn ribbon [32]. The amino-terminal portion of these proteins comprises a predicted globular domain that also occurs as a stand-alone protein in some of the euryarchaea and contains a conserved signature GxDDXD preceded by a predicted β strand. The combination of this potential enzymatic domain with two predicted DNA-binding domains suggests that members of this protein family are likely to be enzymes with an important role in archaeal DNA metabolism, most probably nucleotidyl transferases or nucleases. The members of COG1444 are multidomain proteins that combine an amino-terminal superfamily I helicase-like ATPase domain with a carboxy-terminal acetyltransferase domain. This domain organization is suggestive of a role in the basal transcription system as a protein-modifying acetyltransferase. COG1094 (KH + S1 domains) and COG1096 (Si+Zn-ribbon domain) are predicted to encode RNA-binding proteins that could be involved in RNA processing or in a translation-related role. COG1293 includes uncharacterized, conserved proteins whose probable ortholog from Streptococcus has been annotated as a fibronectin-binding protein [33]. Their conservation and phyletic distribution is, however, more consistent with a basic core function. Consistent with this, we detected in these proteins a specific version of the helix-hairpin-helix nucleic-acid-binding module, which is specifically similar to those found in ribosomal proteins of the S13/S18 family (L.A., unpublished observations) and suggests a function for the members of this COG as a ribosome-associated, RNA-binding protein or, less likely, an uncharacterized DNA repair system component.
A tally of the COGs that are shared by A. pernix with each of the five euryarchaeal species, to the exclusion of the rest of the euryarchaea, shows a clear prevalence of the association with pyrococci and A. fulgidus (data not shown). Given the larger number of genes in the latter, the relationship with the pyrococci is most notable, in agreement with the clustering data presented above. A. pernix and the pyrococci share some typically bacterial proteins, for example ribonuclease E/G, an RNA-processing enzyme (COG1530), and the glycine cleavage system (COGs 0403, 0404, 0509 and 1003). This is compatible with horizontal gene transfer between these two archaeal lineages subsequent to the acquisition of the respective gene from a bacterium.
A. pernix shows a notable paucity of signaling proteins, resembling in this respect Methanococcus jannaschii and parasitic bacteria. A. pernix and M. jannaschii have no detectable histidine kinase, PAS or GAF domains, unlike Methanobacterium thermoautotrophicum and A. fulgidus, in which these domains comprise the basis of the signaling system. These domains are present in the pyrococci, but in much smaller numbers than in M. thermoautotrophicum and A. fulgidus. A. pernix, M. jannaschii and the pyrococci also encode very few serine/threonine kinases, and those that are present are highly conserved representatives of the Ri01 family, which are probably involved in transcription regulation rather than in typical signaling [34]. It appears therefore that conventional phosphorylation-mediated signaling is selected against in A. pernix and other hyperthermophiles. In contrast, all these archaea, including A. pernix, possess comparable numbers of predicted transcription factors, mainly those of the helix-turn-helix class [32], some of which could directly bind small molecules and convert signals into transcriptional outputs.
Crenarchaeota as a distinct branch of the Archaea
In spite of the rather unexpected clustering with the pyrococci seen in the co-occurrence-based classification (Figure 3), the COG analysis provides evidence for the distinctness of Crenarchaeota as represented by A. pernix. At a quantitative level, this can be illustrated by comparing the number of COGs in which each of the archaeal species is missing but the rest are represented. The number of such COGs is notably greater in the case of A. pernix than in each of the euryarchaeal species (Figure 4). Qualitatively, a considerable number of apparently essential genes conserved in the euryarchaea are missing in A. pernix (Table 6). A. pernix is expected to encode unrelated or distantly related proteins performing these functions (non-orthologous gene displacement [35]). Notably, unlike the euryarchaea with completely sequenced genomes, A. pernix does not encode enzymes of the de novo purine biosynthesis pathway (Table 6). The enzymes for interconversion of IMP, GMP and AMP are present, and it appears likely that A. pernix partly relies on salvage pathways for the formation of purine nucleotides, but also probably imports nucleosides and/or bases into the cell. In this respect, A. pernix is similar to such parasitic bacteria as H. pylori, Borrelia burgdorferi, and Chlamydia that do not possess purine biosynthesis capabilities either and import nucleosides or bases from the surrounding medium. In contrast, the pyrimidine biosynthesis pathway genes are present in A. pernix, as they are in other archaea.
Figure 4.

COGs not represented in each of the archaeal species while including members of the remaining five species. For P. horikoshii and P. abyssi, the absence of the respective second pyrococcal species was allowed. For abbreviations, see the Materials and methods section.
Table 6.
