

Abstract
Activation of the transcription factor FKHR (Forkhead in human rhabdomyosarcoma, FOXO1a) in various established cell lines induces cell cycle arrest followed by apoptosis. These effects are inhibited through activation of the phosphatidylinositol 3-kinase/Akt pathway, resulting in FKHR phosphorylation and its export from the nucleus, thus blocking its pro-apoptotic activity. Here we report that FKHR regulates fusion of differentiating primary myoblasts. We demonstrate that FKHR is localized in the cytoplasm of proliferating myoblasts, yet translocates to the nucleus by a phosphorylation-independent pathway following serum starvation, a condition that induces myoblast differentiation. FKHR phosphorylation during terminal differentiation appears to downregulate its fusion activity, as a dominant-active non-phosphorylatable FKHR mutant dramatically augments the rate and extent of myotube fusion. However, this FKHR mutant exerts its effects only after other events initiated the differentiation pro cess. Conversely, enforced expression of a dominant-negative FKHR mutant blocks myotube formation whereas wild-type FKHR has no effect. We conclude that in addition to the role of FoxO proteins in regulating cell cycle progress and apoptosis, FKHR controls the rate of myotube fusion during myogenic differentiation.
Keywords: Akt/fusion/myoblast/rhabdomyosarcoma
Introduction
The FKHR (FOXO1a), AFX (FOXO4) and FKHR-L1 (FOXO3a) members of the FOXO family of Forkhead transcription factors each contain three conserved Akt/PKB phosphorylation sites, which regulate their activity in established cell lines (Brunet et al., 1999; Alvarez et al., 2001; Brownawell et al., 2001). However, it is likely that Akt is not the only regulator of Forkhead proteins, as these sites can also be phosphorylated by other serine/threonine kinases, including p70S6K or SGK (Blume-Jensen and Hunter, 2001; Brunet et al., 2001; Nakae et al., 2001), which are members of the AGC (protein kinase A, protein kinase G and protein kinase C) kinase subfamily.
In established cell lines, the Forkhead proteins FKHR, AFX and FKHRL1 have been reported to trigger apoptosis by regulating the transcription of genes such as FasL and Bim (Brunet et al., 1999; Dijkers et al., 2000a; Suhara et al., 2002). Similarly, overexpression studies have shown that AFX, FKHR and FKHRL1 induce cell cycle arrest by augmenting the expression of the cyclin-dependent kinase inhibitor p27kip1 (Dijkers et al., 2000b; Medema et al., 2000). Recent studies have also indicated that AFX and FKHRL1 can induce a G2/M checkpoint following oxidative stress (Furukawa-Hibi et al., 2002) and appear to regulate progression through M phase (Alvarez et al., 2001). All of these activities supposedly are held in check by Akt-mediated phosphorylation, which results in export of FOXO members to the cytoplasm (Tang et al., 1999; Nakae et al., 2000; Nakamura et al., 2000; Tomizawa et al., 2000), allowing cells to proliferate and/or survive. In contrast to the apoptosis induced by overexpression of FKHR, AFX and FKHRL1 in established cell lines, overexpression of FKHR in mouse thymocytes in vivo appears to promote cell survival (Leenders et al., 2000). Therefore, each Forkhead member may have cell type-specific functions.
Because FKHR is the target of the recurrent t(2;13) and t(1;13) in alveolar rhabdomyosarcoma (ARMS) (Galili et al., 1993; Davis et al., 1994; Barr, 2001), we wished to address the reason for this specificity and tested whether FKHR plays a role in normal muscle growth and differentiation. In addition, we assessed the role of the phosphatidylinositol 3-kinase (PI3K)/Akt pathway in regulating Fkhr activity in myogenic precursor cells. If reduction in FKHR activity were important for myogenic cell growth and differentiation, we reasoned that loss of one allele in ARMS might contribute to the tumorigenic phenotype. Here, we report that FKHR plays an essential role in myotube fusion, an important step during myogenic differentiation, and that the activity of FKHR is regulated independently of the PI3K/Akt pathway in primary myoblasts.
Results and discussion
FKHR undergoes nuclear translocation during myoblast differentiation
Fluorescent immunohistochemistry of endogenous Forkhead proteins (Fkhr, Afx and FkhrL1) revealed that low levels of Fkhr were present in the cytoplasm of primary mouse myoblasts proliferating in 20% serum, while Afx and FkhrL1 were expressed at high levels in the nucleus (Figure 1A). Transfer of the primary myoblasts to medium containing 2% serum induced terminal differentiation, which resulted in the rapid accumulation of high levels of Fkhr in the nucleus. In contrast, the levels of Afx and FkhrL1 were diminished and largely re-localized to the cytoplasm (Figure 1A). Therefore, the steady-state levels and localization of Fkhr and Afx/FkhrL1 are inversely regulated by signals that provoke differentiation of primary myoblasts.

Fig. 1. FKHR subcellular localization and transcriptional activity are regulated during muscle cell differentiation. (A) Immunofluorescence analysis of endogenous Fkhr, Afx and FkhrL1 expression was performed on proliferating (day 0, high serum) and differentiating (day 2, low serum) myoblasts. (B) Transient transfection assays were used to determine the FKHR transcriptional activity during myoblast differentiation, using the 6FBD luciferase reporter. Myoblasts were transfected with the pGL3-basic reporter (pGL3-reporter), the 6FBD reporter (6FBD) or the 6FBD reporter with AFX (6FBD + AFX) or wild-type FKHR (6FBD + FKHR). The activation of the 6FBD reporter in the presence of co-transfected FKHRL1 was indistinguishable from that of the 6FBD reporter alone (data not shown). In proliferating myoblasts, the reporter was also partially responsive to transfected AFX. Means of three independent experiments are shown for each transfection. Error bars show the variance at each data point. (C) Immunoblot analysis of Fkhr, Afx and FkhrL1 localization in proliferating (day 0) versus differentiating (day 2) myoblasts. Endogenous Fkhr, Afx and FkhrL1 were detected by western blotting. Cytoplasmic and matrix-bound actin served as a control.
We also performed similar experiments with freshly isolated mouse embryonic fibroblasts (MEFs), as well as with immortalized (NIH-3T3) and transformed cell lines (HEK 293 and HeLa). Fluorescent immunostaining of the endogenous Forkhead proteins revealed that, similarly to mouse primary myoblasts, MEFs showed accumulation of Fkhr in the nucleus following serum starvation, but there was no Afx or FkhrL1 present in these cells (see figure 1 of the Supplementary data, available at The EMBO Journal Online). Surprisingly, expression of the Forkhead proteins appeared grossly altered in the immortalized counterpart. NIH-3T3 cells did not express Fkhr, while a diffuse nuclear/cytoplasmic FkhrL1 was highly expressed in proliferating as well as in serum-starved cells (supplementary figure 1). HEK 293 and HeLa cell lines expressed variable amounts of all Forkhead proteins, which was quite different from that in primary myoblasts and fibroblasts. We do not know whether this expression pattern is the result of their transformed state since we did not study the expression pattern of Forkhead proteins in their primary counterparts (kidney cells and cervix epithelial cells, respectively). Taken together, our data suggest that established cell lines might have altered expression and regulation of Forkhead proteins as a result of their immortalized phenotype.
