

Abstract
We have applied in vivo intracellular antibody capture (IAC) technology to isolate human intrabodies which bind to the oncogenic RAS protein. IAC facilitates the capture of antibody fragments, in this case single-chain Fvs (scFvs), which tolerate reducing environments, such as the cytoplasm of cancer cells. Three anti-RAS scFvs with different affinity, solubility and intracellular binding activity were characterized. The anti-RAS scFvs with highest affinity were expressed relatively poorly in mammalian cells, and greater soluble expression was achieved by mutating the antibody framework to canonical consensus scaffolds, previously derived from IAC, without losing antigen specificity. Mutagenesis experiments showed that the consensus scaffolds are functional as intrabody fragments without an intra-domain disulfide bond. Furthermore, we could convert an intrabody which does not bind RAS in mammalian cells into a high-affinity reagent capable of inhibiting RAS-mediated NIH 3T3 transformation by exchanging VH and VL complementarity-determining regions onto its consensus scaffold. These data show that the consensus scaffold is a robust framework by which to improve intrabody function.
Keywords: cancer therapy/CDR/framework/intrabody/oncogene
Introduction
Antibodies are used extensively in bioscience as in vitro tools for recognizing target antigens and for medical applications such as diagnosis or therapeutics. Recently, gene cloning technologies have allowed the genes for coding antibodies to be manipulated and expressed intracellularly (Cattaneo and Biocca, 1999). Intracellular antibodies or intrabodies with specific and high-affinity binding properties have great potential for application in the therapy of human diseases in which target proteins or protein interactions are found only inside the target cell. A suitable form for intrabody expression is the single-chain Fv antibody, also known as single-chain variable fragment or scFv (Marasco et al., 1993; Biocca et al., 1994; Cohen, 2002), which is composed of the heavy and light chain variable domains and a flexible linker peptide to fuse them (Bird et al., 1988; Huston et al., 1988). Applications of functional scFvs as intrabodies have been exploited and achieved in several fields. There is potential for their use in cancer cells, where chromosomal translocations or somatic mutations occur, effectively producing tumour-specific intracellular proteins (Rabbitts, 1994; T.H.Rabbitts and M.R.Stocks, manuscript submitted). As the protein products are inside the cell, rather than exposed on the cell surface, conventional antibody therapy is not an option.
The scFv format is suitable for intracellular use because of its optimal size and ease of expression from vectors since the VH and VL segments are present on a single macromolecule, and thus require no inter-chain disulfide linkage to hold together the two chains. Several such antibody fragments have been demonstrated to be effective in targeting proteins in vivo (Biocca et al., 1993; Tavladoraki et al., 1993; Rondon and Marasco, 1997). However, there remain few antibodies which work effectively in intracellular reducing environments because there are often problems with correct folding and the resulting lack of function, low expression and short half-life (Cattaneo and Biocca, 1999). Indeed, it has been found generally that most of the scFvs which are derived from hybridomas do not function effectively in vivo, regardless of their having sufficient high affinity and antigen specificity in vitro.
The intra-domain disulfide bond does not form in scFvs expressed in the cytoplasm of eukaryotic cells (Biocca et al., 1995), but some scFvs have been shown to tolerate the absence of this bond in vitro (Proba et al., 1998; Worn and Pluckthun, 1998a). At this time, there is no general rule or prediction of the requirements for soluble and stable intracellular antibodies. There is an interest in selecting or engineering frameworks suitable for intracellular expression, and several approaches have been adopted to solve this problem. These include the modification of the sequence of VH and VL domains utilizing random mutation to replace the need for disulfide bonds to stabilize scFvs with high intrinsic stability (Proba et al., 1998; Worn and Pluckthun, 1998b) or use of frameworks which prove to be effective in vivo (Ohage and Steipe, 1999; Ohage et al., 1999; Wirtz and Steipe, 1999; Desiderio et al., 2001; Tse et al., 2002b; Visintin et al., 2002). We recently have developed a selection method to isolate intracellular antibodies which primarily depends on their function inside yeast and mammalian cells, described as intracellular antibody capture (IAC) technology (Tse et al., 2002b; Visintin et al., 2002). An advantage of this genetic selection approach is to isolate functional scFvs from a diverse mixture of antibodies, which can tolerate reducing cellular environments.
We have now used IAC methods to isolate intrabodies binding in cells to the RAS protein as a model to evaluate a consensus scaffold derived from IAC methods. RAS is a membrane-bound GTP/GDP-binding G protein that serves as a molecular switch, converting signal transduction from the membrane to the nucleus (reviewed in Lowy and Willumsen, 1993). The mutated RAS gene encodes constitutively activated GTP-bound protein, thereby stimulating cell proliferation and inhibiting apoptosis in tumorigenesis (Adjei, 2001). Mutation in the RAS oncogene has been identified in ∼30% of human cancers, making it an important target for the development of anti-cancer drugs. Using the IAC method, we were able to isolate and characterize three different anti-RAS scFvs which have different characteristics, e.g. affinity, solubility in mammalian cells, and protein yields when expressed in bacteria. We found that poor solubility and function of anti-RAS scFvs could be improved by mutating the framework to versions of a consensus framework defined by previous use of IAC technology (Tse et al., 2002a; Visintin et al., 2002) and, in addition, the mutated consensus scFvs can interact with antigen without the intra-domain disulfide bond. Our results demonstrate that the framework scFv scaffold derived by IAC technology adopts a robust in vivo structure and can become the basis for de novo generation of bespoke human intracellular scFv libraries.
Results
Isolation of specific intracellular antibody fragments which recognize RAS protein in vivo
We have applied the IAC technique (Tse et al., 2002a; Visintin et al., 2002) to the isolation of anti-RAS intrabodies. The sequential steps comprise initial in vitro phage scFv library panning with purified RAS protein and in vivo antigen–antibody two-hybrid interaction screening to isolate specific intracellular antibodies. For the in vitro phage antibody screen, purified C-terminal truncated human HRASG12V was used as antigen, bound to 5′-guanylylimidodi-phosphate (GppNp, a non-hydrolysable analogue of GTP). After one round of in vitro panning, ∼1.18 × 106 antigen-bound phage were recovered from 2.7 × 1013 initial phage (Figure 1). A sublibrary was prepared as phagemid DNA and cloned into a yeast pVP16* transcriptional activation domain (AD) vector to make an anti-RAS scFv-VP16-AD library (∼4 × 106 clones). This yeast sublibrary was transfected into a yeast strain (L40 with HIS3 and β-galactosidase reporter genes) expressing the fusion protein bait comprising the pBTM116 LexA-DNA-binding domain (DBD) fused to HRASG12V. Approximately 8.45 × 107 yeast colonies were screened (Figure 1). A total of 428 colonies grew in the absence of histidine, and these clones also showed activation of β-gal. The scFv-VP16-AD plasmids were isolated from the histidine-independent, β-gal-positive clones and sorted by their DNA restriction patterns. More than 90% of these scFv-VP16-AD plasmids had an identical DNA fingerprinting pattern, and 20 had identical DNA sequences. Those scFvs with differing DNA fingerprint patterns were co-transformed with the pBTM116-HRASG12V bait in fresh yeast and assayed for histidine-independent growth and β-gal activation. Three anti-RAS scFvs, designated 33, J48 and I21, were thus identified (Figure 1). The specificity of these scFvs for binding to RAS in yeast was verified further by their lack of interaction with the LexA-DBD (made from the empty pBTM116 vector) and a non-relevant antigen (β-gal) (data not shown).

