

Abstract
Several Bartonella species have now been implicated as human pathogens. The recovery of these fastidious organisms in the clinical microbiology laboratory remains difficult, and current methods are still relatively insensitive. Thus, the bartonellae are good candidates for detection by PCR. We have developed a PCR assay which uses a single primer pair targeting the riboflavin synthase gene (ribC) and detected six Bartonella species that have been implicated in human disease, B. henselae, B. quintana, B. bacilliformis, B. clarridgeiae, B. elizabethae, and B. vinsonii subsp. berkhoffii. Species identification is achieved simply by restriction enzyme digestion of the amplicon. This PCR assay appears to be specific for the Bartonella genus because it failed to amplify DNA from several other bacterial species.
The genus Bartonella, which until 1993 comprised only one species, has been redefined recently and now incorporates 19 species (3, 9, 22, 25) which were previously ascribed to other genera such as Rochalimea, Grahamella, and Rickettsia. Of these, eight have now been implicated in human disease.
B. bacilliformis, the original member of the genus, is the etiologic agent of Oroya fever and verruga peruana (1, 3,22). B. henselae, which frequently causes chronic bacteremia in cats (7, 14), is the most common etiologic agent of cat scratch disease and, in immunocompromised hosts, of bacillary angiomatosis and peliosis hepatitis (1, 3,22). It also causes some cases of endocarditis (1, 22). B. quintana (formerly Rochalimea quintana and Rickettsia quintana) is the etiologic agent of trench fever and has also been associated with bacteremia, endocarditis, and some cases of bacillary angiomatosis (1, 22). B. clarridgeiae also causes chronic bacteremia in cats (7, 14) and has been associated with cat scratch disease (13). B. elizabethae, B. vinsonii subsp. berkhoffii, and B. vinsonii subsp. arupensis have been implicated in endocarditis (6, 20, 23). B. grahamii has been recovered in a case of neuroretinitis (12).
Although they are fastidious organisms, Bartonella species can be cultured in both axenic media and cell culture systems (1, 15, 22). However, successful isolation requires 2 to 6 weeks of incubation or longer (1), and currently neither of these two procedures appears completely satisfactory (15). Therefore, a PCR-based approach appears to be indicated for rapid diagnosis of Bartonella infections.
Several different PCR methods have been used for the detection and species identification of Bartonella. Ideally, a practical method for the clinical laboratory should use a single primer pair, detect several species implicated in human diseases, and allow simple species identification. For example, Matar and coworkers (17) described a PCR method targeting the 16S rRNA gene coding region, which can detect four species of Bartonella and identify them with a panel of two restriction enzymes; Jensen and coworkers (10) proposed a method targeting the 16S-23S intergenic spacer region, which detected six Bartonella species and identified them by the size of the amplicon. However, PCR assays based on 16S sequences or on the 16S-23S spacer region are often complicated by the intraspecies heterogeneity observed in these regions of the genome (5, 17, 19), although of course this is advantageous for molecular epidemiology studies. Another scheme has been proposed by Norman and coworkers, who designed a PCR assay based on the citrate synthase gene sequence. They were able to detect and identify B. henselae and B. quintana but not other Bartonella species. They also obtained an amplicon of similar size by using DNA from R. prowazekii as the template (18). PCR targeting the 60-kDa heat shock protein gene (groEL) sequence or the cell division protein gene (ftsZ) have also been used for phylogenetic studies (16, 24, 25).
In 1999, Bereswill and coworkers (4) proposed the riboflavin synthesis genes as a suitable target for PCR, pointing out their high evolutionary conservation among bacteria and their absence from the genome of vertebrates. The latter property is ideal for PCR assays performed on human clinical specimens. They performed a detailed sequence analysis of these genes for B. bacilliformis, B. henselae, B. quintana, and B. clarridgeiae and designed species-specific primer pairs in the riboflavin synthase gene (ribC). We chose a different approach and took advantage of the sequences and analysis provided by Bereswill and coworkers (4) to design a unique primer pair highly homologous to segments of the ribC gene conserved among the bartonellae but not to the corresponding segments in the genome of unrelated bacteria. We successfully amplified DNA from B. henselae, B. quintana B. bacilliformis, B. clarridgeiae, B. elizabethae, and B. vinsonii subsp. berkhoffii. For the last two species, we also sequenced the amplicons. We designed a species identification method based on digestion of the amplicons with the restriction enzyme TaqI, supplemented for some species by digestion with EarI. We illustrated the feasibility of using this method in the clinical laboratory by testing a small number of samples from patients for whom cat scratch disease was part of the differential diagnosis.