To evaluate the consequences of the changes in the level and localization on the transcriptional activity of Fkhr in primary myoblasts, we transiently transfected proliferating cells with an FKHR-responsive luciferase reporter bearing six concatemerized FKHR-binding sites (Furuyama et al., 2000). A striking 4- to 6-fold increase in luciferase activity was evident as early as 5 h following the exposure of the cells to differentiation medium (Figure 1B). Moreover, co-transfection of exogenous wild-type FKHR further augmented this transcriptional response but only in differentiating myoblasts. In proliferating myoblasts, overexpressed FKHR remained inactive and confined to the cytoplasm (Figures 1B and 2A). Overexpression of exogenous AFX and FKHRL1 in differentiating myoblasts recapitulated what was observed for the endogenous proteins (Figures 1A and 2B): both overexpressed proteins were exported from the nucleus efficiently (Figure 2B).

Fig. 2. Localization of FKHR, AFX and FKHRL1 in proliferating versus differentiating mouse primary myoblasts. (A) Immunofluorescence of myoblasts transfected with FLAG-tagged FKHR using a DNA stain (DAPI), a FLAG antibody (anti-TAG) or the FKHR antibody. (B) Immunofluorescence of myoblasts transfected with AFX or FKHRL1 using DAPI or indirect immunufluorescence with the respective antibodies. Transduced and untransduced cells are indicated.
To confirm that the nuclear translocation of Fkhr in differentiating myoblasts coincided with its binding to chromatin, we performed subcellular fractionation experiments with primary myoblasts before and after their transfer to differentiation medium (Figure 1C). Indeed, FKHR was only bound to chromatin after the induction of differentiation, while Afx and FkhrL1 were not associated with chromatin in these cells. In contrast, we found Afx to be partially bound to chromatin in proliferating myoblasts, and FkhrL1 was never bound (Figure 1C). In accordance with the subcellular fractionation experiments, co-transfection of exogenous AFX or FLHRL1 with the FKHR-responsive reporter elicited little (AFX) or no (FKHRL1) transcriptional response (Figure 1B). Therefore, Fkhr transcriptional activity is tightly and selectively associated with the induction of the myoblast differentiation program, which provokes the accumulation of Fkhr in the nucleus. Importantly, the activation of endogenous or overexpressed FKHR during differentiation had no effect on the apoptotic index of the myoblasts, which remained below 5% as measured by TUNEL assays (data not shown). Therefore, FKHR activation does not trigger apoptosis of primary myoblasts.
To determine whether the apparent increase in Fkhr levels in differentiating myoblasts was due to transcriptional upregulation of the gene, we compared the mRNA levels of the three Forkhead genes using specific primers and hot-stop quantitative RT–PCR (Uejima et al., 2000). The mRNA levels of Fkhr, Afx and FkhrL1 were equivalent and remained largely unchanged following the induction of differentiation (Figure 3). We therefore assessed whether the accumulation of Fkhr in the nucleus was due to an increase in the half-life of the protein. Primary myoblasts were transduced with a retroviral vector expressing His6-tagged FKHR, and a [35S]methionine pulse–chase analysis was performed to determine the half-life of the protein during exponential growth versus that during the second day of differentiation of the culture. The half-life of His6-FKHR in proliferating myoblasts was short (t1/2 = 15 min), but its stability increased at least 4- to 6-fold by the second day of differentiation (Figure 4B), strongly suggesting that this was the reason for its accumulation in the nucleus. Despite this finding, the steady-state level of Fkhr protein on immunoblots did not increase significantly during myoblast differentiation (Figure 4D). This discrepancy could be due to the lysis conditions used to perform immunoblot analyses. In particular, the nuclear form of FKHR might be poorly extracted under our conditions, because of its tight association with chromatin (Figure 1C). Alternatively, modified forms of FKHR might be less well detected by the FKHR antibody due to, for example, phosphorylation of FKHR, which would be in agreement with the fact that the migration of FKHR alters following differentiation (Figure 4D). Indeed, the FKHR band we detected in the half-life experiments in differentiating myoblasts (Figure 4B) co-migrated with the higher molecular weight form of FKHR seen in differentiating myoblasts by immunoblot analysis (Figure 4D).
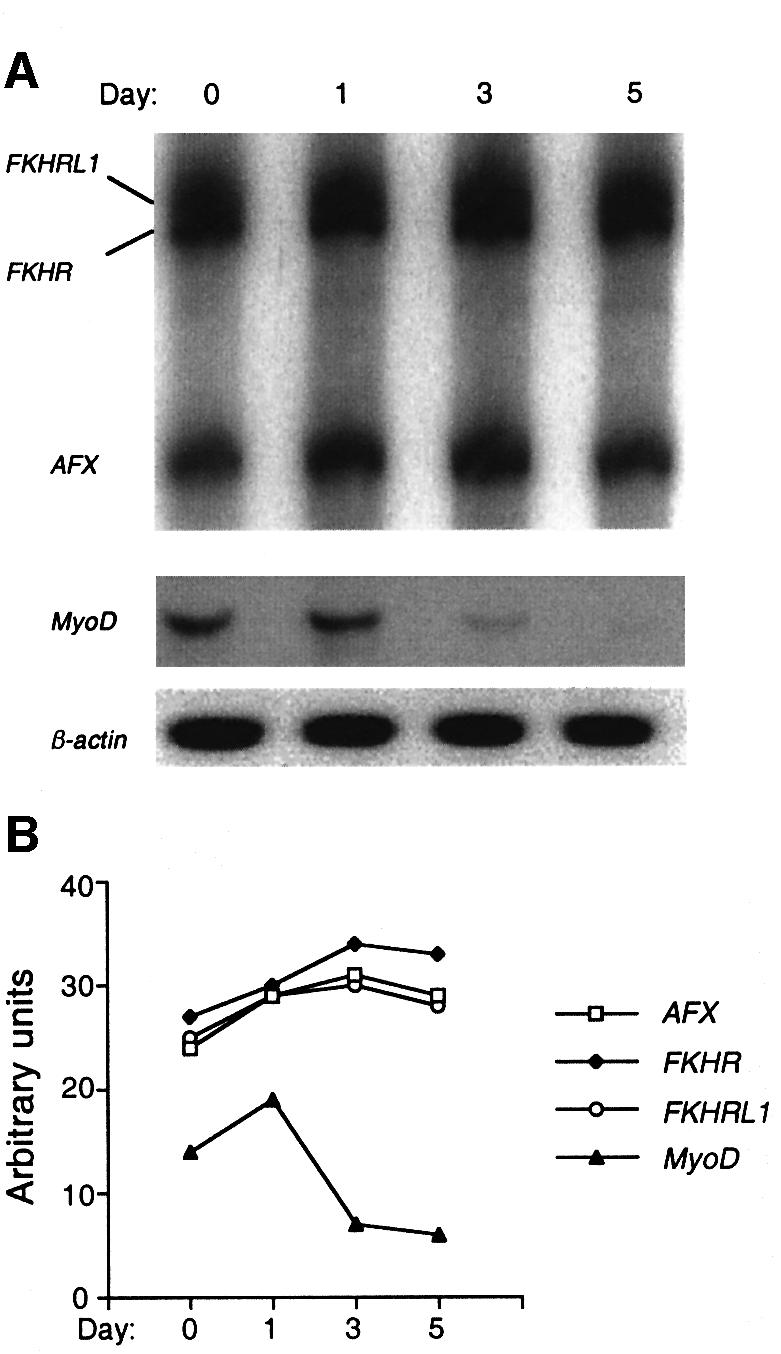
Fig. 3. Hot-stop quantitative RT–PCR analysis of Fkhr, FkhrL1 and Afx expression during myoblast differentiation. (A) Detection of Fkhr, FkhrL1 and Afx transcripts following RT–PCR using a 4% polyacrylamide gel. MyoD was used as a control to validate the sensitivity of this technique. β-actin was used as a loading control. (B) Relative amounts of Fkhr, FkhrL1, Afx and MyoD mRNA after normalization for the amount of β-actin mRNA. While the levels of MyoD transcripts dropped dramatically during myoblast differentiation, levels of Fkhr, FkhrL1 and Afx transcripts only increased marginally (10–20%).