Fig. 1. Intracellular antibody capture of anti-RAS scFvs. A total of 2.7 × 1013 clones from three different phage libraries (Sheets et al., 1998; de Wildt et al., 2000) (total diversity 7.0 × 109) were screened with purified HRASG12V antigen in vitro. A total of 1.18 × 106 phage were recovered, phagemid DNA was prepared and scFv fragments cloned into the yeast vector pVP16* to make a sublibrary of 4.13 × 106 clones. Yeast clones (8.45 × 107) were screened in the yeast L40 strain expressing the LexA-HRASG12V bait; 428 colonies grew on histidine selective plates and showed strong activation of the lacZ gene, determined by β-gal filter assay. All prey plasmids were isolated from histidine-independent and β-gal-positive yeast colonies and were fingerprinted by digestion with restriction enzymes BstNI, MspI, MboI, RsaI or HinfI to identify the differing scFv clones. Subsequently, 57 scFv clones which had different DNA fingerprinting patterns were re-tested in yeast with LexA-HRASG12V bait, and three scFvs (which originated from different libraries) were isolated. Of these three anti-RAS scFvs, only two detectably bound RAS protein in a mammalian reporter assay.
The efficacy of the anti-RAS intrabodies was confirmed using a mammalian cell reporter assay and in vivo antigen co-location assays (Figure 2). The mammalian cell assay used was luciferase production from a luciferase reporter gene. The three scFvs were shuttled into a mammalian expression vector, pEF-VP16, which has the elongation factor-1a promoter (Mizushima and Nagata, 1990) and the VP16-AD (Triezenberg et al., 1988). The scFvs were cloned in-frame with the VP16 segment, on its N-terminal side. The HRASG12V antigen was cloned into the pM1 vector (Sadowski et al., 1992) which has the Gal4-DBD as an N-terminal fusion with antigen (pM1-HRASG12V). pEF-scFv-VP16 and pM-HRASG12V were co-transfected into COS7 cells with the luciferase reporter plasmid. More than 10-fold activation was observed when scFv33 or the scFvJ48-VP16 fusion were expressed with the bait antigen HRASG12V (Figure 2A), but none with a non-relevant antigen β-gal. However, no activation was observed when the yeast anti-RAS scFvI21 was co-expressed with the HRASG12V bait (Figure 2A). Similar results were obtained in other mammalian cell lines, i.e. HeLa and CHO cells. The failure of scFvI21 to interact with antigen in this mammalian cell assay, as opposed to yeast, may be due simply to it having insufficient affinity, or may reflect the relative insensitivity of mammalian assays compared with yeast, perhaps due to factors such as transfection efficiency, reporter gene activation requiring access to endogenous transcription factors and/or the expression level of antigen or antibody.

Fig. 2. Interaction of anti-RAS scFv with RAS protein in mammalian cells. (A) Luciferase assay. COS7 cells were transiently co-transfected with various scFv-VP16-AD fusions and the Gal4-DBD bait plasmid pM1-HRASG12V (closed boxes) or pM1-lacZ (open boxes), together with the firefly luciferase reporter plasmid pG5-Luc and an internal Renilla luciferase control plasmid pRL-CMV. scFv-VP16 prey vectors were used expressing anti-RAS scFv33, J48 and I21 or anti-β-gal scFvR4 (Martineau et al., 1998). The luciferase activities were measured 48 h after transfection using the Dual Luciferase Assay System (Promega) and a luminometer. The luciferase activities of each assay were normalized to the Renilla luciferase activity (used as internal control for the transfection efficiency). The fold luciferase induction level is shown, with the activity of each scFv-VP16 with non-relevant bait taken as baseline. (B) In situ immunofluorescence study. COS7 cells were transiently co-transfected with pEF/myc/nuc-scFvJ48 (anti-RAS scFv) or scFvR4 (anti-β-gal scFv) and pHM6-HRASG12V vectors expressing the RAS antigen. After 48 h, cells were fixed and stained with 9E10 monoclonal antibody (detecting the myc-tagged scFv) and rabbit anti-HA tag polyclonal serum, followed by secondary fluorescein-conjugated anti-mouse and Cy3-conjugated anti-rabbit antibodies, respectively. The staining patterns were examined using a BioRadiance confocal microscope. Co-location of antigen and intrabody fluorescence was found for scFvJ48 co-expressed with RAS. Green (fluorescein) = fluorescence of scFv; red (Cy3) = fluorescence of antigen.
The observed interaction of scFv33 and scFvJ48 in a yeast system expressing LexA-DBD and a mammalian system expressing Gal4-DBD is a good indicator that the scFvs interact with a native epitope of the RAS antigen, rather than an artificial one due to fusion of RAS and a DBD in the bait. Additional evidence for this was obtained from co-location assays in which the native RAS antigen was expressed together with the scFvs to which nuclear localization signals (nls) had been added. COS7 cells were co-transfected with a RAS expression vector with a haemagglutinin (HA) epitope tag and scFv expression vectors encoding scFv with a myc epitope tag. After 48 h, RAS antigen was detected with anti-HA tag antibody and scFv with anti-myc tag antibody (Figure 2B). When the RAS antigen was expressed alone or with a non-relevant scFv (scFvR4; Martineau et al., 1998), the antigen was detected in the cytoplasm and antibody in the nucleus (Figure 2B, lower panels), whereas if the antigen was co-expressed with the anti-RAS scFvJ48 with an nls, co-location of RAS antigen and scFv was observed in the nucleus. This means that the anti-RAS intrabody has sufficient expression and affinity to bind RAS antigen in vivo and cause re-location within the cell (similar results were found with anti-RAS scFv33, data not shown).
The sequence and bacterial expression of anti-RAS scFvs
The anti-RAS scFvs (33, J48 and I21) were sequenced and the derived protein sequence was aligned (Figure 3). All three scFvs belong to the VH3 (IGHV3) subgroup joined to the JH5 (IGHJ5) and to the Vκ1 (IGKV1) subgroup. Our previous data on anti-BCR and anti-ABL scFvs (Tse et al., 2002b), which were isolated only from the library of Sheets et al. (1998), also belong to the VH3 (IGHV3) and Vκ1 (IGKV1) subgroup. In our previous study, we were able to define a consensus framework which was derived by comparing the anti-BCR and anti-ABL scFvs (Tse et al., 2002b), and an analogous study was conducted with anti-TAU intrabodies (Visintin et al., 2002). We concluded that a framework composed of VH3 (IGHV3) and the Vκ1 (IGKV1) is highly amenable for scFv function inside the cell, and the consensus defined a basic sequence on which to design other intrabodies. The anti-RAS intrabodies described here help to refine this concept.

Fig. 3. Sequence of anti-RAS intracellular scFvs. The nucleotide sequences were obtained and the derived protein translations (shown as the single letter code) were aligned. Dashes in the framework regions (FRs) represent identities with the consensus (Con) sequence (derived from anti-BCR and anti-ABL scFvs isolated by the IAC method; Tse et al., 2002). The numbers indicate the reference positions of the residues, according to the system of Lefranc and Lefranc (2001) (top column number, indicated as IMGT) and Kabat et al. (1991) (second column, Kabat). The 15 residues of the linker, (GGGGS)3, between the heavy chain domain (VH) and light chain variable domain (VL) are not shown. The complementarity-determining regions (CDRs) are highlighted on a grey background and demarcated from FRs. The three anti-RAS intracellular scFvs are designated 33, J48 and I21. All anti-RAS scFvs belong to the VH3 subgroup of heavy chains and Vκ1 subgroup of light chains shown in the middle (designed VH3 or Vκ1) from the Kabat database (Kabat et al., 1991) or IGHV3 and IGKV1 from the Lefranc database (Lefranc and Lefranc, 2001). The mutated anti-RAS scFvs are shown designated as I21K33, I21R33, I21R33VHI21VL, con33 and I21R33VH (VHC22S;C92S) or (VHC23S;C104S) (Kabat or IMGT nomenclatures, respectively). I21K33 comprises the six CDRs of scFv33 in the I21 framework, and I21R33 is identical except for a mutation Lys94(106)Arg; I21R33VHI21VL comprises the VH domain of I21R33 fused to the VL domain of I21; con33 has all six CDRs of scFv33 in the canonical consensus framework (Tse et al., 2002b); I21R33 (VHC22S;C92S) or (VHC23S;C104S) is a mutant of clone I21R33 with the mutations Cys22(23)Ser and Cys92(104)Ser of the VH domain. There are only four amino acid differences (at positions H1, H5, L0 and L3) between consensus and I21R framework regions. ScFv 33 and J48 belong to IGHV3-48 with IGHJ4 and IGHD1-26 (VH domain), and to IGKV1-39 with IGKJ4 (VL domain) according to the IMGT database (Lefranc and Lefranc, 2001). ScFvI21 belongs to IGHV3-23 with IGHJ4 and IGHD1-8 (VH domain) and IGKV1D-39 with IGKJ1 (VL domain).