MATERIALS AND METHODS
Bacterial strains and DNA.
The following strains were purchased from the American Type Culture Collection (Manassas, Va.): B. bacilliformis (ATCC 35685), B. clarridgeiae (ATCC 51734), B. elizabethae (ATCC 49927), and B. vinsonii subsp. berkhoffii (ATCC 51672). B. quintana was provided by Kathryn Bernard, National Microbiology Laboratory, Winnipeg, Canada. Afipia felis was provided by Frances Jamieson, Laboratory Branch, Ministry of Health, Ontario, Canada. Clinical isolates of Staphylococcus aureus, Streptococcus pneumoniae, Enterococcus faecalis, Moraxella catarrhalis, Neisseria meningitidis, Clostridium perfringens, Bacillus fragilis, Escherichia coli, Pseudomonas aeruginosa, Brucella melitensis, and Candida albicans were obtained from the Microbiology Laboratory at the Hospital for Sick Children and were identified by standard methods. For Brucella melitensis, the identification was further confirmed by 16S rRNA sequencing. Purified, quantified DNA from B. henselae was purchased from the American type Culture Collection (ATCC 49882)
Culture.
Bartonella strains were grown on Trypticase soy agar with 5% defibrinated sheep blood at the temperature and atmosphere recommended for each different species.
Extraction of DNA.
DNA from suspensions of bacterial colonies or from blood or tissue samples was extracted with the blood and tissue QIAamp kits (Qiagen) as per the manufacturer's recommendations.
Primers.
Primers for Bartonella PCR were designed to target segments of the ribC gene that were well conserved among Bartonella species but not with unrelated microorganisms, based on an alignment of ribC sequences from B. bacilliformis, B. quintana, B. henselae, and B. clarridgeiae (4). We used the primer BARTON-1 (5′-TAACCGATATTGGTTGTGTTGAAG-3′) as the sense primer and BARTON-2 (5′-TAAAGCTAGAAAGTCTGGCAACATAACG-3′) as the antisense primer.
PCR.
Each reaction was performed in a 0.6-ml tube (Diamed PRE 050) in a total volume of 50 μl overlaid with 50 μl of mineral oil. Each reaction mix contained 5 μl of 10× Cetus buffer II (Perkin Elmer), 5 μl of 25 mM MgCl2, 5 μl of deoxynucleoside triphosphate mix (Pharmacia; each deoxynucleoside triphosphate at 2 mM), 25 pmol of each primer, 0.5 μl (2.5 U) of Amplitaq Gold (Perkin Elmer), and molecular grade double distilled water to a volume of 40 μl. The master mix was then aliquoted in tubes, to which 10 μl of template DNA, dissolved in double distilled water, was added. PCR was performed on a Stratagene Robocycler 40. The cycling parameters consisted of an initial denaturing step of 10 min at 95°C, followed by 37 cycles consisting of denaturation at 95°C for 1 min, annealing at 51°C for 1 min, and elongation at 72°C for 1 min, and a final incubation at 72°C for 3 min.
Precautions against PCR contamination.
Extensive precautions against PCR contamination, as previously described (11), were strictly observed.
PCR controls.
For each clinical sample, extracted DNA was recovered in a defined volume of double distilled water. One aliquot of 10 μl was tested in a PCR “as is,” and a second aliquot of 10 μl was tested in a PCR spiked with a defined amount of template to control the integrity of the reaction mix and to rule out the presence of PCR inhibitors originating from the sample. The spike consisted of approximately 10−3 ng of B. henselae genomic DNA. In addition, for each PCR run of up to 10 samples, an aliquot of phosphate-buffered saline was also subjected to DNA extraction as a negative control. The extracts, along with a positive control (same as for the spike) and a water aliquot as an additional control to rule out contamination of the reagents or by aerosols, were then subjected to PCR as described above.