Fig. 4. FKHR shuttling, half-life and phosphorylation are regulated during the differentiation of primary mouse myoblasts. (A) Inhibition of Fkhr nuclear export by Crm1 using leptomycin B (400 nM) in proliferating myoblasts. Cells were incubated for 2 h with leptomycin B, and FKHR was detected by immunofluorescence. (B) The half-life of a His6-tagged FKHR in proliferating (day 0) and differentiating (day 2) myoblasts was determined by immunoprecipitation analyses after a 15 min pulse labeling with [35S]methonine, followed by a 5–120 min chase with cold methionine. (C) Myoblasts treated with the LY294002 (PI3K IC50 = 1.5 µM) inhibitor did not show significant nuclear accumulation of Fkhr even at high doses. However, high doses of wortmannin (100 nM, PI3K IC50 = 5 nM) inhibited 50% of Fkhr nuclear export in growing myoblasts. The effects of the vehicle dimethylsulfoxide (DMSO) are shown as the negative control. (D) Western blot analysis of phosphorylated and non-phosphorylated Fkhr and Akt in proliferating (day 0) and differentiating (day 1 and day 2) myoblasts. Actin was used as a loading control. (E) The phosphorylation status of a His6-tagged FKHR in proliferating (day 0) and differentiating (day 2) myoblasts was determined by immunoprecipitation after a 2 h labeling with [32P]orthophosphate.
FKHR shuttles between the cytoplasm and nucleus in proliferating myoblasts
The translocation of Fkhr to the nucleus in differentiating myoblasts suggested that it normally resides in the cytoplasm or it continuously shuttles between the nucleus and cytoplasm, with the equilibrium of this reaction far to the cytoplasmic side. We could distinguish between these possibilities by culturing the proliferating cells in the presence of leptomycin B (Ossareh-Nazari et al., 1997), a specific inhibitor of the the nuclear export transporter Crm1, which caused rapid accumulation of FKHR in the nucleus. This suggested that the protein is exported continuously from the nucleus to the cytoplasm in proliferating myoblasts in a Crm1-dependent manner.
In overexpression studies using established cell lines, FKHR activity is subject to negative regulation by the PI3K/Akt pathway whereby phosphorylated FKHR is exported to the cytoplasm (Biggs et al., 1999; Brunet et al., 1999; Tang et al., 1999). If this scenario applies to proliferating myoblasts, inhibition of PI3K enzyme activity by the specific inhibitor LY294002 (PI3K IC50 = 1.5 µM) (Collado et al., 2000) should result in Fkhr dephosphorylation and nuclear accumulation. However, addition of the LY294002 inhibitor to proliferating myoblasts had little effect on Fkhr localization (Figure 4C), and we observed a mere 20% nuclear accumulation of FKHR at a concentration of 100 µM, which is 66-fold above the IC50 of LY294002 for PI3K (Vlahos et al., 1994). This suggested that a PI3K-initiated pathway does not control Fkhr nuclear translocation. To address this issue further, we repeated the experiment using the wider spectrum serine/threonine kinase inhibitor wortmannin (PI3K IC50 = 5 nM). Only at doses of wortmannin exceeding those required for inhibiting PI3K by 10- to 50-fold (Virbasius et al., 1996) did we observe a 50% accumulation of Fkhr in the nucleus (Figure 4C). These data suggested that an as yet unknown kinase, inhibited by a high dose of wortmannin but poorly responsive to LY294002, regulates nuclear translocation of Fkhr in proliferating myoblasts.
The shift in migration of FKHR to a slower migrating form in differentiating myoblasts (Figure 4D) suggested that FKHR phosphorylation might be involved in the accumulation, localization and activation of Fkhr transcriptional activity. Indeed, immunoprecipitation of FKHR from [32P]orthophosphate-labeled cells confirmed that the protein was not phosphorylated in proliferating myoblasts but became phosphorylated in differentiating cells (Figure 4E). To address whether phosphorylation of FKHR on its known Akt kinase sites (Thr24/Ser256/Ser319) (Rena et al., 1999) played any role in its nuclear localization and transcriptional activation during myogenic differentiation, myoblasts were transduced with a retroviral vector harboring a mutant form of FKHR (FKHR-3A), in which all three Akt phosphorylation sites were mutated to alanines (Rena et al., 1999). Fluorescent immunostaining showed that the nuclear localization of FKHR-3A in differentiation medium was unaffected (Figure 5A), demonstrating that phosphorylation of Fkhr at these sites is not essential for nuclear localization.