The levels of expression of three anti-RAS scFvs were examined initially by bacterial periplasmic expression. These scFvs were subcloned into pHEN2, which has the PelB leader sequence 5′ to the scFv, allowing the periplasmic expression of soluble scFv protein. Periplasmic scFv extracts were purified by immobilized metal ion affinity chromatography (IMAC) and protein preparations separated by SDS–PAGE (Figure 4). The scFvI21 accumulated mainly in the soluble fraction, when secreted to the periplasm at 30°C, and the periplasmic expression yield was ∼3 mg/l of culture. The other anti-RAS scFvs (33 and J48) were expressed at <0.1 mg/l. Comparison of the anti-RAS scFv sequences with the consensus intrabody sequence (Figure 3) reveals only four differences in the VH framework residues of 33 and J48, one of which is position 7 in VH framework region 1 (FR1). This residue is one of three which influence the conformation of this region (Jung et al., 2001) and may thus influence scFv33 and J48 solubility. ScFvI21 conforms to the consensus in positions VH FR1 6, 7 and 10 (positions 6, 7 and 11 by IMGT; Lefranc and Lefranc, 2001).
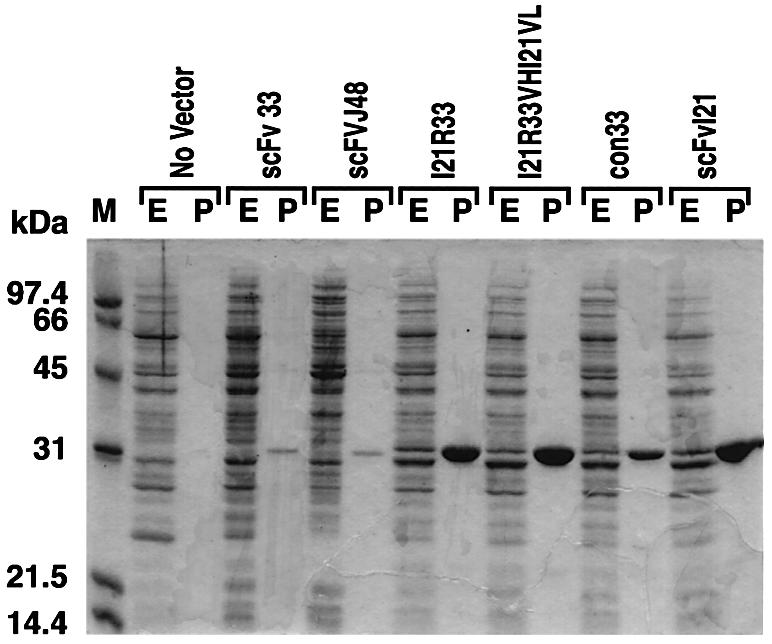
Fig. 4. Periplasmic expression and purification of anti-RAS scFvs. The scFvs with the PelB leader sequence at the N-terminus and a His tag and myc tag at the C-terminus were expressed periplasmically from the pHEN2-scFv vector in E.coli HB2151 using 1 mM IPTG for 2 h at 30°C in 1 l of 2× TY medium with 100 µg/ml ampicillin and 0.1% glucose. After induction, the cells were harvested and extracted in 4 ml of ice-cold 1× TES buffer (0.2 M Tris–HCl pH 7.5, 0.5 mM EDTA, 0.5 M sucrose), and a further 6 ml of 1:5 TES buffer were added. The supernatants of cell extracts were used as the soluble periplasmic fraction. The His-tagged scFvs were purified by immobilized Ni2+ ion chromatography and fractionated by 15% SDS–PAGE, and proteins were revealed by Coomassie Blue staining. The approximate yields of purified anti-RAS scFv33 and J48 were <100 µg/l of culture; those of scFvI21R33, I21R33VHI21VL and I21 were >3 mg/l; and that of con33 was 1 mg/l. E = complete periplasmic extracts; P = purified scFv; M = molecular weight markers.
Biochemical and biophysical characterization of anti-RAS scFv
The properties of the intrabodies isolated in our work were also characterized using two in vitro assays. The interaction of the scFvs with RAS antigen was investigated with enzyme-linked immunosorbent assay (ELISA) and biosensor assay using purified scFvs made in bacteria. HRASG12V-GppNp was coated as antigen onto ELISA plates, challenged with purified scFv, and bound scFv was detected using horseradish peroxidase (HRP)-conjugated anti-polyhistidine tag antibody (Figure 5). All three anti-RAS scFvs produced significant signals with RAS antigen compared with bovine serum albumin (BSA), and the signals were inhibited by pre-incubation with HRASG12V antigen, as a measure of specificity of the interaction. These results further suggest that these anti-RAS scFvs may interact only with the native form HRASG12V-GppNp.

Fig. 5. Specific antigen binding and competition ELISA of anti-RAS scFvs. Purified HRASG12V-GppNp (4 µg/ml, ∼200 nM; black boxes) or BSA (30 mg/ml, ∼450 µM; grey boxes) were coated onto ELISA plates for 1.5 h at room temperature. For both sets of wells, 3% BSA in PBS was added for blocking and, subsequently, purified scFv (450 ng per well) was added and incubated overnight at 4°C. After washing with PBS–0.1% Tween-20, bound scFv was detected with HRP-conjugated anti-polyhistidine antibody (HIS-1, Sigma) and signals quantitated using an Emax microplate reader (Molecular Devices). For competition assays (indicated in the figure as +), scFvs were pre- incubated with HRASG12V-GppNp (8 µg/ml; ∼400 nM) for 30 min at room temperature before addition to the ELISA well.
The affinities of binding of anti-RAS scFvs to antigen were measured by binding kinetics in the BIAcore (Figure 6). The Kds of scFv33 and J48 were determined to ∼9.97 and 2.6 nM, respectively (Figure 6B). The affinity difference of their scFvs may reflect the differences of the complementarity-determining region 1 (CDR1) sequence in the VH domain. The scFvI21 had a Kd of 1.3 µM, about three orders of magnitude weaker than scFv33 or J48. This weak affinity of scFvI21, in the micromolar range, is consistent with its weak β-gal reporter gene activation in yeast in vivo antigen–antibody interaction assays and lack of detectable binding in mammalian cell assays.

Fig. 6. Affinity measurements of anti-RAS scFvs using BIAcore. Biosensor measurements were made using the BIAcore2000. Purified scFvs from bacterial cultures were used. (A) Sensograms showing the binding of anti-RAS scFv with HRASG12V-GppNp antigen (immobilized 1500 RU). The purified scFvs (10–2000 nM) were loaded on two channels of the chip, containing either immobilized HRASG12V-GppNp or no antigen. The sensograms of each measurement were normalized by the resonance of the channel without antigen. (B) The table summarizes the values of the association rate (Kon), dissociation rate (Koff) and calculated equilibrium dissociation constants (Kds) by BIAevaluation 2.1 software.