Examples of COGs conserved in euryarchaea but missing in A. pernix
| COG number | Phylogenetic pattern* | Function | Comments |
| 0101 | amtks-yqvdcebrhujwgpolinx | Pseudouridylate synthase (tRNA psi55) | This RNA modification enzyme hitherto has been considered ubiquitous and essential [54] |
| 1549 | amtks- - - - - - - - - - - - - - - - - - - - | Archaeosine tRNA-ribosyltransferase, contains PUA domain | See text |
| 2036 | amtks-y- - - - - - - - - - - - - - - - - - | Histones H3 and H4 | In the crenarchaeon Sulfolobus solfataricus, the role of histones in maintaining DNA supercoiling appears to be relegated to the DNA-binding protein Sso7d [55]. However, A. pernix does not encode an ortholog of this protein, and the mechanism of chromatin packaging remains unclear |
| 1933 | amtks- - - - - - - - - - - - - - - - - - - - | Unique archaeal DNA polymerase, large subunit | This DNA polymerase is highly conserved among the euryarchaea but so far has not been seen in other taxa [15] |
| 1311 | amtks-y- - - - - - - - - - - - - - - - - - | Small subunit of DNA polymerase, predicted phosphatase (calcineurin-like superfamily) | The absence of this subunit, which is represented by (predicted) active phosphatases in archaea and inactivated forms in eukaryotes [37] correlates with the absence of the large subunit |
| 1111 | amtks-y- - - - - - - - - - - - - - - - - | ERCC4-like helicase | A typical archaeal-eukaryotic repair protein, a predicted active helicase in euryarchaea and an inactivated form in eukaryotes [39] |
| 1107 | amtks- - - - - - - - - - - - - - - - - - - - | Archaea-specific RecJ-like exonuclease, ontains DnaJ-type Zn finger domain | |
| 1243 | amtks-y-v- - - - - - - - - - - - - - - - | Transcription elongation factor ELP3 | Consists of an amino-terminal biotin-synthase-like domain and a carboxy-terminal histone acetylase domain (only the biotin-synthase-like domain is represented in some bacteria) |
| 0206 | amtks- -qvdcebrhuj-gpol-nx | Cell division GTPase FtsZ | A central component of bacterial and archaeal cell-division machinery which is homologous (although only weakly similar) and functionally analogous to eukaryotic tubulins [56]. So far, among prokaryotes, FtsZ has been found missing only in Chlamydia [57]. |
| 0455 | amtks- -qvdcebr-uj- - -ol-n- | ATPases involved in chromosome partitioning | There may be a correlation between the absence of FtsZ and the presence of only one chromosome-partitioning ATPase (the ortholog of bacterial Mrp) in A. pernix |
| 1149 | amtks- - -v- - - - - - - - - - - - - - - | MinD superfamily P-loop ATPases containing an inserted ferredoxin domain | |
| 0065 | amtks-yqvdcebrh-j- - - - - -n- | 3-Isopropylmalate dehydratase large subunit | A. pernix does not encode committed enzymes of leucine biosynthesis, indicating that, unlike all other free-living organisms whose genomes have been sequenced so far, it cannot produce leucine. It might depend on other archaea and/or bacteria as the source of leucine, or perhaps synthesize leucine by a distinct, completely uncharacterized pathway |
| 0066 | amtks-yqvdcebrh-j- - - - - -n- | 3-isopropylmalate dehydratase small subunit | |
| 0473 | amtks-yqvdcebrh-j- - - - - -nx | 3-isopropylmalate dehydrogenase | |
| 0119 | amtks-yqvdcebrh-j- - - - - -n- | Isopropylmalate/homocitrate/citramalate synthase | |
| 0015 | amtks-yqvdcebrhuj- - - - - -n- | Adenylosuccinate lyase | Most of the enzyme of the de novo pathway of purine biosynthesis are missing in A. pernix. It appears likely that, in this organism, all purine nucleotides are produced via the salvage pathway. |
| 0104 | amtks-yqvdcebrhuj- - - - - -n- | Adenylosuccinate synthase | |
| 0034 | amtks-yqvdcebrh-j- - - - - -n- | Glutamine phosphoribosyl-pyrophosphate amidotransferase | |
| 0151 | amtks-yqvdcebrhuj- - - - - -n- | Phosphoribosylamine-glycine ligase | |
| 0150 | amtks-yqvdcebrh-j- - - - - -n- | Phosphoribosylamino-imidazol (AIR) synthetase PurM | |
| 0152 | amtks-yqvdcebrh-j- - - - - -nx | Phosphoribosylamino-imidazolesuccino-carboxamide (SAICAR) synthase | |
| 0041 | amtks-yqvdcebrh-j- - - - - -n- | Phosphoribosylcarboxy-aminoimidazole (NCAIR) mutase PurE | |
| 0047 | amtks-yqvdcebrh-j- - - - - -n- | Phosphoribosylformyl-glycinamidine (FGAM) synthase, glutamine amidotransferase | |
| domain | |||
| 0046 | amtks-yqvdcebrh-j- - - - - -n- | Phosphoribosylformyl-glycinamidine (FGAM) synthase, synthetase domain | |
| 0340 | amtks-yqvdcebrhuj- - - -linx | Biotin-(acetyl-CoA carboxylase) ligase | A. pernix apparently does not encode any enzymes of biotin synthesis or biotin-utilizing enzymes |
| 0511 | amtks-yqvdcebrhuj- - - -linx | Biotin carboxyl carrier protein of acetyl-CoA carboxylase | |
| 0157 | amtks-yqv-cebrhu- - - - - - -n- | Nicotinate-nucleotide pyrophosphorylase |
*In the phylogenetic patterns, each letter indicates that a particular genome is represented in the given COG, and a dash indicates the absence of a representative from the corresponding genome. The one-letter code for genomes is as follows: a, Archeoglobus fulgidus; m, Methanococcus jannaschii; t, Methanobacterium thermoautotrophicum; k, Pyrococcus horikoshii; s, Pyrococcus abyssi; z, Aeropyrum pernix; y, Saccharomyces cerevisiae; q, Aquifex aeolicus; v, Thermotoga maritima; d, Deinococcus radiodurans; c, Synechocystis sp; e, Escherichia coli; b, Bacillus subtilis; r, Mycobacterium tuberculosis; h, Haemophilus influenzae; u, Helicobacter pylori; j, Campylobacter jejuni; w, Ureaplasma urealyticum; g, Mycoplasma genitalium; p, Mycoplasma pneumoniae; o, Borrelia burgdorferi; l, Treponema pallidum; i, Chlamydia trachomatis and C. pneumoniae; n, Neisseria meningitidis; x, Rickettsia prowazekii.