Fig. 5. Effects of FKHR and FKHR mutants on primary myoblast fusion. (A) Fluorescent immunohistochemistry of FKHR in proliferating (day 0) versus differentiating (day 2) myoblasts expressing FKHR, FKHR-3A and FKHRΔTA using an FKHR antibody. (B) Morphology of proliferating (day 0) and differentiating (day 2) myoblasts expressing FKHR, FKHR-3A or FKHRΔTA. Empty vector, DN-Akt- and PTEN-transduced myoblasts were morphologically indistinguishable from those expressing FKHR (data not shown). (C) The number of nuclei per cell was counted at day 0 (black column) and day 2 (white column) for myoblasts expressing IRES–GFP, FKHR-3A and FKHRΔTA mutants. Means of at least three independent fields are given for each construct. Error bars show the variance at each data point. The number of nuclei for the FKHR-3A mutant is an underestimate, since these cultures often consisted of large syncytia that covered the entire culture dish. (D) The percentage of apoptotic cells is given following PI staining and FACS analysis on myoblasts expressing various FKHR constructs at day 0 and day 2 of differentiation. ND: not determined, since this type of analysis could not be performed with differentiated myoblasts expressing wild-type FKHR or dominant active FKHR (FKHR-3A), because the size of the cells is too large to pass through the FACS.
FKHR regulation is independent of PI3K/Akt activity in primary myoblasts
To exclude further the PI3K pathway in the regulation of FKHR during differentiation in primary myoblasts, we also checked the activation status of Akt, a downstream effector of PI3K. Immunoblotting with an Fkhr S256 phospho-specific antibody during myoblast differentiation confirmed the increase in Ser256 phosphorylated Fkhr (Figures 1C and 4D), a known substrate site of Akt (Guo et al., 1999). However, this increase in phosphorylated Ser256 of Fkhr did not correlate with the activation of Akt during myogenic differentiation, because the level of phosphorylation of Akt on Thr308 and Ser473 (Bellacosa et al., 1998), both markers detecting activated Akt, remained level in proliferating and differentiating myoblasts when we probed the same blots with Akt Thr308 and Ser473 phospho-specific antisera (Figure 4D). Collectively, these data suggested that not PI3K/Akt but an independent pathway regulates the phosphorylation and subcellular localization of FKHR.
Fkhr regulates the rate of fusion of differentiating primary myoblasts
To evaluate the functional relevance of Fkhr phosphorylation during later steps of myogenic differentiation, proliferating primary myoblasts were transduced with the MSCV-internal ribosome entry site (IRES)–green fluorescent protein (GFP) retroviral vector or with this vector also encoding wild-type FKHR or the FKHR-3A mutant. To evaluate also the consequences of loss of function of Fkhr on myogenic differentiation, we transduced myoblasts with a retroviral vector expressing a dominant-negative FKHR mutant lacking the transactivation domain (FKHRΔTA). GFP-positive myoblasts were sorted using a fluorescence-activated cell sorter (FACS), expanded in culture, and then differentiated in 2% serum. We confirmed that all forms of FKHR were localized to the cytoplasm in proliferating myoblasts, and underwent nuclear translocation within 48 h following transfer to low serum (Figure 5A). Compared with vector-transduced myoblasts, overexpression of wild-type FKHR (Figure 5A and B) had no effect on the rate of myoblast fusion following the shift to differentiation medium. However, both FKHR mutants displayed dramatic and opposing effects on myotube formation. Expression of the FKHR-3A mutant markedly accelerated myotube fusion, and after 2 days these cultures consisted of several syncytia covering large areas of the culture dish (Figure 5B and C). Conversely, overexpression of the FKHRΔTA mutant completely impaired myoblast fusion (Figure 5B and C), and we never observed any multinucleated myotubes, even after leaving the cells in differentiation medium for up to 8 days. Counting the number of nuclei per cell after 2 days of differentiation demonstrated that GFP-only or FKHR-virus-transduced myotubes typically contained on average 4–6 nuclei, while FKHR-3A-expressing myotubes averaged >220 nuclei. FKHRΔTA-expressing myoblasts contained only one nucleus per cell (Figure 5C). We excluded the possibility that this lack of nuclei was due to excessive apoptosis, because staining these cells with propidium iodide and analysis of their DNA content by FACS (Figure 5D) showed no increase in the number of apoptotic cells. This demonstrated that reducing FKHR transcriptional activity (see below) inhibits myoblast fusion without affecting cell survival. We also transduced myoblasts with retroviral vectors encoding the FKHR-Ser256Ala, FKHR-Thr24Ala, FKHR-Ser319Ala, FKHR-Thr24Ala/Ser256Ala, FKHR-Ser256Ala/Ser319Ala and FKHR-Thr24Ala/Ser319Ala mutants. Differentiation assays revealed that expression of the dual Thr24Ala/Ser319Ala mutant did not affect the rate of myotube fusion during differentiation. Increased fusion was only observed in cells expressing the Ser256Ala mutant alone or in combination with the Thr24Ala or the Ser319Ala mutation (Figures 5C and 6), but the effect was clearly not as dramatic as with cells expressing the FKHR-3A mutant (Figure 6). These findings support the notion that phosphorylation of Ser256 is a pre-requisite for FKHR inactivation (Nakae et al., 1999).

Fig. 6. Summary of myoblast phenotypes generated by expression of different FKHR mutants. Top left panels show the differentiation of wild-type, FKHR mutants, PTEN- and DN-Akt-expressing myoblasts at 0 and 24 h of differentiation. No difference in the extent of fusion between any of these populations was observed. Lower left panels show the temporal expression of early (MyoD), intermediate (myogenin) and late (myosin heavy chain) myogenic markers during 2 days of differentiation, which was the same for all samples. Right panels show the complete range of fusion phenotypes generated by the different FKHR mutants.
The fact that overexpression of wild-type FKHR does not phenocopy the effects of FKHR-3A suggests that the inactivating kinase effectively neutralizes the increased levels of nuclear FKHR.
To disprove further the involvement of the PI3K/Akt pathway in FKHR regulation, we overexpressed PTEN or a dominant-negative form of Akt (DN-Akt) in primary myoblasts by retroviral transduction. Overexpression of PTEN or DN-Akt had no noticeable effect on the rate of myoblast fusion and also did not affect the localization or the transcriptional activity of endogenous Fkhr (Figures 6 and 7B, and data not shown). This was despite the fact that DN-Akt was expressed at high levels (Figure 7B, lower panel, αAKT) and effectively suppressed the kinase activity of endogenous Akt, as measured by the reduction of the levels of Ser9 phosphoryated GSK3-β (Figure 7B), a known Akt substrate (Cross et al., 1995). Thus, a Fkhr-dependent, but PI3K/Akt-independent, pathway regulates the fusion of myoblasts.