The functional improvement of anti-RAS scFvs by modification of the scFv framework sequences
There is an excellent quantitative correlation between stability and yield of scFv when expressed in bacterial and mammalian cells, in which scFvI21 showed a higher expression yield in bacteria (Figure 4) and in mammalian cell cytoplasm (Figure 7) compared with the other two anti-RAS scFvs. While all scFvs tested in COS7 cells showed significant amounts of ‘insoluble’ scFv (Figure 7B), the best expression levels were apparent for scFvI21 (Figure 7A). The ability to improve solubility and expression of anti-RAS scFv33 in vivo, as well as its utility in vitro, was assessed by mutating the framework of scFv33, to include some or all of the 13 amino acid differences between it and scFvI21 (Figure 3). When scFv33 was mutated in the VH FRs to make it equivalent to I21 (but including arginine at the end of FR3 rather than lysine), excellent in vivo solubility was found (Figure 7A, I21R-33). In addition, mutation of both cysteine residues, needed for intra-domain disulfide bonds, to serine [Figure 7, scFv I21R-33(VHC22S;C92S) or (VHC23S; C104S) (Kabat or IMGT nomenclatures, respectively)] had only a small effect on soluble expression levels.

Fig. 7. Influence of framework residues on the solubility of expressed scFvs in COS7 cells. COS7 cells were transiently transfected with pEF/myc/cyto-scFv expression clones as indicated. Soluble and insoluble proteins were extracted, as described in Materials and methods, and fractionated by 15% SDS–PAGE. After electrophoresis, proteins were transferred to membranes and incubated with the anti-myc tag monoclonal antibody 9E10. The migration molecular weight markers (in kDa) are shown on the left. Arrows on the right indicate the scFv fragment band.
The in vivo interaction of the various mutants of the scFvs was assessed in COS7 cells using the luciferase reporter assay (Figure 8). Figure 8A shows expression and luciferase reporter data of various modifications of the scFv33 framework compared with levels of scFv33 itself, scFvR4 (anti β-gal negative control; Martineau et al., 1998) or scFvI21 which does not give significant luciferase activity. One notable mutation of scFv33 is Arg94Lys (Kabat et al., 1991) (position 106 according to IMGT; Lefranc and Lefranc, 2001), which completely eliminated reporter response (Figure 8A) even though the expression of this scFv-VP16 is increased compared with the original scFv33 (Figure 8A). The arginine residue at position 94 (or position 106 by IMGT; Lefranc and Lefranc, 2001) is very close to the antigen-binding site (CDR3 of the heavy chain) and may be involved in interaction with RAS antigen directly. Alternatively, the residue at this position may form a surface bridge across the CDR3 loop through its positively charged side chain with the carboxyl group of the aspartic acid at position H101 (H116 by IMGT; Morea et al., 1998), and the substitution (arginine to lysine) may affect the critical conformation of CDR3. The other mutant scFv33 variants generally maintained their binding ability with RAS antigen as judged by the luciferase reporter assay (Figure 8A). Interestingly, three mutant variants, VH(A74S + S77T), VL(I85T) and VH(Q1E + V5L + A7S +S28T) +VL(G100Q + L104V), were increased 1.5- to 2.5-fold in reporter gene activity, accompanied by an increase of scFv-VP16 in the soluble fraction.

Fig. 8. Improvement of the intracellular interaction between anti-RAS intrabodies and RAS antigen by the mutation of framework sequences. Mammalian two-hybrid antibody–antigen interaction assays were performed in COS7 cells. (A) COS7 were transfected with the pEF-scFv-VP16 vectors and pM1-HRASG12V, together with the luciferase reporter clones, and luciferase levels were determined as described in Materials and methods. Upper panel: the normalized fold induction of luciferase signals (zero being taken as the signal from prey plasmid without scFv) for scFv-VP16 binding RAS antigen bait. Lower panel: a western blot of COS7 cell extracts after the expression of scFv-VP16 fusion proteins. ScFv-VP16 fusion proteins were detected by western blot using anti-VP16 (14-5, Santa Cruz Biotechnology) monoclonal antibody and HRP-conjugated anti-mouse IgG antibody. ScFv used as a control was anti-β-gal scFvR4 (Martineau et al., 1998). scFv33 mutants were numbered according to Kabat et al. (1991) and the numbers in parentheses indicate numbering according to Lefranc and Lefranc (2001) (see Figure 33): VH(A74S+S77T), substitutions of Ala74(83)Ser and Ser77(86)Thr of VH; VH(D84A), substitution of Asp84(96)Ala of VH; VH(R94K), substitution of Arg94(106)Lys of VH; VL(0T + V3Q), addition of threonine between the linker and VL domain plus substitution of Val3(3)Gln of VL; VL(F10S), substitution of Phe10(10)Ser of VL; VL(I85T), substitution of Ile85(100)Thr of VL; VH(Q1E + V5L + A7S + S28T) + VL(G100Q + V104L), substitutions of Gln1(1)Glu, Val5(5)Leu, Ala7(7)Ser and Ser28(29)Thr of VH plus Gly100(120)Gln and Val104(124)Leu of VL. (B) COS7 cell two-hybrid antibody– antigen interaction assay using scFvs with framework mutations to convert to consensus sequence scaffolds. The various scFv-VP16 prey constructs shown were transiently transfected with pM1-HRASG12V bait plasmid in COS7 cells, and the luciferase activities were measured 48 h after transfection. The fold luciferase activity levels are shown in the histogram, with the activity of no scFv (prey plasmid without scFv) as baseline. The expression levels of scFv-VP16 in the soluble fraction of COS7 cells are shown in the lower panel. The bands were visualized by western blot using anti-VP16 (14-5) antibody and HRP-conjugated anti-mouse IgG antibody.
The mutation of scFv33 into the framework of scFvI21 was performed, except that arginine at position 94 (106 by IMGT) in VH was maintained (I21R33). Two further scFv33 variants were constructed, one in which scFv33 was converted to the intrabody consensus framework (con33) and one in which mutation of only the VH frameworks was carried out (I21R33VHI21VL, Figure 3). In the mammalian reporter assay, a 2- to 3-fold increase of reporter gene activity was observed with I21R33 and con33, but with dramatically improved solubility compared with the original scFv33 (Figure 8B). These data show that the consensus, or I21, frameworks are the most suitable scaffolds for intracellular antibody expression and, furthermore, intrabody function can be improved using these frameworks.
Activity of anti-RAS scFvs lacking conserved cysteine residues in the VH domain
The mutated anti-RAS scFvI21R33 interacts specifically with RAS antigen in COS7 cells, even though, in this reducing environment, scFvs mostly cannot form disulfide bonds (Tavladoraki et al., 1993; Biocca et al., 1995). Perhaps a small population of overexpressed scFvs does form disulfide bonds in the cytoplasm and interacts with antigen in vivo, such as the anti-β gal scFvR4, some of which is disulfide bonded in the cytoplasm of bacteria (Martineau et al., 1998). Thus a small population could be detectable in vivo using our antigen–antibody interaction assay, if the scFv has a high affinity for the antigen. However, in vitro studies have demonstrated that some scFvs can be made which are disulfide free but fold correctly (Proba et al., 1998; Worn and Pluckthun, 1998a). Therefore, to test the requirement for an intra-domain disulfide bond, an expression vector encoding a mutant scFv lacking the cysteine residues at position 22 and 92 (Kabat numbering, or 23 and 104 in IMGT numbering) was constructed. This scFvI21R33(VHC22S;C92S) or (VHC23S;C104S) (Kabat or IMGT nomenclatures, respectively), based on the I21R33 sequence, had the two cysteine codons mutated to serine. A vector encoding this protein was tested in our mammalian reporter assay (Figure 8B). The scFv protein was expressed at high levels, roughly comparable with those of I21R33 and I21, and the ability to activate the luciferase reporter was similar to that of the 12R33 scFv. These results show that anti-RAS scFvI21R33(VHC22S;C92S) can fold adequately without an intra-domain disulfide bond and function inside cells in this condition.