A. pernix lacks certain conserved RNA-modifying enzymes such as the tRNA-specific pseudouridine synthase (COG0101), which has so far been considered ubiquitous, and the tRNA archaeosine transglycosylase (COG1549). The absence of the latter enzyme suggests that archaeosine could be a euryarchaea-specific RNA modification.
Interestingly, A. pernix lacks several conserved proteins and features of domain architecture that are specifically shared by euryarchaea and eukaryotes. A particularly notable absence is that of the two subunits of the euryarchaeal DNA polymerase (Table 6). The large subunit is present only in the euryarchaea [36], whereas the small subunit, which belongs to the calcineurin superfamily of phosphoesterases and is predicted to possess phosphatase activity, is conserved in euryarchaea and eukaryotes [37]. Unlike the euryarchaea, the replicative DNA polymerases in A. pernix are represented only by three paralogous members of the B family (one of them possibly inactivated; COG0417), which is shared by archaea and eukaryotes [38]. The replication factor A (RPA) ortholog (COG1599) from A. pernix contains a single OB-fold domain whereas euryarchaea and eukaryotes encode forms with multiple tandem repeats of the OB-fold that are more similar to each other. The eukaryotic DNA repair component ERCC4 and its ortholog in euryarchaea contain fused super-family II helicase and nuclease domains (COG1111 and COG1948 [39]). In contrast, the crenarchaea A. pernix and Sulfolobus possess only the nuclease domain (also represented in A. fulgidus), with no counterpart to the helicase domain. Two predicted helicases with a potential function in DNA repair (COG1112, a superfamily I helicase, and COG1205, a superfamily II helicase fused to a predicted metal-dependent nuclease domain) are also shared by the euryarchaea and eukaryotes, to the exclusion of A. pernix. The transcription machinery of A. pernix also shows several deviations from the general euryarchaeal-eukaryotic pattern. The transcription factor TFIIB from both A. pernix and Sulfolobus (COG1405) contains a disrupted amino-terminal Zn ribbon domain. However, A. pernix encodes a stand-alone version of the TFIIB Zn ribbon (APE0508 in COG1405) that could substitute for the disrupted version in trans. Importantly, the histones that function in the chromosomal structure maintenance and possibly also in transcription in euryarchaea (COG2036) are lacking in A. pernix (see Table 6 for more details). The RNA polymerase elongation factor ELP3, which combines a biotin synthase domain with a histone acetylase domain and is shared by euryarchaea and eukaryotes (COG1243), is missing in A. pernix.
The current COG collection was not well suited for detecting COGs that are represented exclusively in crenarchaea and in eukaryotes because only one species from each of these taxa is represented. Our additional analysis, however, revealed very few such genes in A. pernix and no trends supporting a possible ancestral relationship between Crenarchaeota and eukaryotes were detected (L.A. and E.V.K., unpublished observations). The current status of archaeal genome analysis, with more 'eukaryotic' features seen in euryarchaea than in crenarchaea, offers no support for the 'eocyte hypothesis', which postulates origin of eukaryotes from crenarchaea [28]. For a more definitive interpretation of the evolutionary relationships between archaea and eukaryotes, however, additional genome sequences, particularly those from other crenarchaea, are required.
Aeropyrum genes without homologs in Euryarchaeota and acquisition of bacterial genes by horizontal transfer
The 48 COGs that include a representative from A. pernix but not from euryarchaea are likely to reflect acquisition of bacterial genes by crenarchaea or loss of ancestral genes early in the evolution of euryarchaea (Table 7). Rigorously distinguishing between these two possibilities may be difficult, but in particular cases there are indications in favor of one of them when there is a distinct similarity between an A. pernix protein and orthologs from a particular bacterial lineage (Table 7). Generally, horizontal gene transfer appears to be the most plausible scenario for the origin of these genes in A. pernix because none of the genes in these 48 COGs is found in all bacteria and eukaryotes, and none shows specific affinity with eukaryotic orthologs. The converse would have suggested ancestral provenance.
Table 7.