Fig. 7. FKHR activity during the differentiation of primary mouse myoblasts. (A) Luciferase reporter-based analyses of FKHR transcriptional activity of myoblasts transfected with pGL3, 6FBD, 6FBD + FKHR, 6FBD + FKHR-3A and 6FBD + FKHRΔTA during 4 days of differentiation. Co-expression of PTEN or DN-Akt produced luciferase responses that were identical to those of myoblasts transfected with 6FBD alone. Means of at least three independent experiments are given for each transfection. Error bars show the variance at each data point. (B) Western blot analyses of primary myoblasts transduced with IRES-GFP, FKHR-3A, FKHRΔTA and DN-Akt retroviral vectors, which have no effect on the levels and timing of expression of early (MyoD), intermediate (myogenin) and late (myosin) markers during muscle cell differentiation. Overexpression of DN-Akt was confirmed using an Akt antibody. The efficacy of DN-Akt in repressing known targets of Akt was assessed by determining the level of phospho-GSK-3b Ser9, which was significantly underphosphorylated in myoblasts overexpressing DN-Akt. GFP levels were used as a loading control. FKHR-3A and FKHRΔTA were detected with an antibody to the N-terminal His6 tag, and DN-Akt with an antibody to the N-terminal HA tag. The expression patterns of these markers in PTEN- and FKHR-transduced myoblasts were indistinguishable from those transduced with the IRES-GFP vector (data not shown). In all lanes, 50 µg of protein lysate was loaded, but 100 µg of FKHR-3A was required for its detection with the His6 antibody. Exposure times for the different samples detected by the same antibody were identical. The exposure time to detect FKHR-3A was 40 times longer than that to detect the TAG of FKHRΔTA and DN-Akt.
Proliferating myoblasts transiently co-transfected with the Fkhr-responsive luciferase reporter and expressing FKHR-3A displayed a 20% increase in luciferase activity compared with myoblasts co-transfected with the reporter and overexpressing wild-type FKHR (Figure 7A). Thus, despite the mainly cytoplasmic localization of FKHR-3A in proliferating myoblasts, the small amount that resides in the nucleus is sufficient to provoke an increase in the transcription of the reporter. Proliferating myoblasts expressing FKHR-3A also displayed obvious morphological changes (Figure 5B), suggesting that Fkhr activity can affect myoblast morphology even in the absence of signals that initiate differentiation. In contrast, overexpression of the FKHRΔTA mutant reduced the response of the reporter by approximately half (Figure 7A), although this effect was only observed in differentiating myoblasts. Therefore, a relatively modest change in Fkhr transcriptional activity appears sufficient to block myoblast fusion. We thus speculate that the hemizygosity of ARMS cells for the FKHR locus reduces their level of FKHR expression, which in turn might contribute to the non-fusion phenotype of these tumor cells. We currently are testing this hypothesis.
To determine which aspect of the myogenic differentiation program (Arnold and Winter, 1998) was disrupted by the FKHR-3A and FKHRΔTA mutants, we performed immunoblots at different time points during differentiation and analyzed the temporal expression of early (MyoD), intermediate (myogenin) and late (myosin heavy chain) myogenic markers (Figures 6 and 7B). Surprisingly, the overexpression of these mutants did not affect the temporal expression of these markers, and their expression was also not affected in myoblasts engineered to overexpress DN-Akt (Figure 7B). As expected, western blot analysis also revealed that the FKHR-3A mutant protein did not undergo the post-translational modification that produces a slower migrating band for wild-type Fkhr on SDS–PAGE during myoblast differentiation (Figure 7B). This suggested that the slower migrating form of Fkhr is due to phosphorylation of Thr24/Ser256/Ser319, and that these modifications downregulate FKHR activity (Figure 7B, lower panel, α-TAG). At present, it is unclear how the phosphorylation of these sites represses Fkhr activity. It is unlikely to increase the half-life of the protein, as the level of FKHR-3A that can be overexpressed in these cells is only one-tenth (Figure 7B) that of the level of overexpressed wild-type FKHR (as detected with an antibody for the epitope tag). It is thus conceivable that Thr24/Ser256/Ser319 phosphorylation directly affects the DNA-binding activity of FKHR and/or its interaction with coactivators, two possibilities that remain to be tested. It is also possible that the FKHR antibody has a weak affinity for this mutated form of FKHR and is therefore less well recognized. It needs to be stressed that expression of FKHR-3A in proliferating myoblasts does not cause cells to fuse (Figure 5A) and thus acts downstream of signals that initiate differentiation. Therefore, Fkhr does not affect the progress of myogenic differentiation per se but specifically regulates the rate of myotube fusion. To our knowledge, Fkhr is the first transcription factor to be implicated in this process in vertebrates.
To determine the effects of FKHR on gene expression during myogenic differentiation, we performed microarray analysis of primary myoblasts expressing the FKHR-3A mutant and compared this with myoblasts expressing only GFP (MSCV-IRES-GFP). Analysis of RNA samples prepared from differentiating (48 h) myoblasts showed that the most significantly upregulated genes were those involved in cell fusion (prosaposin, frizzled-4, slow-myosin HC) and those that regulate extracellular matrix remodeling (procollagen type V and XVIII, fibulin-2, tenascin-C, ankyrin-3; Table I). The overexpression of these latter genes is not surprising since myoblasts undergo massive extracellular matrix reorganization in order to accomplish fusion. The identification of the prosaposin, frizzled-4 and slow myosin HC genes as possible targets is particularly relevant to the process of myotube fusion. Overexpression of prosaposin in rat L6 myoblasts has recently been demonstrated to induce myotube fusion, an effect that could be eliminated by adding neutralizing anti-prosaposin antibody to the cultures (Rende et al., 2001). Frizzled-4 knockout mice lack skeletal muscle in the lower esophagus, which was attributed to a fusion defect (Wang et al., 2001). We further confirmed the upregulation of these genes in myoblasts expressing FKHR-3A and their downregulation in myoblasts expressing FKHRΔTA using quantitative SYBR green real-time Taqman assays (Table I). Furthermore, sequence database analysis revealed that the promoter regions of these genes all contain multiple copies of the consensus FKHR-binding site (Furuyama et al., 2000), suggesting that these genes might be direct transcriptional targets of FKHR. In agreement with the induced expression of pro-collagens in FKHR-3A-expressing myoblasts, they acquired the ability to attach to and grow on non-collagen-coated plates, whereas myoblasts transduced with empty vector or FKHRΔTA did not (data not shown). Notably, no genes were significantly downregulated in FKHR-3A-expressing cells, indicating that this mutant indeed behaves as a dominant active transcription factor. These data further underscore the concept that Fkhr plays an essential role in regulating the rate of myotube fusion.