Conversion of intrabodies into anti-RAS reagents to block tumorigenic transformation
In the experiments discussed above, we sought to improve the effectiveness of anti-RAS intrabodies by mutational analysis of the VH and VL FRs to make them equivalent to the canonical IAC consensus (Tse et al., 2002b; Visintin et al., 2002). A further test of the utility of our pre-determined consensus frameworks was carried out by assessing the ability of anti-RAS sequences to inhibit oncogenic RAS transformation of NIH 3T3 cells. We evaluated this by taking as a starting point the scFvI21 clone, which was isolated from the yeast screening (Figure 1) using RAS as a bait. Mutagenesis of the scFv33 to I21R33 (i.e. the I21 framework with VH and VL CDRs of scFv33) gives a well expressed protein able to activate the luciferase reporter gene (Figure 8B). We have used this scFv in competitive transformation assays in which NIH 3T3 cells were transfected with a plasmid expressing activated HRAS alone (HRASG12V) to yield transformed foci (non-contact-inhibited colonies) which can grow in multilayers and show a swirling appearance of spindle-shaped cells (Figure 9A, HRASG12V + empty scFv vector). When the NIH 3T3 cells were co-transfected with the HRASG12V vector together with one expressing scFvI21, essentially no difference from control was observed (Figure 9A and B), in keeping with the observed lack of activation of the RAS-dependent luciferase reporter assays. On the other hand, when HRASG12V was expressed with the mutated I21 clone, scFvI21R33, the number of transformed foci was reduced to 30%, presumably due to interaction of the scFv with the HRASG12V-expressed protein, preventing its function. Thus the consensus scaffolds provide a basis for creation of functional scFvs.

Fig. 9. Inhibition of RAS-dependent NIH 3T3 cell transformation activity by anti-RAS scFv. Mutant HRASG12V cDNAs were subcloned into the mammalian expression vector pZIPneoSV(X), and anti-RAS scFv into the pEF-FLAG-Memb vector which has a plasma membrane targeting signal at the C-terminus of scFv and a FLAG tag at the N-terminus. A 100 ng aliquot of pZIPneoSV(X)-HRASG12V and 2 µg of pEF-FLAG-Memb-scFv were co-transfected into NIH 3T3 cells clone D4. Two days later, the cells were transferred to 10 cm plates and grown for 14 days in DMEM containing 5% donor calf serum with penicillin and streptomycin. Finally, the plates were stained with crystal violet, and foci of transformed cells were counted. (A) Representative photograph of stained plates. Empty vector in the left top panel indicates co-transfection of pZIPneoSV(X) without HRASG12V, and pEF-FLAG-Memb without scFv as negative control. No foci formation was observed. The right top panel indicates pZIPneoSV(X)-HRASG12V with pEF-FLAG-Memb without scFv as positive control. In the other plates, the HRASG12V vector was co-transfected with either pEF-FLAG-Memb-scFvI21 or pEF-FLAG-Memb-scFvI21R33. (B) The relative percentage of transformed foci was determined as the number of foci normalized to the focus formation induced by pZIPneoSV(X)-HRASG12V and pEF-Memb empty vector, which was set at 100. Results represent one experiment with each transfection performed in duplicate. Two additional experiments yielded similar results.
Discussion
The most important requirements for intracellular antibodies as therapeutic or bioscience research tools is that these antibodies (or antibody fragments) exhibit good expression levels and are functional within any compartment of mammalian cells. Ideally, these also work as functional reagents for in vitro use. There are severe limitations, and few scFv fragments derived from hybridomas are stable under a reducing environment without modification, even if they have good affinity in vitro. The IAC technology described here, and previously (Tse et al., 2002b; Visintin et al., 2002), overcomes these difficulties as it is based on an in vivo genetic screen for the direct isolation of functional scFvs.
The IAC approach has several advantages compared with other screening methods. It is based on the yeast two-hybrid in vivo assay (Fields and Song, 1989), which works as direct cytoplasmic selection of scFvs (Visintin et al., 1999). In addition, it theoretically allows the selection of antibody fragments (in the experiments described here scFvs) against any expressed antigen, including post-transcriptionally modified proteins or especially protein complexes, as it allows targeting of antigen in its native form. A further consideration is that the screening process involves verification of candidate intracellular scFvs in mammalian cells. By adopting these different bait and reporter systems, false-positive scFvs are eliminated. In addition, the mammalian antigen–antibody interaction assay is performed at 37°C, compared with 30°C in yeast or at room temperature (or at 4°C) for an in vitro phage screen. This step from yeast to mammalian cells makes it possible to select more thermally tolerant intracellular scFvs and, because the isolation involves a mammalian assay, higher affinity interactions may be selected which are suitable for competitive binding of the target antigen with endogenous dimerization molecules.
In this work, we have applied IAC technology against the oncogenic protein RAS and have isolated specific anti-RAS scFvs which bind to this antigen in the cell cytoplasm. Sequence analysis demonstrates that all anti-RAS scFvs belong to the VH3 and Vκ1 subgroup defined from the Kabat database (Kabat et al., 1991) or the IGHV3 and IGKV1 subgroup defined from the IMGT database (Lefranc and Lefranc, 2001). Most intrabodies selected for binding to the antigens BCR or ABL (Tse et al., 2002b) also belong to the same subgroup, supporting the notion that these subgroups of VH and Vκ framework can function intracellularly. This observation allowed a consensus framework to be defined (Tse et al., 2002b). In this screening for anti-RAS scFvs, one scFv (scFvI21) shows high yields in bacterial periplasm, high solubility in mammalian cells but poor affinity of interaction in mammalian cells, whilst other scFvs (scFv33 and J48) have high affinity but relatively low yields of soluble expressed protein. The I21 scFv framework sequence, however, conforms closely with the consensus framework (Tse et al., 2002b) in both the VH and VL domains. In support of the utility of this consensus, we found that mutation of scFv33 to the consensus framework (con33) or to the I21 framework (I21R33) improved this function, including solubility and binding. Moreover, improved anti-RAS scFv function did not require intra-domain disulfide bond formation. Finally, when the scFv33 was mutated to the I21R consensus framework, retaining the scFv33 CDR sequences, the intrabodies were able to perform the crucial biological function of inhibiting oncogenic HRASG12V transformation of NIH 3T3 cells. This illustrates the versatility of our approach in generating effective intrabodies for mammalian cell use.
In the light of our improvements in intrabody expression and function using the consensus framework, it is reasonable to expect that intrabody libraries could be constructed based on the consensus intrabody scaffold and of sufficient diversity to allow primary screening directly in yeast cell assays, without recourse to a preliminary in vitro phage antibody screen with protein antigen. This would provide very clear technical advantages, i.e. that antigens would not need to be expressed in vitro to provide for protein, and the requirement for the yeast bait expression for IAC selection is merely DNA sequence. This opens up the possibility of using IAC technology to select intrabodies which bind to any protein predicted from genome sequence analysis of any organism, e.g. from the human genome sequencing programme.
A key question is the degree of library diversity required to contain effective intracellular antibodies and thus whether sufficient antibody diversity can be made and screened in yeast. In the work described herein, we have screened three different human scFv libraries (Sheets et al., 1998; de Wildt et al., 2000), each of which has sufficient diversity for screening against many different antigens. The success of the current IAC requires starting with in vitro phage antibody screening of highly diverse libraries. However, such phage antibodies do not necessarily work as intracellular antibodies without modification (because of solubility and folding problems in a reducing environment), meaning that the effective diversity of these phage scFv libraries, as intracellular antibody libraries, is likely to be less than expected. The construction of specially designed human intracellular antibody libraries using randomized CDRs on the fixed consensus framework should increase effective intrabody diversity. This will make it possible to screen the library for intrabodies without the need for the preliminary phage panning step, while keeping full effective diversity and accelerating the screening for intracellular antibodies.
Materials and methods
RAS antigen
Recombinant oncogenic HRAS (G12V; residues 1–166) was expressed in bacterial cells harbouring expression plasmids based on pET11a (Novagen) and purified by ion-exchange chromatography and gel filtration described elsewhere (Pacold et al., 2000). To prepare the active form of RAS antigen, 3 mg of purified HRASG12V protein was loaded with 2 mM GppNp (Sigma), a non-hydrolysable analogue of GTP, using the alkaline phosphatase protocol (Herrmann et al., 1996). This GppNp-bound HRASG12V was used as antigen throughout.