A. pernix proteins conserved in a wide range of organisms but missing in euryarchaea (examples)
| A. pernix gene/COG number | Phylogenetic pattern* | Function | Comments | |
| APE1618/1048 | - - - - -zyq-d-ebr- - - - - - - - - -x | Aconitase A | Unlike other archaea with sequenced genomes, A. pernix is an aerobe and possesses the complete TCA cycle. APE1618 belongs to a distinct family of (otherwise) bacterial aconitases (e.g E. coli AcnA and B. subtilis CitB). APE1816 also belongs to a specific family of bacterial fumarases. | |
| APE1816/0114 | - - - - -zy- -dcebrhu- - - - - -i-x | Fumarase | ||
| APE1677/1071 | - - - - -zy- -dc-br- - - -gp- -i-x | pyruvate dehydrogenase E1 component, α-subunit | ||
| APE1674/0022 | - - - - -zy- -dc-br- - - -gp- -i-x | pyruvate dehydrogenase E1 component, β-subunit | ||
| APE1671/0508 | - - - - -zy- -dcebrh- - -gp- -i-x | Dihydrolipoamide acyltransferase | ||
| APE1725/1290 | - - - - -z-q-dc-br-uj- - - - - -nx | Cytochrome b | As an aerobe, A. pernix encodes specific electron-transport chain components. APE1623 is closely related to the ortholog from Aquifex aeolicus. | |
| APE1623, APE0793_1/0843 | - - - - -z-q-dcebr-uj- - - - - -nx | Cytochrome c oxidase, heme b and copper-binding subunit | ||
| APE0793_2/ | - - - - -z-q-dcebr- - - - - - - - - -x | Cytochrome oxidase, subunit 3 | ||
| 1845 | ||||
| APE1498/1171 | - - - - -zy-vdcebrh- - - - - - - - -x | Threonine dehydratase | Specifically related to a subfamily of bacterial catabolic threonine dehydratases (e.g. E. coli TdcB) | |
| APE1038/0295 | - - - - -zy-vd-ebrh- -wgpo- - - - | Cytidine deaminase | ||
| APE1353/0514 | - - - - -zy-dceb-h- - - - - -l- - - | DNA helicase (RecQ family) | APE1353 differs from other members of the RecQ family by the presence of long amino-terminal extension that probably form a non-globular domain. APE1353 shows no specific affinity to any of the bacterial orthologs. | |
| APE2450/0260 | - - - - -z-q-dcebrhu-wgp- -i-x | Leucyl aminopeptidase | ||
| APE0137/0405 | - - - - -zy- -dcebr-u- - - - - - - - - | Gamma-glutamyltranspeptidase | ||
| APE2464/ | - - - - -z-qvdcebr- - - - - - - - - - - | Phosphate starvation-inducible protein | ||
| COG1702 | PhoH, Predicted ATPase | |||
| APE0993/>0813 | - - - - -z- - -d-eb-hu-wgp-l- - - | Purine-nucleoside phosphorylase | Correlates with the absence of de novo purine biosynthesis and the probable importance of salvage pathways | |
| APE2105/0813a | - - - - -z- - - - - -e- -h- - - - -l- - - | Uridine phosphorylase | ||
| APE0033/1866 | - - - - -zy- -d-eb-h- - - - - - - - - - | Phosphoenolpyruvate carboxykinase |
*The designations are as in Table 6.
The most notable group of genes that are found in A. pernix, but not in euryarchaea, reflects its aerobic metabolism. As an aerobe, A. pernix encodes the complete set of the tricarboxylic acid cycle (TCA) cycle enzymes, in contrast to the anaerobic euryarchaea which possess a truncated version of this pathway (Table 7). In addition, A. pernix encodes accessory enzymes that are required for the formation of the pyruvate dehydrogenase complex, such as lipoate synthase (COG0320) and lipoate-protein ligase (COG0095). The set of A. pernix proteins that are related to respiration and not seen in euryarchaea additionally includes specific electron-transfer chain components such as cytochrome b, cytochrome oxidase, nitrate reductase, NADH-ubiquinone oxidoreductase, NADPH:quinone reductase and Rieske Fe-S protein.
A set of 23 COGs is shared by A. pernix, yeast and a subset of bacteria, to the exclusion of the euryarchaea. These cases seem to be readily explained by lateral acquisition of genes from a bacterial source in both eukaryotes (largely from mitochondria) and crenarchaea. Respiration-related enzymes mentioned above are an obvious case in point, but this explanation could also apply to at least some of the remaining few COGs in this set, for example, a RecQ-family helicase (COG0514).
Detecting interspecies differences in Pyrococcus abyssi and P. horikoshii
In the case of two closely related genomes, such as those of P. abyssi and P. horikoshii, the COG analysis provides for straightforward genome subtraction (Table 8). Given the high level of sequence conservation between orthologous proteins from these two species (typically over 70% identity), it seems somewhat unexpected that 80 COGs include proteins from P. abyssi, but not P. horikoshii, whereas the inverse is seen in 46 COGs. Many of these differences are likely to reflect differential gene loss, whereas others are probably due to horizontal gene acquisition. The greater number of COGs that P. abyssi is represented in, to the exclusion of P. horikoshii, seems to reflect the greater metabolic endowment of the former. In particular, the entire aromatic amino acid and cysteine biosynthesis pathways are present in P. abyssi but not in P. horikoshii (Table 8). In the case of the aromatic amino acid pathway, the direction of evolution seems to be clear - loss of the respective genes by P. horikoshii in the course of its adaptation to the heterotrophic lifestyle which seems to have gone further than in P. abyssi. The case of the cysteine pathway is, however, particularly interesting because, among all archaea whose genomes are currently available, it is shared only by A. pernix and P. abyssi; the mechanism of cysteine formation in other euryarchaea remains a mystery [19]. In this case, acquisition of the respective genes from bacteria via horizontal transfer seems to be the most likely possibility because a gene-loss scenario would require several independent events in euryarchaea. Interestingly, probable horizontal acquisition of bacterial genes encoding the cytosine biosynthesis pathway enzymes has been described also in the euryarchaeon Methanosarcina barkeri [40]. Other COGs with differential representation of the two Pyrococcus species tend to include genes that are inherently mobile such as restriction-modification systems (Table 8).
Table 8.