Table I. FKHR activation augments the expresson of genes involved in myotube fusion.
| Gene product | Function | 3A versus IRES–GFP | 3A versus IRES–GFP | ΔTA versus IRES–GFP | FKHR- |
|---|---|---|---|---|---|
| Affymetrix | Real-time PCR | Real-time PCR | binding sites | ||
| Slow-myosin HC β | Sarcomeric protein | 2.8× | ND | ND | NA |
| Tenascin-C | Extracellular matrix | 3.0× | 1.7× | 0.1× | 2 |
| Procollagen α2 (type V) | Extracellular matrix | 3.1× | ND | ND | 3 |
| Prosaposin | Myotrophic factor | 3.4× | 3.4× | 0.2× | 3 |
| Frizzled-4 | Skeletal muscle fate | 3.6× | 7.7× | 0.6× | 6 |
| Ankyrin-3 | Membrane protein | 4.0× | 1.5× | 0.6× | 9 |
| Fibulin-2 | Extracellular matrix | 4.0× | ND | ND | NA |
| Procollagen α1 (type XVIII) | Extracellular matrix | 4.6× | ND | ND | NA |
Genes displaying the largest transcriptional variation during differentiation in FKHR-3A-expressing primary myoblasts (day 2) compared with myoblasts transduced with vector only. Using the mouse U74 genechip, all genes identified by Affymetrix analysis were overexpressed. Most genes (eight out of 16) are involved in extracellular matrix remodeling, fusion and muscle differentiation, demonstrating the essential role played by FKHR in muscle terminal differentiation. Induction or repression of these genes was confirmed using real-time RT–PCR in myoblasts expressing FKHR-3A (3A) or FKHRΔTA (ΔTA). Expression of target genes was normalized for GAPDH expression and displayed as fold change relative to myoblasts transduced with empty vector (IRES–GFP). The number of FKHR-binding sites (TTGTTTAC, forward and/or reverse) in the 10 kb upstream of the ATG is given for each gene. Sequence data were downloaded from the ENSEMBL database. Microarray analysis and real-time RT–PCR were performed using standard protocols. ND, not determined; NA, not available.
Conclusions
Our results suggest a novel function and regulation of Fkhr in myoblasts. In contrast to its accepted role in established cell lines using overexpression studies, Fkhr activation does not stimulate apoptosis in primary myoblasts and its activity is not subject to regulation by Akt. Rather, Fkhr plays a critical role in regulating myoblast fusion, indicating that Fkhr functions are more diverse than previously suggested and may well depend on cell context.
Our studies revealed that FKHR activity is minimally regulated at two different levels by two independent pathways (Figure 8). First, by regulation of its Crm1-mediated export from the nucleus, a process that is independent of phosphorylation of the three Akt sites in FKHR, and secondly by phosphorylation of the Akt sites by an as yet unknown kinase in the nucleus, leading to inactivation of transcriptionally active FKHR.

Fig. 8. Model for regulation of FKHR function in primary myoblasts. Two regulatory events control the localization and function of FKHR prior to (nuclear export pathway) and during (fusion control pathway) primary myoblast differentiation. Once activated, FKHR activates genes that are involved in extracellular matrix remodeling and myotube fusion.
More in-depth characterization of Fkhr targets during muscle differentiation may provide insights into the molecular events that govern muscle cell fusion, and may also reveal how disruption of this pathway could contribute to ARMS pathogenesis.
Materials and methods
Mouse primary myoblast isolation and culture, and culture of other cell lines
Primary myoblasts were isolated from 2- to 5-day-old C57Bl6/J mice using a modification of the procedure described by Rando and Blau (1994). In brief, cells were isolated from dissected muscle by dispase/collagenase P (Gibco-BRL) treatment, and were pre-plated onto tissue culture plastic twice for 1 h to purge the cell suspension of fibroblasts. This 1 h purging was repeated during the following three passages. After growth in F10 medium supplemented with basic fibroblast growth factor (b-FGF) and 20% fetal calf serum on collagen-treated (BD Bioscences) dishes, the myogenic cells were harvested after a 5 min incubation in varsene (Gibco-BRL). Cell pools prepared in this fashion typically consisted of 99% myogenic cells as determined by their expression of MyoD and desmin (data not shown). The cells were expanded and differentiated as described (Rando and Blau, 1994). Myoblasts were never frozen or cultured for more than 40 days or seven passages after their date of isolation. MEFs, NIH-3T3 cells, HEK 293 and HeLa cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM), 10% fetal bovine serum (FBS) following standard procedures.
Immunofluorescence and protein analysis
Immunofluorescence, western blotting, [35S]methionine pulse–chase (15 min) labeling and cell fractionation experiments were performed using standard protocols (Ausubel et al., 2001). Quantitative PCR for Afx, Fkhr and FkhrL1 was performed using the hot-stop technique (Uejima et al., 2000). To avoid any discrepancies in PCR efficiency introduced by the use of different primer sets, we used universal primers amplifying the highly conserved forkhead DNA-binding domain (FKH/F 5′-sca.gat. cta.cga.rtg.gat.ggt-3′; FKH/R 5′-atg.kwm.tcc.atg.tcr.cab.tcc.a-3′). Size polymorphism was used to discriminate between the products of the three genes (1086 bp for AFX, 1299 bp for FKHR and 1338 bp for FKHRL1). PCR products were resolved by non-denaturing PAGE (Ausubel et al., 2001). MyoD (MyoD/F 5′-agg.ctct.gct.gcg.cga.cc-3′; MyoD/R 5′-tgc.agt.cga.tct.ctc.aaa.gca.cc-3′) and β-actin (β-actin/F 5′-cgt.tga.cat.ccg. taa.aga.cct.cta-3′; β-actin/R 5′-taa.aac.gca.gct.cag.taa.cag.tcc.g-3′) were used as PCR controls. Phospho-FKHR (Ser256), FKHR, phospho-GSK-3β (Ser9), phospho-Akt (Thr308), phospho-Akt (Ser473) and Akt antibodies were purchased from Cell Signaling Technology, FKHRL1 antibody from Upstate biotechnology, MyoD and myogenin antibodies from BD PharMingen, and actin antibody from Sigma Bioscience. AFX and myosin heavy-chain antibodies were gifts from B.Burgering and D.Fischman, respectively.
Luciferase assays
The FKHR luciferase reporter (6FBD) was generated by inserting six concatemerized FKHR-binding sites upstream of the minimal promoter of the pGL3-basic firefly luciferase vector (Furuyama et al., 2000) (Promega). Myoblasts were transiently transfected with the 6FBD reporter using FuGene6 following the Gibco-BRL protocol. This reporter was responsive to activated FKHR, minimally responsive to AFX and not responsive to FKHRL1 (Figure 1B). Luciferase detection was performed using the Luciferase Assay System from Promega following the manufacturer’s recommendations. An APR-SEAP reporter was co-transfected (100 ng per transfection) to normalize for transfection efficiency, and SEAP activity was determined as previously described (Berger et al., 1988). In co-transfection experiments, the molar amount of reporter was 10-fold (33 fmol) lower than the molar amount of FKHR wild-type or mutant vectors. FKHR and FKHR mutants were cloned into the pcDNA3 expression vector (Invitrogen).