In vitro scFv phage library screening and preparation of a specific scFv-VP16 yeast library
The IAC screening of three different scFv libraries (Sheets et al., 1998; de Wildt et al., 2000) was performed as described (Tse et al., 2002a,b) (see also a link within the Laboratory of Molecular Biology website http://www.mrc-lmb.cam.ac.uk) with slight modifications. In brief, a first panning step using phage antibody library screens was performed using 50 µg/ml HRASG12V-GppNp antigen in phosphate-buffered saline (PBS) containing 1 mM MgCl2, bound to immunotubes. Anti-RAS-bound phage were rescued and amplified in Escherichia coli TG1. The scFv DNA fragments were subcloned into the pVP16* yeast vector (Hollenberg et al., 1995; Visintin et al., 1999) and 4.13 × 106 clones used for yeast screening. The RAS bait was prepared by cloning truncated HRASG12V cDNA into the EcoRI–BamHI site of pBTM116 vector (LexA-DBD). The pBTM116-HRASG12V bait vector (Trp+) was transfected into Saccharomyces cerevisiae L40 using the lithium acetate/polyethylene glycol method (Tse et al., 2002a), and colonies growing on Trp– plates were selected. The expression of LexA-RAS fusion protein was confirmed by western blot using anti-pan RAS (Ab-3, Oncogene Research Product) monoclonal antibody. For library screening, 100 µg of yeast scFv-VP16 library DNA were transformed into the L40 clone stably expressing antigen. Positive colonies were selected for His prototropy and confirmed by β-gal activity by filter assay. For the isolated individual clones, false-positive clones were eliminated, and true-positive clones were confirmed by re-testing of His-independent growth and β-gal activation.
Purification of scFv for in vitro assay
Periplasmic bacterial expression of scFvs was as described (Tse et al., 2002a). The scFvs were cloned into pHEN2 vector (see www.mrc-cpe.cam.ac.uk for map) and expressed for 2 h at 30°C with 1 mM isopropyl-β-d-thiogalactopyranoside (IPTG) in a 1 l culture of E.coli HB2151 cells. The cells were harvested and periplasmic proteins were extracted with TES buffer (Tris–HCl pH 7.5, EDTA, sucrose). The periplasmic proteins were dialysed overnight against 2.5 l of PBS including 10 mM imidazole. IMAC of periplasmic scFvs was carried out at 4°C for 1 h with 4 ml of Ni-NTA–agarose (QIAGEN). The agarose was washed four times with 20 ml of PBS with 20 mM imidazole. The polyhistidine-tagged scFvs were eluted with 4 ml of 250 mM imidazole in PBS. The eluate was dialysed overnight against 2.5 l of 20 mM Tris–HCl pH 7.5, including 10% glycerol at 4°C. Purified scFv was concentrated to 1–5 mg/ml using a Centricon concentrator (YM-10, Amicon), and the aliquots were stored at –70°C. Protein concentrations of purified scFvs were measured using the Bio-Rad Protein assay Kit (Bio-Rad).
ELISAs
The ELISA plate wells were coated with 100 µl of purified HRASG12V-GppNp antigen (4 µg/ml, ∼200 nM) in PBS overnight at 4°C. Wells were blocked with 3% BSA–PBS for 2 h at room temperature. The purified scFvs (∼450 ng) were diluted in 90 µl of 1% BSA–PBS and allowed to bind for 1 h at 37°C. After washing three times with PBS containing 0.1% Tween-20 (PBST), HRP-conjugated anti-polyhistidine (HIS-1, Sigma) monoclonal antibodies, which were diluted 1:2000 in 1% BSA–PBS, were allowed to bind for 1 h at 37°C. After washing six times with PBST, HRP activity was visualized using the 3,3′,5,5-tetramethylbenzidine (TMB) liquid substrate system according to the manufacturer’s instructions. The reaction was stopped with 0.5 M hydrosulfate, and data were collected with a microtitre plate reader (450–650 nm filter). To verify the specificity of scFv with antigen, competitive ELISA was also performed. scFvs were pre-incubated with HRASG12V-GppNp antigen (8 µg/ml) for 30 min at room temperature, before adding the mixture to the antigen-coated ELISA wells. All measurements were performed in duplicate.
Surface plasmon resonance analysis
The BIAcore2000 (Pharmacia Biosensor) was used to measure the binding kinetics of scFv with antigen. To immobilize antigen on a CM5 sensorchip, the sensorchip was first activated by flowing 40 µl of a mixture of EDC/NHS [N-ethyl-N-(dimethylaminopropyl) carbodiimide hydrochloride/N-hydroxysuccinimide] at 10 µl/min flow rate. Then, 100 µg/ml of purified HRASG12V-GppNp in 10 mM sodium acetate pH 3.5 was injected and immobilized until ∼1500 RU. After immobilization, the chip was inactivated with 40 µl of ethanolamine-HCl. Purified scFvs (10–2000 nM) were loaded at flow rate of 20 µl/min at 25°C using running buffer HBS-EP [0.01 M HEPES pH 7.4, 0.15 M NaCl, 3 mM EDTA, 0.005% (v/v) polysorbate 20 plus 2 mM MgCl2] on two channels of the chip containing either immobilized HRASG12V-GppNp or no antigen, for the determination of the binding affinity of scFvs. Each determination was performed in duplicate. The antigen-immobilized surface on the sensorchip after binding scFvs was regenerated by rinsing with 10 mM HCl until the starting baseline was achieved. The kinetic rate constants, kon and koff, were evaluated using the BIAevaluation 2.1 software supplied by the manufacturer. Kd values were calculated from koff and kon rate constants (Kd = koff/kon).
Mammalian in vivo antigen–antibody interaction assay
The scFvs were cloned into the SfiI–NotI site of the pEF-VP16 expression vector (Tanaka et al., 2003). The HRAS expression plasmid (pM1-HRASG12V) expressing HRASG12V in-frame with Gal4-DBD was made by subcloning HRASG12V cDNA (codons 1–166) into the EcoRI–BamHI site of the pM1 vector (Sadowski et al., 1992). The bait pM1-β-gal and the prey pEF-scFvR4-VP16 (anti-β-gal scFv), used as positive or negative controls, have been described (Tse and Rabbitts, 2000). COS7 cells were transiently co-transfected with 500 ng of pG5-Luc reporter plasmid (de Wet et al., 1987), 50 ng of pRL-CMV (Promega), 500 ng of pEF-scFv-VP16 and 500 ng of pM1-antigen bait with 8 µl of LipofectAMINE™ transfection reagent (Invitrogen, according to the manufacturer’s instructions). At 48 h after transfection, the cells were washed once with PBS and lysed in 500 µl of 1× passive lysis buffer (Promega) at room temperature for 15 min with gentle shaking. A 20 µl aliquot of cell lysate was assayed using the Dual-Luciferase Reporter Assay System (Promega) in a luminometer. Transfection efficiency was normalized with the Renilla luciferase activity. The fold luciferase activity was calculated by dividing the normalized firefly luciferase activity of the sample containing the vector alone. The data represent two experiments performed in duplicate.