Electrophoretic analysis of the PCR.
A 10-μl volume of each reaction mix was subjected to electrophoresis on 1.5% agarose gels containing ethidium bromide. The gels were visualized on a UV transilluminator and photographed.
Restriction enzyme analysis.
Each reaction mixture in which amplicons were detected was subjected to digestion with the restriction enzyme TaqI (New England Biolabs). For B. clarridgeiae, B. elizabethae, and B. vinsonii subsp. berkhoffii, digestion with the restriction enzyme EarI (New England Biolabs) was also performed. The digestion mixture consisted of 10 μl of PCR mixture, 1.5 μl of the appropriate enzyme buffer, 1 μl of enzyme, and 2.5 μl of double distilled water for a total of 15 μl. The reaction mixtures were incubated at 65°C (TaqI) or 37°C (EarI) for 1 h and analyzed by agarose gel electrophoresis as above.
16S rRNA PCR.
The 16S rRNA PCR was performed with a PCR master mix as above except that the primers used consisted of the last 20 nucleotides of FD1 (21) and 806R (8). The cycling parameters consisted of denaturation at 95°C for 10 min, annealing at 45°C for 2 min, and elongation at 72°C for 3 min, followed by 35 cycles of denaturation at 95°C for 1 min, annealing at 45°C for 2 min, and elongation at 72°C for 3 min.
Sequencing.
Amplicons obtained from B. elizabethae and B. vinsonii subsp. berkhoffii were subjected to automated sequencing, for both strands, with the BARTON-1 and BARTON-2 primers as sequencing primers. The amplicon obtained from Brucella melitensis by 16S PCR was sequenced with primer FD1 as the sequencing primer. Sequencing was performed by the DNA Sequencing Facility, Centre for Applied Genomics, Hospital for Sick Children.
Clinical samples.
Lymph node biopsies and lymph node aspirates (submitted fresh or frozen) from patients for whom cat scratch disease was included in the differential diagnosis of their lymphadenitis were submitted over the course of 1 year to our laboratory and analyzed with our PCR assay. Blood samples from cats were obtained as 200 μl of blood in EDTA from the remainders of samples obtained for clinical purposes from cats in a veterinary practice and submitted under code.
RESULTS
Detection of Bartonella species by PCR.
With DNA extracted from colonies of B. henselae, B. quintana, B. bacilliformis, and B. clarridgeiae, we obtained the 585- to 588-bp amplicons predicted by the DNA sequence of the ribC gene of these organisms (4). From the DNA extracted from colonies of B. elizabethae and B. vinsonii subsp. berkhoffii, we obtained amplicons of similar size, which were then sequenced. This demonstrated that for these two organisms, the amplicon size was 585 bp. The 533-bp sequences internal to the primers were deposited in GenBank (accession numbers AF548030 and AF548031).
Sensitivity and specificity.
The sensitivity was determined with a 10-fold serial dilution of B. henselae genomic DNA. We were able to obtain an amplicon from as little as 10−4 ng of DNA. To characterize the specificity of our assay, we performed PCR with the DNA extracted from several bacterial species (Brucella melitensis, Afipia felis, Staphylococcus aureus, Streptococcus pneumoniae, Enterococcus faecalis, Moraxella catarrhalis, Neisseria meningitidis, Clostridium perfringens, Bacillus fragilis, Escherichia coli, and Pseudomonas aeruginosa) and from Candida albicans as the template and did not observe any amplicon. Lastly, as our practice consists of running samples in two duplicate reactions, one of which is spiked, we demonstrated that the presence of human genomic DNA in the reaction did not inhibit the PCR.
Species identification.
Based on the sequences of the amplicons, we designed a rapid identification method with a panel of two restriction enzymes (Table 1). As shown in Fig. 1, digestion with TaqI unambiguously identified B. henselae, B. quintana, and B. bacilliformis. The patterns obtained with TaqI from B. clarridgeiae, B. elizabethae, and B. vinsonii subsp. berkhoffii (Fig. 1) were markedly different from the first three Bartonella species but were similar and may potentially be confused with one another if the electrophoresis is not done meticulously. If any of these three species is encountered, additional digestion of the amplicon with EarI provides an easier identification (Fig. 2).