Virus production, myoblast transduction and cell sorting
E.Vanin, of St Jude Children’s Research Hospital, provided the MSCV-IRES-GFP vector. MSCV-PTEN-IRES-GFP and MSCV-dominant-negative Akt-IRES-GFP were gifts from S.Baker and J.Cleveland, respectively. DN-Akt is a kinase-dead (K179M) mutant, which competes with wild-type Akt when overexpressed. All FKHR point mutations were introduced using standard molecular biology protocols (Ausubel et al., 2001). For virus production, the retroviral vector constructs were transiently transfected into Phoenix-Eco cells (Swift et al., 1999). Myoblasts were transduced using standard techniques (Ausubel et al., 2001), and 2 days after transduction cells were FACS sorted for GFP expression. To reduce cell clumping during FACS sorting, myoblasts were harvested in F10 medium supplemented with 10% (w/v) collagen. While varsene (Gibco-BRL) was used to detach myoblasts from plates for passaging; 10 µl of trypsin-EDTA (Gibco-BRL) was added to harvest the more avidly attaching FKHR-3A-expressing myoblasts.
Gene expression profiling analysis
RNA was extracted from primary myoblasts using the Trizol reagent, and the RNA integrity was assessed by using an Agilent 2100 Bioanalyzer (Agilent, Palo Alto, CA). cDNA was synthesized using a T-7 linked oligo(dT) primer, and cRNA was then synthesized with biotinylated UTP and CTP. The labeled RNA was then fragmented and hybridized to U74 oligonucleotide arrays (Affymetrix Incorporated, Santa Clara, CA) according to Affymetrix protocols. Arrays were scanned using a laser confocal scanner (Agilent), and the expression value for each gene was calculated using Affymetrix Microarray software v.4.0. The average intensity difference values were normalized across the sample set.
Real-time quantitative RT–PCR
For real-time quantitative RT–PCR, 2 µg of total RNA was reverse transcribed using Superscript II (Life Technologies) and a mix of random hexamer primers and oligo(dT). The SYBR-green master mix was used to detect accumulation of PCR product during cycling in the SDS9700 apparatus (Applied Biosystems). Expression of target genes was normalized for GAPDH expression and displayed as fold change relative to myoblasts transduced with empty vector (MSCV-IRES-GFP). Sequences of primers are available from the authors upon request.
Supplementary data
Supplementary data are available at The EMBO Journal Online.
Acknowledgments
Acknowledgements
We thank Boudewijn Burgering for AFX antibody and AFX and FKHRL1 expression plasmids, Frédérique Zindy and Martine Roussel for providing MEFs, Suzie Baker for MSCV-PTEN-IRES-GFP, and John Cleveland for MSCV-DN-Akt-IRES-GFP. We are also indebted to John Cleveland for critical reading of the manuscript. This research was supported in part by NCI grant CA-71907, the cancer center (CORE) support grant CA-21765, and by the American Lebanese Syrian Associated Charities (ALSAC) of St Jude Children’s Research Hospital. P.B. is a fellow of the Van Vleet foundation of Memphis.
References
- Alvarez B., Martinez,A.C., Burgering,B.M. and Carrera,A.C. (2001) Forkhead transcription factors contribute to execution of the mitotic programme in mammals. Nature, 413, 744–747. [DOI] [PubMed] [Google Scholar]
- Arnold H.H. and Winter,B. (1998) Muscle differentiation: more complexity to the network of myogenic regulators. Curr. Opin. Genet. Dev., 8, 539–544. [DOI] [PubMed] [Google Scholar]
- Ausubel F.M., Brent,R., Kingston,R.E., Moore,D.D., Smith,J.A. and Struhl,K. (2001) Current Protocols in Molecular Biology. John Wiley and Sons, Harvard.
- Barr F.G. (2001) Gene fusions involving PAX and FOX family members in alveolar rhabdomyosarcoma. Oncogene, 20, 5736–5746. [DOI] [PubMed] [Google Scholar]
- Bellacosa A., Chan,T.O., Ahmed,N.N., Datta,K., Malstrom,S., Stokoe,D., McCormick,F., Feng,J. and Tsichlis,P. (1998) Akt activation by growth factors is a multiple-step process: the role of the PH domain. Oncogene, 17, 313–325. [DOI] [PubMed] [Google Scholar]
- Berger J., Hauber,J., Hauber,R., Geiger,R. and Cullen,B.R. (1988) Secreted placental alkaline phosphatase: a powerful new quantitative indicator of gene expression in eukaryotic cells. Gene, 66, 1–10. [DOI] [PubMed] [Google Scholar]
- Biggs W.H. 3rd, Meisenhelder,J., Hunter,T., Cavenee,W.K. and Arden,K.C. (1999) Protein kinase B/Akt-mediated phosphorylation promotes nuclear exclusion of the winged helix transcription factor FKHR1. Proc. Natl Acad. Sci. USA, 96, 7421–7426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blume-Jensen P. and Hunter,T. (2001) Oncogenic kinase signalling. Nature, 411, 355–365. [DOI] [PubMed] [Google Scholar]
- Brownawell A.M., Kops,G.J., Macara,I.G. and Burgering,B.M. (2001) Inhibition of nuclear import by protein kinase B (Akt) regulates the subcellular distribution and activity of the forkhead transcription factor AFX. Mol. Cell. Biol., 21, 3534–3546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunet A. et al. (1999) Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell, 96, 857–868. [DOI] [PubMed] [Google Scholar]
- Brunet A., Park,J., Tran,H., Hu,L.S., Hemmings,B.A. and Greenberg,M.E. (2001) Protein kinase SGK mediates survival signals by phosphorylating the forkhead transcription factor FKHRL1 (FOXO3a). Mol. Cell. Biol., 21, 952–965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collado M. et al. (2000) Inhibition of the phosphoinositide 3-kinase pathway induces a senescence-like arrest mediated by p27Kip1. J. Biol. Chem., 275, 21960–21968. [DOI] [PubMed] [Google Scholar]
- Cross D.A., Alessi,D.R., Cohen,P., Andjelkovich,M. and Hemmings, B.A. (1995) Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature, 378, 785–789. [DOI] [PubMed] [Google Scholar]
- Davis R.J., D’Cruz,C.M., Lovell,M.A., Biegel,J.A. and Barr,F.G. (1994) Fusion of PAX7 to FKHR by the variant t(1;13)(p36;q14) translocation in alveolar rhabdomyosarcoma. Cancer Res., 54, 2869–2872. [PubMed] [Google Scholar]