Immunofluorescence assays
scFv DNA fragments were cloned into the NcoI–NotI site of pEF/nuc/myc (Invitrogen) with the nls signal and myc tag at the C-terminus of expressed scFv. For expression of RAS antigen, full-length HRASG12V cDNA was cloned into the KpnI–EcoRI site of pHM6 vector (Boehringer Mannheim) to encode RAS with an HA tag at the N-terminus and a His tag at the C-terminus. The day before transfection, 1.2 × 104 COS7 cells were seeded on a Lab-Tek II chamber slide (Nalge Nunc International). The plasmids were co-transfected using LipofectAMINE™ and, 48 h after transfection, cells were washed twice with PBS, permeabilized with 0.5% Triton X-100 in PBS and fixed with 4% paraformaldehyde in PBS. Cells were stained with anti-c-myc mouse monoclonal antibody (9E10, Santa Cruz) and anti-HA rabbit polyclonal serum (sc-805; Santa Cruz) both at dilutions of 1:100. Secondary antibodies, fluorescein-linked sheep anti-mouse antibody and Cy3-linked goat anti-rabbit antibody (Amersham Pharmacia Biotech), were used at dilutions of 1:200 for staining. After several washes with PBS, the slides were overlaid with coverslips, and staining patterns were studied using a Bio-Radiance confocal microscope (Bio-Rad).
Western blot analysis
To evaluate the expression level and solubility of scFvs in mammalian cells, the scFvs or scFv-VP16 fusion proteins were expressed in COS7 cells. For scFv expression, scFv DNA fragments were cloned into the Nco1–NotI sites of the pEF/myc/cyto expression vector (Invitrogen). The day before transfection, COS7 cells were seeded at ∼2 × 105 per well in a 6-well culture plate (Nalge Nunc International). A 1 µg aliquot of pEF/myc/cyto-scFv or pEF-scFv-VP16 was transiently transfected with 8 µl of LipofectAMINE™. At 48 h after transfection, the cells were washed once with PBS, lysed for 30 min in ice-cold extraction buffer [10 mM HEPES pH 7.6, 250 mM NaCl, 5 mM EDTA, 0.5% NP-40, 1 µg/ml leupeptin, 1 µg/ml pepstatin A, 0.1 mg/ml aprotinin, 1 mM phenylmethylsulfonyl fluoride (PMSF)] and centrifuged for 10 min at 13 000 r.p.m. at 4°C. The pellets (‘insoluble’ fraction) and the supernatants (‘soluble’ fraction) were analysed by SDS–PAGE, followed by western blot using anti-myc (9E10) monoclonal antibody (for detection of scFv) or anti-VP16 (14-5, Santa-Cruz) monoclonal antibody (for the scFv-VP16-AD fusion) as primary antibody and HRP-conjugated rabbit anti-mouse IgG antibody (Amersham Pharmacia Biotech) as secondary antibody. The blots were visualized by an enhanced chemiluminescence (ECL) detection kit (Amersham Pharmacia Biotech).
Mutation of framework residues for anti-RAS scFv
The construct I21R33 (sequence shown in Figure 3), which comprises FRs of anti-RAS scFvI21 and the CDRs of anti-RAS scFv33, was made using stepwise site-specific mutagenesis of scFv33, cloned in pEF-VP16, as primary template using PCR mutagenesis (Hoogenboom and Winter, 1992; Tanaka et al., 2003). Pfu DNA polymerase was used throughout. I21R33 (VHC22S;C92S), con33 and I21R33VHI21VL (Figure 3) were also constructed by mutations of I21R33 using PCR mutagenesis with appropriate oligonucleotides (Hoogenboom and Winter, 1992; Tanaka et al., 2003). All scFv constructs were digested with SfiI or NcoI, and Not1 and subcloned into pEF-VP16 (for in vivo antigen antibody interaction assay) and pEF/myc/cyto vector (for expression of scFv in mammalian cells). All mutated scFv constructs were verified by DNA sequencing.
Transformation assays in NIH 3T3 cells
RAS protein is functionally localized to the plasma membrane of cells and, therefore, in order to localize scFv to cell membrane, we generated the pEF-Memb and pEF-FLAG-Memb vectors, with farnesylation sites for membrane localization of proteins. The pEF-Memb expression plasmid was constructed by introducing the coding region for the C-terminal 20 amino acid residues of HRAS into the NotI–XbaI site of pEF/myc/cyto vector (Invitrogen). A FLAG tag peptide-coding sequence (MDYKDDDDK) and an SfiI–NotI cloning site was introduced into pEF-Memb to make pEF-FLAG-Memb. The scFvs were subcloned into SfiI–NotI sites of pEF-FLAG-Memb. For expression of HRASG12V, mutant HRASG12V cDNA was subcloned into the expression vector pZIPneoSV(X) (Cepko et al., 1984). Low passage NIH 3T3 cells clone D4 (a kind gift from Dr Chris Marshall) were seeded at 2 × 105 cells per well in 6-well plates, and 2 µg of each pEF-FLAG-Memb-scFv plus 100 ng of pZIPneoSV(X)-HRASG12V vector were used with 12 µl of LipofectAMINE™ for transfection. Two days later, the cells were transferred to 10 cm plates and grown for 2 weeks in Dulbecco’s modified Eagle’s medium (DMEM) containing 5% donor calf serum (Invitrogen) together with penicillin and streptomycin. The plates were finally stained with crystal violet and the number of foci counted.
Acknowledgments
Acknowledgements
We thank Professor James Marks and Dr Ian Tomlinson for the phage antibody libraries, Dr Chris Marshall for NIH 3T3 cells, clone D4, Dr Lawrence A.Quilliam for pZIPneoSV (X) vector, Professor Alan Hall for the RASG12V cDNA clone, Drs Roger Williams, Olga Perisic and Michael Pacold for pEF11a-RAS expression vector and invaluable help and advice on protein purification, and Dr Ruud de Wildt for help with the BIAcore experiment. This work was supported by the Medical Research Council. T.T. is supported partly by the Medical Research Council, the University of Kyoto and the National Foundation for Cancer Research.
References
- Adjei A.A. (2001) Blocking oncogenic Ras signaling for cancer therapy. J. Natl Cancer Inst., 93, 1062–1074. [DOI] [PubMed] [Google Scholar]
- Biocca S., Pierandrei-Amaldi,P. and Cattaneo,A. (1993) Intracellular expression of anti-p21ras single chain Fv fragments inhibits meiotic maturation of Xenopus oocytes. Biochem. Biophys. Res. Commun., 197, 422–427. [DOI] [PubMed] [Google Scholar]
- Biocca S., Pierandrei-Amaldi,P., Campioni,N. and Cattaneo,A. (1994) Intracellular immunization with cytosolic recombinant antibodies. Biotechnology, 12, 396–399. [DOI] [PubMed] [Google Scholar]
- Biocca S., Ruberti,F., Tafani,M., Pierandrei-Amaldi,P. and Cattaneo,A. (1995) Redox state of single chain Fv fragments targeted to the endoplasmic reticulum, cytosol and mitochondria. Biotechnology, 13, 1110–1115. [DOI] [PubMed] [Google Scholar]
- Bird R.E. et al. (1988) Single-chain antigen-binding proteins. Science, 242, 423–426. [DOI] [PubMed] [Google Scholar]
- Cattaneo A. and Biocca,S. (1999) The selection of intracellular antibodies. Trends Biotechnol., 17, 115–121. [DOI] [PubMed] [Google Scholar]
- Cepko C.L., Roberts,B.E. and Mulligan,R.C. (1984) Construction and applications of a highly transmissible murine retrovirus shuttle vector. Cell, 37, 1053–1062. [DOI] [PubMed] [Google Scholar]
- Cohen P.A. (2002) Intrabodies. Targeting scFv expression to eukaryotic intracellular compartments. Methods Mol. Biol., 178, 367–378. [DOI] [PubMed] [Google Scholar]
- Desiderio A., Franconi,R., Lopez,M., Villani,M.A., Viti,F., Chiaraluce,R., Consalvi,V., Neri,D. and Benvenuto,E. (2001) A semi-synthetic repertoire of intrinsically stable antibody fragments derived from a single-framework scaffold. J. Mol. Biol., 310, 603–615. [DOI] [PubMed] [Google Scholar]
- de Wet J.R., Wood,K.V., DeLuca,M., Helinski,D.R. and Subramani,S. (1987) Firefly luciferase gene: structure and expression in mammalian cells. Mol. Cell. Biol., 7, 725–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Wildt R.M., Mundy,C.R., Gorick,B.D. and Tomlinson,I.M. (2000) Antibody arrays for high-throughput screening of antibody–antigen interactions. Nat. Biotechnol., 18, 989–994. [DOI] [PubMed] [Google Scholar]
- Fields S. and Song,O. (1989) A novel genetic system to detect protein–protein interactions. Nature, 340, 245–246. [DOI] [PubMed] [Google Scholar]
- Herrmann C., Horn,G., Spaargaren,M. and Wittinghofer,A. (1996) Differential interaction of the ras family GTP-binding proteins H-Ras, Rap1A and R-Ras with the putative effector molecules Raf kinase and Ral-guanine nucleotide exchange factor. J. Biol. Chem., 271, 6794–6800. [DOI] [PubMed] [Google Scholar]
- Hollenberg S.M., Sternglanz,R., Cheng,P.F. and Weintraub,H. (1995) Identification of a new family of tissue-specific basic helix–loop–helix proteins with a two-hybrid system. Mol. Cell. Biol., 15, 3813–3822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoogenboom H.R. and Winter,G. (1992) By-passing immunisation. Human antibodies from synthetic repertoires of germline VH gene segments rearranged in vitro. J. Mol. Biol., 227, 381–388. [DOI] [PubMed] [Google Scholar]
- Huston J.S. et al. (1988) Protein engineering of antibody binding sites: recovery of specific activity in an anti-digoxin single-chain Fv analogue produced in Escherichia coli. Proc. Natl Acad. Sci. USA, 85, 5879–5883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung S., Spinelli,S., Schimmele,B., Honegger,A., Pugliese,L., Cambillau,C. and Pluckthun,A. (2001) The importance of framework residues H6, H7 and H10 in antibody heavy chains: experimental evidence for a new structural subclassification of antibody V(H) domains. J. Mol. Biol., 309, 701–716. [DOI] [PubMed] [Google Scholar]
- Kabat E.A., Wu,T.T., Perry,H.M., Gottesman,K.S. and Foeller,C. (1991) Sequences of Proteins of Immunological Interest. NIH, Bethesda, MD, USA.