Examples of differential genome display of Pyrococcus abyssi and Pyrococcus horikoshii using the COG approach
| Gene/COG Number | Phylogenetic pattern* | Function | Comment |
| Present in P.abyssi but not P. horikoshii | |||
| PAB2044/0547 | amt-szyqvdcebrhuj- - - - - -n- | Anthranilate phosphoribosyltransferase | The entire branched pathway for aromatic amino acid biosynthesis appears to be present in P. abyssi but not in P. horikoshii |
| PAB2045/0147 | amt-szyqvdcebrhuj- - - - - -n- | Anthranilate synthase component I | |
| PAB2046/0512 | amt-szyqvdcebrhuj- - - - - -n- | Anthranilate synthase component II | |
| PAB0307/0082 | - - - -szyqvdceb-huj- - - - -inx | DAHP synthase | |
| PAB2049/0159 | amt-szyqvdcebrhuj- - - - -in- | Tryptophan synthase α subunit | |
| PAB2048/0133 | amt-szyqvdcebrhuj- - - - -in- | Tryptophan synthase β subunit | |
| PAB0250/0031 | - - - -szyqvdcebrhuj- - - - - -n- | Cysteine synthase | P. abyssi appears to be the only euryarchaeon that encodes the typical cysteine biosynthesis pathway which it shares with A. pernix |
| PAB1595/2046 | a- - -szyq-dc-b- - -j- - - - - - - - | ATP sulfurylase | |
| PAB0781/0529 | a- - -szyq-dcebr- -j- - - - - -n- | Adenylylsulfate kinase | |
| PAB1839/0035 | - -t-szyqvdcebrh-jwgp-l-n- | Uracil phosphoribosyltransferase | |
| PAB2246/0827 | -m- -s- - - - - - -brhu-w-p- - - - - | Adenine-specific DNA methyltransferases | |
| PAB2154/0610 | amt-s- - - - - -e- -hujw-p- - -n- | Restriction enzymes type I helicase subunit | |
| Present in P. horikoshii but not P. abyssi | |||
| PH0369/0153 | - - -k- -y-v- -ebrh- - - - - -I--- | Galactokinase | |
| PH1048, PH1046/2309 | - - -k-z-q-d- -b- - - - - - -o- - - - | Leucyl aminopeptidase (aminopeptidase T) | |
| PH0365/1085 | a- -k- -y-v- -e-rh- - - - - - - - - - | Galactose-1-phosphate uridylyltransferase | |
| PH0896/1230 | - - -k- -yqvd-eb- - -j- - - - - -n- | Co/Zn/Cd efflux system component | |
| PH0162/1353 | amtk- - -qv- - - -r- - - - - - - - - - - | Predicted hydrolase of the HD superfamily | |
| PH1032/0338 | -m-k- - - - - -ce- -hu- - - - -I- - - | Site-specific DNA methylase dam | |
| PH0873/1401 | - -tk- - -q-dceb-uj- - - - - - - - | GTPase subunit (McrB) of a restriction endonuclease | |
*The designations are as in Table 6.
Conclusions
The annotation of a new genome is likely to be as good as the database(s) to which it is compared. The COG database was constructed on the phylogenetic principle of protein classification, namely clustering by (probable) orthology. In addition, considerable effort has been invested in the functional characterization and classification of the COGs. As a result, using the COGs for annotating new genomes of organisms that do not belong to already well-characterized groups provides for numerous functional predictions that are not readily attained in more routine annotation protocols. Furthermore, taking advantage of the structure of the COG database, it is possible to reveal the main functional systems of an organism and its probable evolutionary affinities, and to systematically uncover sets of genes whose presence or absence in the given genome is unexpected and informative from an evolutionary standpoint.
Materials and methods
Sequence data and databases
The genome sequences and the sets of annotated proteins from Aeropyrum pernix and Pyrococcus abyssi were retrieved from the Genomes division of the Entrez system [41]. The COG database, as of 1 June, 2000, consisted of 2,112 COGs that included 26,919 out of the total of 43,897 proteins from 21 completely sequenced bacterial, archaeal and eukaryotic genomes. This release of the COG database included the following genomes. Bacteria: Aquifex aeolicus (Aa), Bacillus subtilis (Bs), Borrelia burgdorferi (Bb), Chlamydia trachomatis (Ct), Chlamydia pneumoniae (Cp), Escherichia coli (Ec), Haemophilus influenzae (Hi), Helicobacter pylori (Hp), Mycoplasma genitalium (Mg), Mycoplasma pneumoniae (Mp), Mycobacterium tuberculosis (Mtu), Synechocystis PCC6803 (Ssp), Thermotoga maritima (Tm), Treponema pallidum (Tp). Archaea: Archaeoglobus fulgidus (Af), Methanobacterium thermoautotrophicum (Mth), Methanococcus jannaschii (Mj) and Pyrococcus horikoshii (Ph). Eukaryotes: yeast, Saccharomyces cerevisiae (Sc). The following recently sequenced genomes were included in the present analysis, to become available in the new release of the COGs. Bacteria: Campylobacter jejunii (Cj), Deinococcus radiodurans (Dr), Neisseria meningitidis (Nm) and Ureaplasma urealyticum (Uu). Archaea: Aeropyrum pernix (Ap) and Pyrococcus abyssi (Pa). In addition to the COG database, the nonredundant (NR) database of protein sequences at the National Center for Biotechnology Information (NIH, Bethesda) was used for sequence similarity searches.
Sequence analysis and assignment of proteins to COGs
Protein sequence similarity searches were performed using the gapped BLASTP program or, for detecting subtle similarities, the position-specific iterative BLAST (PSI-BLAST) program, with either an individual protein or a collection of pre-computed position-specific scoring matrices used as a query [42]. Searches of nucleotide sequences translated in six frames were performed using the gapped TBLASTN program [42]. Regions of low complexity in protein sequences were identified using the SEG program [43].
Proteins were assigned to COGs using the COGNITOR program essentially as previously described [12,15]. Briefly, each protein sequence from the analyzed genomes was compared to the protein sequences that comprise the COGs using the gapped BLASTP program and genome-specific best hits (BeTs) were registered. A protein was included in a COG when at least three BeTs were to the members of the given COG. In cases when two or more COGs met this criterion, the COG with the greater alignment scores was given priority. If no COGs satisfied this condition, an ambiguous result was reported. The new version of the COGNITOR program used in this analysis detects position-specific BeTs so that distinct domains of multidomain proteins were assigned to different COGs when justified by the above criteria.