TABLE 1.
Characteristics of amplicons as predicted by their DNA sequences
| Species | Length (bp) | TaqI fragments (bp) | EarI fragments (bp) |
|---|---|---|---|
| B. henselae | 588 | 387, 122, 79 | 448, 140 |
| B. quintana | 588 | 466, 122 | 588 |
| B. bacilliformis | 585 | 522, 63 | 588 |
| B. clarridgeiae | 585 | 304, 281 | 401, 184 |
| B. elizabethae | 585 | 325, 260 | 585 |
| B. vinsonii subsp. berkhoffii | 585 | 284, 260, 41 | 323, 184, 78 |
FIG. 1.

Restriction enzyme digestion patterns obtained with TaqI. Digestion mixtures were electrophoresed on 1.5% agarose gels. On each panel, the gel is flanked by a 100-bp ladder (Invitrogen Life Technologies) (lane MW). (A) The three species illustrated are unambiguously identified by the TaqI pattern. However, for the species illustrated in panel B, the TaqI patterns are similar to one another, although clearly distinct from that of the species in panel A.
FIG. 2.
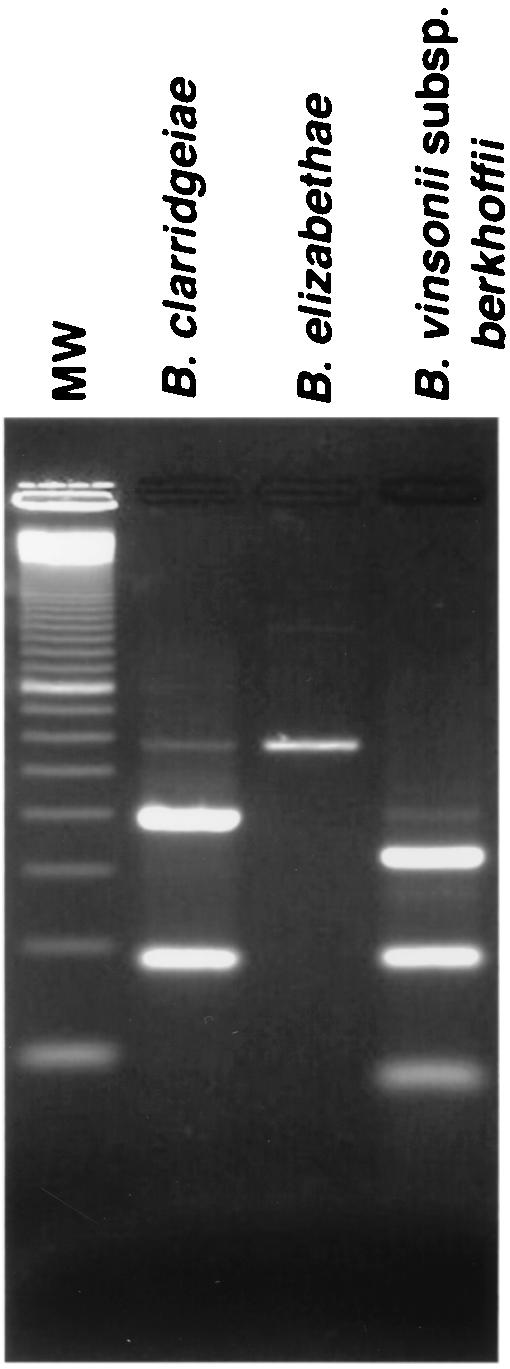
Restriction enzyme digestion patterns obtained with EarI. For B clarridgeiae, B. elizabethae, and B. vinsonii subsp. berkhoffii, positive identification is facilitated by obtaining the digestion pattern with EarI in addition to TaqI.
Detection of Bartonella spp. in clinical samples.