- Dijkers P.F., Medema,R.H., Lammers,J.W., Koenderman,L. and Coffer,P.J. (2000a) Expression of the pro-apoptotic Bcl-2 family member Bim is regulated by the forkhead transcription factor FKHR-L1. Curr. Biol., 10, 1201–1204. [DOI] [PubMed] [Google Scholar]
- Dijkers P.F. et al. (2000b) Forkhead transcription factor FKHR-L1 modulates cytokine-dependent transcriptional regulation of p27(KIP1). Mol. Cell. Biol., 20, 9138–9148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furukawa-Hibi Y., Yoshida-Araki,K., Ohta,T., Ikeda,K. and Motoyama,N. (2002) FOXO Forkhead transcription factors induce G2–M checkpoint in response to oxidative stress. J. Biol. Chem., 277, 26729–26732. [DOI] [PubMed] [Google Scholar]
- Furuyama T., Nakazawa,T., Nakano,I. and Mori,N. (2000) Identification of the differential distribution patterns of mRNAs and consensus binding sequences for mouse DAF-16 homologues. Biochem. J., 349, 629–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galili N., Davis,R.J., Fredericks,W.J., Mukhopadhyay,S., Rauscher, F.J.D., Emanuel,B.S., Rovera,G. and Barr,F.G. (1993) Fusion of a fork head domain gene to PAX3 in the solid tumour alveolar rhabdomyosarcoma [published erratum appears in Nat. Genet. 1994 Feb;6(2):214]. Nat. Genet., 5, 230–235. [DOI] [PubMed] [Google Scholar]
- Guo S., Rena,G., Cichy,S., He,X., Cohen,P. and Unterman,T. (1999) Phosphorylation of serine 256 by protein kinase B disrupts transactivation by FKHR and mediates effects of insulin on insulin-like growth factor-binding protein-1 promoter activity through a conserved insulin response sequence. J. Biol. Chem., 274, 17184–17192. [DOI] [PubMed] [Google Scholar]
- Leenders H., Whiffield,S., Benoist,C. and Mathis,D. (2000) Role of the forkhead transcription family member, FKHR, in thymocyte differentiation. Eur. J. Immunol., 30, 2980–2990. [DOI] [PubMed] [Google Scholar]
- Medema R.H., Kops,G.J., Bos,J.L. and Burgering,B.M. (2000) AFX-like Forkhead transcription factors mediate cell-cycle regulation by Ras and PKB through p27kip1. Nature, 404, 782–787. [DOI] [PubMed] [Google Scholar]
- Nakae J., Park,B.C. and Accili,D. (1999) Insulin stimulates phosphorylation of the forkhead transcription factor FKHR on serine 253 through a wortmannin-sensitive pathway. J. Biol. Chem., 274, 15982–15985. [DOI] [PubMed] [Google Scholar]
- Nakae J., Barr,V. and Accili,D. (2000) Differential regulation of gene expression by insulin and IGF-1 receptors correlates with phosphorylation of a single amino acid residue in the forkhead transcription factor FKHR. EMBO J., 19, 989–996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakae J., Kitamura,T., Ogawa,W., Kasuga,M. and Accili,D. (2001) Insulin regulation of gene expression through the forkhead transcription factor Foxo1 (Fkhr) requires kinases distinct from Akt. Biochemistry, 40, 11768–11776. [DOI] [PubMed] [Google Scholar]
- Nakamura N., Ramaswamy,S., Vazquez,F., Signoretti,S., Loda,M. and Sellers,W.R. (2000) Forkhead transcription factors are critical effectors of cell death and cell cycle arrest downstream of PTEN. Mol. Cell. Biol., 20, 8969–8982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ossareh-Nazari B., Bachelerie,F. and Dargemont,C. (1997) Evidence for a role of CRM1 in signal-mediated nuclear protein export. Science, 278, 141–144. [DOI] [PubMed] [Google Scholar]
- Rando T.A. and Blau,H.M. (1994) Primary mouse myoblast purification, characterization and transplantation for cell-mediated gene therapy. J. Cell Biol., 125, 1275–1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rena G., Guo,S., Cichy,S.C., Unterman,T.G. and Cohen,P. (1999) Phosphorylation of the transcription factor forkhead family member FKHR by protein kinase B. J. Biol. Chem., 274, 17179–17183. [DOI] [PubMed] [Google Scholar]
- Rende M. et al. (2001) Prosaposin is immunolocalized to muscle and prosaptides promote myoblast fusion and attenuate loss of muscle mass after nerve injury. Muscle Nerve, 24, 799–808. [DOI] [PubMed] [Google Scholar]
- Suhara T., Kim,H.S., Kirshenbaum,L.A. and Walsh,K. (2002) Suppression of Akt signaling induces Fas ligand expression: involvement of caspase and Jun kinase activation in Akt-mediated Fas ligand regulation. Mol. Cell. Biol., 22, 680–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swift S., Lorens,J., Achacoso,P. and Nolan,G.P. (1999) Rapid production of retroviruses for efficient gene delivery to mammalian cells using 293T cell-based systems. In Coligan,J.E., Kruisbeek,A.M., Margulies,D.H., Shevach,E.M. and Strober,W. (eds), Current Protocols in Immunology. John Wiley and Sons, Harvard, Vol. 2, p. 10.28. [DOI] [PubMed]
- Tang E.D., Nunez,G., Barr,F.G. and Guan,K.L. (1999) Negative regulation of the forkhead transcription factor FKHR by Akt. J. Biol. Chem., 274, 16741–16746. [DOI] [PubMed] [Google Scholar]
- Tomizawa M., Kumar,A., Perrot,V., Nakae,J., Accili,D., Rechler,M.M. and Kumaro,A. (2000) Insulin inhibits the activation of transcription by a C-terminal fragment of the forkhead transcription factor FKHR. A mechanism for insulin inhibition of insulin-like growth factor-binding protein-1 transcription. J. Biol. Chem., 275, 7289–7295. [DOI] [PubMed] [Google Scholar]
- Uejima H., Lee,M.P., Cui,H. and Feinberg,A.P. (2000) Hot-stop PCR: a simple and general assay for linear quantitation of allele ratios. Nat. Genet., 25, 375–376. [DOI] [PubMed] [Google Scholar]
- Virbasius J.V., Guilherme,A. and Czech,M.P. (1996) Mouse p170 is a novel phosphatidylinositol 3-kinase containing a C2 domain. J. Biol. Chem., 271, 13304–13307. [DOI] [PubMed] [Google Scholar]
- Vlahos C.J., Matter,W.F., Hui,K.Y. and Brown,R.F. (1994) A specific inhibitor of phosphatidylinositol 3-kinase, 2-(4-morpholinyl)-8-phenyl-4H-1-benzopyran-4-one (LY294002). J. Biol. Chem., 269, 5241–5248. [PubMed] [Google Scholar]
- Wang Y., Huso,D., Cahill,H., Ryugo,D. and Nathans,J. (2001) Progressive cerebellar, auditory and esophageal dysfunction caused by targeted disruption of the frizzled-4 gene. J Neurosci., 21, 4761–4771. [DOI] [PMC free article] [PubMed] [Google Scholar]