Proteins from the analyzed genomes that could not be included in the pre-existing COGs were subjected to the original COG construction procedure [12]. Specifically, a new COG was formed when a triangle of consistent reciprocal BeTs was identified, which occurs when one protein from each of three distinct species gives as a genome-specific best hit the other two proteins, and vice versa. In the present analysis, only such elementary new COGs could be identified because those COGs that would include a greater number of species have already been created.
Manual validation of preliminary COG assignments
Each preliminary assignment was checked for validity, and adjustments were made as necessary. An assignment of a particular protein to a given COG was considered correct if: (1) the genome-specific best hits for that protein were members of the COG; (2) the size of the query protein was similar to the sizes of the COG members; (3) the region of similarity between the query and the COG members was extensive; (4) the similarity scores indicated statistical significance.
For cases that did not fit some of the straightforward criteria given above, other criteria or methods were used to confirm an assignment. For example, PSI-BLAST would be used to determine if the query is a diverged member of the COG, or sequence alignments would be examined to determine if the query shares conserved motifs with the COG members.
False positives that required reassignment generally fulfilled the criteria above, but failed to meet either condition (1) or condition (2). In the former case, some of the genome-specific best hits were to a different COG. In the latter case, the query protein merely contained a single domain of a COG whose members were composed of multiple domains. These proteins were assigned to COGs that contain the single domain only.
Certain false positives required removal from COGs. Although fulfilling the three-BeT criterion for inclusion, these assignments probably arose by chance as indicated by several lines of evidence: first, the genome-specific hits that caused the apparently incorrect assignment had lower alignment scores than those for non-COG proteins; second, the statistical significance of the hits was very low; third, the hits were non-reciprocal; fourth, the alignments with the COG members did not include diagnostic motifs of the respective protein family; and fifth, the protein was assigned to a COG based on hits to low-complexity (typically, coiled-coil) regions.
Proteins with multiple domains that hit members from more than one COG were manually divided according to the COGNITOR results. The sub-sequences were given separate FASTA entries in the COG database, and the domains were listed as members of the appropriate COG.
Classification of genomes by co-occurrence in COGs
The table of co-occurrence of genomes in COGs is available on the COG website [44]. These data were used for classifying genomes by cluster analysis and phylogenetic tree construction. The distance between genomes was calculated as Dij = 1 - (Cij/Ni + Nj - Cij), where Cij is the number of COGs in which genomes i and j co-occur, and Ni, Nj are the numbers of COGs that include the genomes i and j, respectively. This formula employs the Jackard co-occurrence coefficient, which is widely used in biometric studies to obtain comparable results for samples of different size [45]. Cluster dendrograms were generated using the UPGMA option of the NEIGHBOR program, and distance-matrix trees were generated using the FITCH program [46] or the Neighbor-Joining [47] option of the NEIGHBOR program. All tree-building programs are parts of the PHYLIP package [48].
Additional data
The results of the analysis of the A. pernix and P. abyssi genomes described here are included, along with those for several recently sequenced bacterial genomes, on the COGs website [44] and a file of a list of COGs is included. A list of A. pernixgenes, in which genes overlapping with COG members and thought to be spurious are flagged, and newly detected genes have been added, and the corresponding FASTA library of protein sequences are available by anonymous ftp (ftp:// ncbi.nlm.nih.gov/pub/koonin/Apernix) and are included as text files.
Supplementary Material
A list of A. pernix genes, in which genes overlapping with COG members are flagged
Acknowledgments
Acknowledgements
We are grateful to Roman Tatusov for maintaining the COG database, running the COGNITOR program in the batch mode and for critical reading of the manuscript and to Kira Makarova for her contribution to different aspects of the COG analysis and useful discussions.
References
- Boguski MS. Biosequence exegesis. Science. 1999;286:453–455. doi: 10.1126/science.286.5439.453. [DOI] [PubMed] [Google Scholar]
- Bork P, Dandekar T, Diaz-Lazcoz Y, Eisenhaber F, Huynen M, Yuan Y. Predicting function: from genes to genomes and back. J Mol Biol. 1998;283:707–725. doi: 10.1006/jmbi.1998.2144. [DOI] [PubMed] [Google Scholar]
- Galperin MY, Koonin EV. Who's your neighbor? New computational approaches for functional genomics. Nat Biotechnol. 2000;18:609–613. doi: 10.1038/76443. [DOI] [PubMed] [Google Scholar]
- Andrade MA, Brown NP, Leroy C, Hoersch S, de Daruvar A, Reich C, Franchini A, Tamames J, Valencia A, Ouzounis C, Sander C. Automated genome sequence analysis and annotation. Bioinformatics. 1999;15:391–412. doi: 10.1093/bioinformatics/15.5.391. [DOI] [PubMed] [Google Scholar]