Eighteen lymph node biopsies and three lymph node aspirates from patients with possible cat scratch disease were submitted to the laboratory during this study. After DNA extraction and PCR, B. henselae was detected in one lymph node biopsy and the three lymph node aspirates (two of which were from the same patient). Blood samples from cats included specimens from four cats that lived indoors without contact with cats living outdoors, which were all negative, and from nine cats that lived outdoors (including five stray cats) or that had contact with cats living outdoors, one of which was positive for B. henselae. It is noteworthy that when PCR was performed on a DNA template from human or cat tissues, we did not observe any amplicons unless a Bartonella species was detected (as verified by restriction enzyme digestion). This observation confirms that the primers do not generate nonspecific amplicons from mammalian genomic DNA.
DISCUSSION
Because of their fastidious nature, Bartonella species are good candidates for detection and species identification by a PCR-based method. Based on the work of Bereswill and coworkers (4), we have designed a PCR assay targeting a segment of the ribC gene. This assay is very sensitive: with commercially available B. henselae genomic DNA, we could detect as little as 10−4 ng. Since the size of the B. henselae genome is approximately 2 × 106 bp (3), our sensitivity is approximately 50 genome copies. Our PCR assay appears to be specific for the Bartonella genus, since the assay failed to generate amplicons from the DNA of a number of bacterial species tested, including A. felis (which frequently coinfects wounds caused by cat scratches along with B. henselae but is not thought to cause cat scratch disease [22]) and Brucella melitensis, which is noteworthy because the brucellae are phylogenetically closely related to the bartonellae. We did not test members of the genus Rickettsia because these metabolically crippled organisms do not have a ribC gene (2).
As noted, and as expected from the fact that mammals do not have a gene coding for riboflavin synthase, we did not observe any amplicons with mammalian DNA as the template unless the tissue sampled was infected by a Bartonella species. We were able to obtain an amplicon from all the Bartonella species that we tested, namely, B. henselae, B. quintana B. bacilliformis, B. clarridgeiae, B. elizabethae, and B. vinsonii subsp. berkhoffii. Species identification in our method is provided by restriction enzyme digestion, producing fragments that are large enough to be analyzed by agarose gel electrophoresis, which is more convenient and rapid than polyacrylamide gel electrophoresis. As shown in Table 1 and Fig. 1, the fragments obtained by digestion with TaqI provided a pattern unique to each species and led to unambiguous identification.
We were concerned, however, that for B. clarridgeiae, B. elizabethae, and B. vinsonii subsp. berkhoffii, the fragment sizes were very similar and may be confused if the electrophoresis is not done meticulously (especially with the optimal amount of DNA). For that reason, in practice an additional digestion of the amplicon with EarI for these three species would provide a definitive and easy identification. Given that the species most likely to be encountered in human samples are B. henselae and B. quintana, we think that the most efficient approach is to digest initially only with TaqI. If a pattern suggestive of B. clarridgeiae, B. elizabethae, or B. vinsonii subsp. berkhoffii is seen, then a supplemental digestion with EarI would be set up. However, since automated sequencing continues to improve in ease of use and affordability, it may well be that in the near future, identification by restriction enzyme digestion will be completely superseded by sequencing of the amplicon.
Of course, a drawback of targeting sequences that are well conserved among the different Bartonella species is that there is not enough sequence variation to permit molecular epidemiology studies, phylogenetic classification, or even differentiation between B. henselae type I (prototype: Houston strain) and type II (prototype: BATF). Nonetheless, the assay as described should be useful for most clinical microbiology laboratories.
The assay was used on clinical specimens from patients for whom cat scratch disease was part of the differential diagnosis. As expected, no artifactual amplicon was produced from human genomic DNA, and our spiked controls demonstrated the good performance of the PCR even in the presence of DNA extracted from clinical samples. We could confirm the presence of B. henselae in four samples with our PCR assay.