- Lefranc M.-P. and Lefranc,G. (2001) The Immunoglobulin Facts Book. Academic Press, London.
- Lowy D.R. and Willumsen,B.M. (1993) Function and regulation of ras. Annu. Rev. Biochem., 62, 851–891. [DOI] [PubMed] [Google Scholar]
- Marasco W.A., Haseltine,W.A. and Chen,S. (1993) Design, intracellular expression and activity of a human anti-human immunodeficient virus type 1 gp120 single-chain antibody. Proc. Natl Acad. Sci. USA, 90, 7899–7893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martineau P., Jones,P. and Winter,G. (1998) Expression of an antibody fragment at high levels in the bacterial cytoplasm. J. Mol. Biol., 280, 117–127. [DOI] [PubMed] [Google Scholar]
- Mizushima S. and Nagata,S. (1990) pEF-BOS, a powerful mammalian expression vector. Nucleic Acids Res., 18, 5322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morea V., Tramontano,A., Rustici,M., Chothia,C. and Lesk,A.M. (1998) Conformations of the third hypervariable region in the VH domain of immunoglobulins. J. Mol. Biol., 275, 269–294. [DOI] [PubMed] [Google Scholar]
- Ohage E. and Steipe,B. (1999) Intrabody construction and expression. I. The critical role of VL domain stability. J. Mol. Biol., 291, 1119–1128. [DOI] [PubMed] [Google Scholar]
- Ohage E.C., Wirtz,P., Barikow,J. and Steipe,B. (1999) Intrabody construction and expression. II. A synthetic catalytic Fv fragment. J. Mol. Biol., 291, 1129–1134. [DOI] [PubMed] [Google Scholar]
- Pacold M.E. et al. (2000) Crystal structure and functional analysis of Ras binding to its effector phosphoinositide 3-kinase γ. Cell, 103, 931–943. [DOI] [PubMed] [Google Scholar]
- Proba K., Worn,A., Honegger,A. and Pluckthun,A. (1998) Antibody scFv fragments without disulfide bonds made by molecular evolution. J. Mol. Biol., 275, 245–253. [DOI] [PubMed] [Google Scholar]
- Rabbitts T.H. (1994) Chromosomal translocations in human cancer. Nature, 372, 143–149. [DOI] [PubMed] [Google Scholar]
- Rondon I.J. and Marasco,W.A. (1997) Intracellular antibodies (intrabodies) for gene therapy of infectious diseases. Annu. Rev. Microbiol., 51, 257–283. [DOI] [PubMed] [Google Scholar]
- Sadowski I., Bell,B., Broad,P. and Hollis,M. (1992) GAL4 fusion vectors for expression in yeast or mammalian cells. Gene, 118, 137–141. [DOI] [PubMed] [Google Scholar]
- Sheets M.D. et al. (1998) Efficient construction of a large nonimmune phage antibody library: the production of high-affinity human single-chain antibodies to protein antigens. Proc. Natl Acad. Sci. USA, 95, 6157–6162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka T., Chung,G.T.Y., Forster,A., Lobato,M.N. and Rabbitts,T.H. (2003) De novo production of diverse intracellular antibody libraries. Nucleic Acids Res., in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tavladoraki P., Benvenuto,E., Trinca,S., De Martinis,D., Cattaneo,A. and Galeffi,P. (1993) Transgenic plants expressing a functional single-chain Fv antibody are specifically protected from virus attack. Nature, 366, 469–472. [DOI] [PubMed] [Google Scholar]
- Triezenberg S.J., Kingsbury,R.C. and McKnight,S.L. (1988) Functional dissection of VP16, the trans-activator of herpes simplex virus immediate early gene expression. Genes Dev., 2, 718–729. [DOI] [PubMed] [Google Scholar]
- Tse E. and Rabbitts,T.H. (2000) Intracellular antibody–caspase mediated cell killing: a novel approach for application in cancer therapy. Proc. Natl Acad. Sci. USA, 97, 12266–12271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tse E., Chung,G. and Rabbitts,T.H. (2002a) Isolation of antigen-specific intracellular antibody fragments as single chain Fv for use in mammalian cells. In Turksen,K. (ed.), Methods in Molecular Biology. Humana Press, Totawa, NJ, 185, 433–446. [DOI] [PubMed]
- Tse E., Lobato,M.N., Forster,A., Tanaka,T., Chung,T.Y.G. and Rabbitts,T.H. (2002b) Intracellular antibody capture technology: application to selection of single chain Fv recognising the BCR-ABL oncogenic protein. J. Mol. Biol., 317, 85–94. [DOI] [PubMed] [Google Scholar]
- Visintin M., Tse,E., Axelson,H., Rabbitts,T.H. and Cattaneo,A. (1999) Selection of antibodies for intracellular function using a two-hybrid in vivo system. Proc. Natl Acad. Sci. USA, 96, 11723–11728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visintin M., Settanni,G., Maritan,A., Graziosi,S., Marks,J.D. and Cattaneo,A. (2002) The intracellular antibody capture technology (IACT): towards a consensus sequence for intracellular antibodies. J. Mol. Biol., 317, 73–83. [DOI] [PubMed] [Google Scholar]
- Wirtz P. and Steipe,B. (1999) Intrabody construction and expression III: engineering hyperstable VH domains. Protein Sci., 8, 2245–2250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Worn A. and Pluckthun,A. (1998a) An intrinsically stable antibody ScFv fragment can tolerate the loss of both disulfide bonds and fold correctly. FEBS Lett., 427, 357–361. [DOI] [PubMed] [Google Scholar]
- Worn A. and Pluckthun,A. (1998b) Mutual stabilization of VL and VH in single-chain antibody fragments, investigated with mutants engineered for stability. Biochemistry, 37, 13120–13127. [DOI] [PubMed] [Google Scholar]