- Gaasterland T, Sensen CW. MAGPIE: automated genome interpretation. Trends Genet. 1996;12:76–78. doi: 10.1016/0168-9525(96)81406-5. [DOI] [PubMed] [Google Scholar]
- Bhatia U, Robison K, Gilbert W. Dealing with database explosion: a cautionary note. Science. 1997;276:1724–1725. doi: 10.1126/science.276.5319.1724. [DOI] [PubMed] [Google Scholar]
- Bork P, Koonin EV. Predicting functions from protein sequences - where are the bottlenecks? Nat Genet. 1998;18:313–318. doi: 10.1038/ng0498-313. [DOI] [PubMed] [Google Scholar]
- Galperin MY, Koonin EV. Sources of systematic error in functional annotation of genomes: domain rearrangement, non-orthologous gene displacement and operon disruption. In Silico Biol. 1998;1:55–67. [PubMed] [Google Scholar]
- Kanehisa M, Goto S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000;28:27–30. doi: 10.1093/nar/28.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Overbeek R, Larsen N, Pusch GD, D'Souza M, Selkov E, Jr, Kyrpides N, Fonstein M, Maltsev N, Selkov E. WIT: integrated system for high-throughput genome sequence analysis and metabolic reconstruction. Nucleic Acids Res. 2000;28:123–125. doi: 10.1093/nar/28.1.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultz J, Copley RR, Doerks T, Ponting CP, Bork P. SMART: a web-based tool for the study of genetically mobile domains. Nucleic Acids Res. 2000;28:231–234. doi: 10.1093/nar/28.1.231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatusov RL, Koonin EV, Lipman DJ. A genomic perspective on protein families. Science. 1997;278:631–637. doi: 10.1126/science.278.5338.631. [DOI] [PubMed] [Google Scholar]
- Fitch WM. Distinguishing homologous from analogous proteins. Syst Zool. 1970;19:99–106. [PubMed] [Google Scholar]
- Fitch WM. Uses for evolutionary trees. Philos Trans R Soc Lond B Biol Sci. 1995;349:93–102. doi: 10.1098/rstb.1995.0095. [DOI] [PubMed] [Google Scholar]
- Tatusov RL, Galperin MY, Natale DA, Koonin EV. The COG database: a tool for genome-scale analysis of protein functions and evolution. Nucleic Acids Res. 2000;28:33–36. doi: 10.1093/nar/28.1.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- National Centre for Sequencing, France http://www.genoscope.cns.fr
- Kawarabayasi Y, Hino S, Horikawa H, Yamazaki S, Haikawa Y, Jin-no K, Takahashi M, Sekine M, Baba S, Ankai A, et al. Complete genome sequence of an aerobic hyper-thermophilic crenarchaeon, Aeropyrum pernix K1. DNA Res. 1999;6:83–152. doi: 10.1093/dnares/6.2.83. [DOI] [PubMed] [Google Scholar]
- Natale DA, Galperin MY, Tatusov RL, Koonin EV. Using the COG database to improve gene recognition in complete genomes. Genetica. 2000. [DOI] [PubMed]
- Makarova KS, Aravind L, Galperin MY, Grishin NV, Tatusov RL, Wolf YI, Koonin EV. Comparative genomics of the Archaea (Euryarchaeota): evolution of conserved protein families, the stable core, and the variable shell. Genome Res. 1999;9:608–628. [PubMed] [Google Scholar]
- Sandigursky M, Franklin WA. Thermostable uracil-DNA glycosylase from Thermotoga maritima a member of a novel class of DNA repair enzymes. Curr Biol. 1999;9:531–534. doi: 10.1016/s0960-9822(99)80237-1. [DOI] [PubMed] [Google Scholar]
- Sandigursky M, Franklin WA. Uracil-DNA glycosylase in the extreme thermophile Archaeoglobus fulgidus. J Biol Chem. 2000;275:19146–19149. doi: 10.1074/jbc.M001995200. [DOI] [PubMed] [Google Scholar]
- Greagg MA, Fogg MJ, Panayotou G, Evans SJ, Connolly BA, Pearl LH. A read-ahead function in archaeal DNA polymerases detects promutagenic template-strand uracil. Proc Natl Acad Sci USA. 1999;96:9045–9050. doi: 10.1073/pnas.96.16.9045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham DE, Overbeek R, Olsen GJ, Woese CR. An archaeal genomic signature. Proc Natl Acad Sci USA. 2000;97:3304–3308. doi: 10.1073/pnas.050564797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitz-Gibbon ST, House CH. Whole genome-based phylogenetic analysis of free-living microorganisms. Nucleic Acids Res. 1999;27:4218–4222. doi: 10.1093/nar/27.21.4218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snel B, Bork P, Huynen MA. Genome phylogeny based on gene content. Nat Genet. 1999;21:108–110. doi: 10.1038/5052. [DOI] [PubMed] [Google Scholar]
- Tekaia F, Dujon B. Pervasiveness of gene conservation and persistence of duplicates in cellular genomes. J Mol Evol. 1999;49:591–600. doi: 10.1007/pl00006580. [DOI] [PubMed] [Google Scholar]
- Lake JA. Optimally recovering rate variation information from genomes and sequences: pattern filtering. Mol Biol Evol. 1998;15:1224–1231. doi: 10.1093/oxfordjournals.molbev.a026030. [DOI] [PubMed] [Google Scholar]
- Rivera MC, Lake JA. Evidence that eukaryotes and eocyte prokaryotes are immediate relatives. Science. 1992;257:74–76. doi: 10.1126/science.1621096. [DOI] [PubMed] [Google Scholar]
- Doolittle RF, Handy J. Evolutionary anomalies among the aminoacyl-tRNA synthetases. Curr Opin Genet Dev. 1998;8:630–636. doi: 10.1016/s0959-437x(98)80030-0. [DOI] [PubMed] [Google Scholar]
- Koonin EV, Mushegian AR, Galperin MY, Walker DR. Comparison of archaeal and bacterial genomes: computer analysis of protein sequences predicts novel functions and suggests a chimeric origin for the archaea. Mol Microbiol. 1997;25:619–637. doi: 10.1046/j.1365-2958.1997.4821861.x. [DOI] [PubMed] [Google Scholar]
- Koonin EV, Wolf YI, Aravind L. Protein fold recognition using sequence profiles and its application in structural genomics. Adv Protein Chem. 2000;54:245–275. doi: 10.1016/s0065-3233(00)54008-x. [DOI] [PubMed] [Google Scholar]