Bartonella species endemic in cats, B. henselae and B. clarridgeiae, are known to cause chronic intermittent bacteremia in cats (7, 14). In cats that were experimentally infected with B. henselae or B. clarridgeiae, bacteremia could be documented intermittently for up to 454 days without apparent disease or symptoms (14), although after euthanasia, histopathologic studies revealed microscopic inflammatory foci in several organs (14). The bacteremia could reach a titer as high as 103/ml, which should be well within the sensitivity of our method, given that we extract DNA from 200 μl of blood. In our study, we could indeed detect bacteremia in one cat. Whereas PCR itself is very sensitive, DNA extraction methods are not well suited for processing large amounts of blood. Because of this, the optimal sample to be submitted for diagnosis of Bartonella infections in humans by PCR would appear to be lymph node or tissue biopsies (including cardiac valves in cases of endocarditis if surgery is performed) or aspirates. If the experimental cat model is any indication, PCR from tissue samples would not always be positive, at least in the immunocompetent host, with considerable differences between organs (14). Because of the limitations in the sample size that can be processed, the concomitant use of serological testing for diagnosis is expected to continue to be helpful, at least in the immunocompetent host (22).
In summary, we have developed a PCR assay that is specific for Bartonella species and permits the convenient detection and identification of six Bartonella species that have been implicated in human disease. The assay is not inhibited by the presence of human genomic DNA from tissue samples and permitted the diagnosis of cat scratch disease from patient samples and of bacteremia in blood samples from cats.
REFERENCES
- 1.Anderson, B. E., and M. A. Neuman. 1997. Bartonella spp. as emerging pathogens. Clin. Microbiol. Rev. 10:203-219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Andersson, S. G. E., A. Zomorodipour, J. O. Andersson, T. Sicheritz-Pontén, U. C. M. Alsmark, R. M. Podowski, A. K. Näslund, A.-S. Eriksson, H. H. Winkler, and C. G. Kurland. 1998. The genome sequence of Rickettsia prowazekii and the origin of mitochondria. Nature 396:133-140. [DOI] [PubMed] [Google Scholar]
- 3.Andersson, S. G. E., and C. Dehio. 2000. Rickettsia prowazekii and Bartonella henselae: differences in the intracellular life styles revisited. Int. J. Med. Microbiol. 290:135-141. [DOI] [PubMed] [Google Scholar]
- 4.Bereswill, S., S. Hinkelmann, M. Kist, and A. Sander. 1999. Molecular analysis of riboflavin synthesis genes in Bartonella henselae and use of the ribC gene for differentiation of Bartonella species by PCR. J. Clin. Microbiol. 37:3159-3166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Birtles, R. J., S. Hazel, K. Bown, D. Raoult, M. Begon, and M. Bennett. 2000. Subtyping of uncultured bartonellae with sequence comparision of 16 S/23S rRNA intergenic spacer regions amplified directly from infected blood. Mol. Cell. Probes 14:79-87. [DOI] [PubMed] [Google Scholar]
- 6.Daly, J. S., M. G. Worthington, D. J. Brenner, C. W. Moss, D. G. Hollis, R. S. Weyant, A. G. Steigerwalt, R. E. Weaver, M. I. Daneshvar, and S. P. O'Connor. 1993. Rochalimaea elizabethae sp. nov. isolated from a patient with endocarditis. J. Clin. Microbiol. 31:872-881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Heller, R., M. Artois, V. Xemar, D. De Briel, H. Gehin, B. Jaulhac, H. Monteil, and Y. Piemont. 1997. Prevalence of Bartonella henselae and Bartonella clarridgeiae in stray cats. J. Clin. Microbiol. 35:1327-1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Heritz, D. M., J.-M. Y. Lacroix, S. D. Batra, K. A. Jarvi, B. Beheshti, and M. W. Mittelman. 1997. Detection of eubacteria in interstitial cystitis by 16S rDNA amplification. J. Urol. 158:2291-2295. [DOI] [PubMed] [Google Scholar]
- 9.Houpikian, P., and D. Raoult. 2001. Molecular phylogeny of the genus Bartonella: what is the current knowledge? FEMS Microbiol. Lett. 200:1-7. [DOI] [PubMed] [Google Scholar]