- Aravind L, Koonin EV. DNA-binding proteins and evolution of transcription regulation in the archaea. Nucleic Acids Res. 1999;27:4658–4670. doi: 10.1093/nar/27.23.4658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Courtney HS, Li Y, Dale JB, Hasty DL. Cloning, sequencing, and expression of a fibronectin/fibrinogen-binding protein from group A streptococci. Infect Immun. 1994;62:3937–3946. doi: 10.1128/iai.62.9.3937-3946.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonard CJ, Aravind L, Koonin EV. Novel families of putative protein kinases in bacteria and archaea: evolution of the "eukaryotic" protein kinase superfamily. Genome Res. 1998;8:1038–1047. doi: 10.1101/gr.8.10.1038. [DOI] [PubMed] [Google Scholar]
- Koonin EV, Mushegian AR, Bork P. Non-orthologous gene displacement. Trends Genet. 1996;12:334–336. [PubMed] [Google Scholar]
- Cann IKO, Komori K, Toh H, Kanai S, Ishino Y. A heterodimeric DNA polymerase: evidence that members of euryarchaeota possess a distinct DNA polymerase. Proc Natl Acad Sci USA. 1998;95:14250–14255. doi: 10.1073/pnas.95.24.14250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aravind L, Koonin EV. Phosphoesterase domains associated with DNA polymerases of diverse origins. Nucleic Acids Res. 1998;26:3746–3752. doi: 10.1093/nar/26.16.3746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cann IK, Ishino S, Nomura N, Sako Y, Ishino Y. Two family B DNA polymerases from Aeropyrum pernix, an aerobic hyper-thermophilic crenarchaeote. J Bacteriol. 1999;181:5984–5992. doi: 10.1128/jb.181.19.5984-5992.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aravind L, Walker DR, Koonin EV. Conserved domains in DNA repair proteins and evolution of repair systems. Nucleic Acids Res. 1999;27:1223–1242. doi: 10.1093/nar/27.5.1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitabatake M, So MW, Tumbula DL, Soll D. Cysteine biosynthesis pathway in the archaeon Methanosarcina barkeri encoded by acquired bacterial genes? J Bacteriol. 2000;182:143–145. doi: 10.1128/jb.182.1.143-145.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benson D, Karsch-Mizrachi AI, Lipman DJ, Ostell J, Rapp BA, Wheeler DL. GenBank. Nucleic Acids Res. 2000;28:15–18. doi: 10.1093/nar/28.1.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wootton JC, Federhen S. Analysis of compositionally biased regions in sequence databases. Methods Enzymol. 1996;266:554–571. doi: 10.1016/s0076-6879(96)66035-2. [DOI] [PubMed] [Google Scholar]
- Clusters of Orthologous Groups of Proteins (COGs) http://www.ncbi.nlm.nih.gov/COG/
- Sneath PHA, Sokal RR. Numerical Taxonomy. San Francisco: WH Freeman; 1973.
- Fitch WM, Margoliash E. Construction of phylogenetic trees. Science. 1967;155:279–284. doi: 10.1126/science.155.3760.279. [DOI] [PubMed] [Google Scholar]
- Saitou N, Nei M. The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol. 1987;4:406–425. doi: 10.1093/oxfordjournals.molbev.a040454. [DOI] [PubMed] [Google Scholar]
- Felsenstein J. Inferring phylogenies from protein sequences by parsimony, distance, and likelihood methods. Methods Enzymol. 1996;266:418–427. doi: 10.1016/s0076-6879(96)66026-1. [DOI] [PubMed] [Google Scholar]
- Wolf YI, Aravind L, Grishin NV, Koonin EV. Evolution of aminoacyl-tRNA synthetases - analysis of unique domain architectures and phylogenetic trees reveals a complex history of horizontal gene transfer events. Genome Res. 1999;9:689–710. [PubMed] [Google Scholar]
- Aravind L, Koonin EV. Novel predicted RNA-binding domains associated with the translation machinery. J Mol Evol. 1999;48:291–302. doi: 10.1007/pl00006472. [DOI] [PubMed] [Google Scholar]
- Aravind L, Koonin EV. DNA polymerase beta-like nucleotidyl-transferase superfamily: identification of three new families, classification and evolutionary history. Nucleic Acids Res. 1999;27:1609–1618. doi: 10.1093/nar/27.7.1609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aravind L. An evolutionary classification of the metallo-beta lactamase fold proteins. In Silico Biol. 1998;1:8. [PubMed] [Google Scholar]
- Holm L, Sander C. An evolutionary treasure: unification of a broad set of amidohydrolases related to urease. Proteins. 1997;28:72–82. [PubMed] [Google Scholar]
- Koonin EV. Pseudouridine synthases: four families of enzymes containing a putative uridine-binding motif also conserved in dUTPases and dCTP deaminases. Nucleic Acids Res. 1996;24:2411–2415. doi: 10.1093/nar/24.12.2411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Garcia P, Knapp S, Ladenstein R, Forterre P. In vitro DNA binding of the archaeal protein Sso7d induces negative supercoiling at temperatures typical for thermophilic growth. Nucleic Acids Res. 1998;26:2322–2328. doi: 10.1093/nar/26.10.2322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margolin W. Self-assembling GTPases caught in the middle. Curr Biol. 2000;10:R328–R330. doi: 10.1016/s0960-9822(00)00458-9. [DOI] [PubMed] [Google Scholar]
- Stephens RS, Kalman S, Lammel C, Fan J, Marathe R, Aravind L, Mitchell W, Olinger L, Tatusov RL, Zhao Q, et al. Genome sequence of an obligate intracellular pathogen of humans: Chlamydia trachomatis. Science. 1998;282:754–759. doi: 10.1126/science.282.5389.754. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
A list of A. pernix genes, in which genes overlapping with COG members are flagged