- 10.Jensen, W. A., M. Z. Fall, J. Rooney, D. L. Kordick, and E. B. Breitschwerdt. 2000. Rapid identification and differentiation of Bartonella species with a single-step PCR assay. J. Clin. Microbiol. 38:1717-1722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Johnson, G., S. Nelson, M. Petric, and R. Tellier. 2000. Comprehensive PCR-based assay for detection and species identification of human herpesviruses. J. Clin. Microbiol. 38:3274-3279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kerkhoff, F. T., A. M. C. Bergmans, A. van der Zee, and A. Rothova. 1999. Demonstration of Bartonella grahamii DNA in ocular fluids of a patient with neuroretinitis. J. Clin. Microbiol. 37:4034-4038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kordick, D. L., E. J. Hilyard, T. D. Hadfield, K. H. Wilson, A. G., Steigerwalt, D. J. Brenner, and E. B. Breitschwerdt. 1997. Bartonella clarridgeiae, a newly recognized zoonotic pathogen causing inoculation papules, fever, and lymphadenopathy (cat scratch disease). J. Clin. Microbiol. 35:1813-1818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kordick, D. L., T. T. Brown, K. Shin, and E. B. Breitschwerdt. 1999. Clinical and pathological evaluation of chronic Bartonella henselae or Bartonella clarridgeiae infection in cats. J. Clin. Microbiol. 37:1536-1547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.La Scola, B., and D. Raoult. 1999. Culture of Bartonella quintana and Bartonella henselae from human samples: a 5-year experience (1993 to 1998). J. Clin. Microbiol. 37:1899-1905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Marston, E. L., J. W. Summer, and R. L. Regnery. 1999. Evaluation of intraspecies genetic variation within the 60-kDa heat shock protein gene (groEL) of Bartonella species. Int. J. Syst. Bacteriol. 49:1015-1023. [DOI] [PubMed] [Google Scholar]
- 17.Matar, G. M., J. E. Koehler, G. Malcolm, M. A. Lambert-Fair, J. Tappero, S. B. Hunter, and B. Swaminathan. 1999. Identification of Bartonella species directly in clinical specimens by PCR-restriction fragment length polymorphism analysis of a 16S rRNA gene fragment. J. Clin. Microbiol. 37:4045-4047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Norman, A. F., R., Regnery, P. Jameson, C. Greene, and D. C. Krause. 1995. Differentiation of Bartonella-like isolates at the species level by PCR-restriction fragment length polymorphism in the citrate synthase gene. J. Clin. Microbiol. 33:1797-1803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Roux, V., and D. Raoult. 1995. Inter- and intraspecies identification of Bartonella (Rochalimaea) species. J. Clin. Microbiol. 33:1573-1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Roux, V., S. J. Eykyn, S. Wyllie, and D. Raoult. 2000. Bartonella vinsonii subsp. berkhoffii as an agent of afebrile blood culture-negative endocarditis in a human. J. Clin. Microbiol. 38:1698-1700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Weisburg, W. G., S. M. Barns, D. A. Pelletier, and D. J. Lane. 1991. 16S ribosomal DNA amplification for phylogenetic study. J. Bacteriol. 173:697-703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Welch, D. F., and L. N. Slater. 1999. Bartonella and Afipia, p. 638-646. In P. R. Murray, E. J. Barron, M. A. Pfaller, F. C. Tenover, and R. H. Yolken (ed.), Manual of clinical microbiology, 7th ed. American Society for Microbiology, Washington, D.C.
- 23.Welch, D. F., K. C. Carroll, E. K. Hofmeister, D. H. Persing, D. A. Robison, A. G. Steigerwalt, and D. J. Brenner. 1999. Isolation of a new subspecies, Bartonella vinsonii subsp. arupensis, from a cattle rancher: identity with isolates found in conjunction with Borrelia burgdorferi and Babesia microti among naturally infected mice. J. Clin. Microbiol. 37:2598-2601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zeaiter, Z., P.-E. Fournier, H. Ogata, and D. Raoult. 2002. Phylogenetic classification of Bartonella species by comparing groEL sequences. Int. J. Syst. E vol. Microbiol. 52:165-171. [DOI] [PubMed] [Google Scholar]
- 25.Zeaiter, Z., Z. Liang, and D. Raoult. 2002. Genetic classification and differentiation of Bartonella species based on comparison of partial ftsZ gene sequences. J. Clin. Microbiol. 40:3641-3647. [DOI] [PMC free article] [PubMed] [Google Scholar]