

Abstract
Excitable cells in many endocrine and neuronal systems display rhythms with periodicities on the order of many minutes. To observe firing patterns that represent the output of these rhythms requires a recording technique that can monitor electrophysiological activity for several hours without affecting cell behavior. A targeted extracellular approach (also known as loose-patch) accomplishes this objective. Because low resistance seals (<20 MΩ) do not influence the cell membrane and because the normal intracellular milieu is maintained, this approach is the least invasive method for monitoring the endogenous electrical activity of single cells. In this report, we detail our use of this technique to record the firing patterns of gonadotropin-releasing hormone (GnRH) neurons in brain slices continuously for several hours.
Keywords: gonadorelin, periodicity, neurons
Introduction
Extracellular recordings of firing patterns of individual cells have been made since the early days of electrophysiology. Also referred to as loose-patch recording, this technique is considered the precursor to modern patch-clamp techniques used commonly today. Due to the low resistance of seals, minimal interaction occurs between the recording electrode and the cell membrane. In addition, the cell membrane is not breached with this method, leaving the intracellular milieu of the cell undisturbed. The targeted extracellular approach is thus one of the least invasive electrophysiological methods available, allowing repeated recordings from the same cell several times without significant damage to the cell (1-2). The targeted extracellular recording technique thus continues to be useful for a variety of experimental purposes. Applications include exploring the distribution of ion channels throughout the surface of a cell, recording from fragile membranes, and making stable long-term recordings (1-3).
Our goal has been to extend the duration of targeted extracellular recordings to several hours in brain slice preparations to monitor the long-term firing patterns of gonadotropin-releasing hormone (GnRH) neurons. GnRH neurons display complex rhythmic patterns of secretion on the order of many minutes to a few hours (4-5) that change continually throughout the ovulatory cycle (6). In order to determine if an electrical correlate to this activity could be observed, we targeted electrodes to record from single GnRH neurons in brain slices (7). The targeted extracellular approach was ideal for this study because we wished to observe the natural firing pattern of GnRH neurons for periods of hours and without disturbing the intracellular milieu of the cell. In this report, we describe in detail our methods for targeting, initiating, and maintaining extracellular recordings for several hours.
Materials and Methods
In writing this manuscript, we have assumed a rudimentary understanding of electrophysiological theory and some experience with equipment and recording methods. If the reader has difficulties with terminology, concepts, or procedures described in this manuscript, there are several resources that provide excellent and detailed descriptions of electrophysiology theory and methods (2, 8-9).
Equipment
The equipment used in these experiments and the manufacturers are listed below:
Digitizer (ITC-18 Computer Interface, Instrutech, Port Washington, NY)
Amplifier (EPC-7 or EPC-8 amplifier, Heka, Germany)
Data Acquisition System (Igor Pro software, Instrutech, Port Washington, NY)
Computer (G4 Macintosh, Apple Computers, Cupertino, CA)
Micromanipulator (MP-285 micromanipulator, Sutter Instruments, Novalto, CA)
Pipette puller (model PP830, Narishige, Japan).
Recording pipettes (catalog #PG52165-4, World Precision Instruments, Sarasota, FL)
Upright microscope equipped with fluorescence and infrared-differential interference contrast microscopy (Olympus BX50WI, Opelco, Dulles, VA)
Monochrome monitor: (13” Sony PVM-13, Opelco)
CCD camera (Cohu 4915-2000/000, Scion Corporation, Fredrick, MD)
Animals, solutions, and tissue preparation
The mice used in this study were transgenically engineered to express green fluorescent protein (GFP) in GnRH neurons (10) using the murine GnRH promoter (generously provided by James Roberts, University of Texas Health Sciences Center, San Antonio, TX). Identification of GnRH neurons thus could be made visually based on GFP fluorescence. Details on solutions and tissue preparation were originally presented in a previous report (7). We have provided additional details for tissue preparation and chemicals needed for solutions in the Supplemental Materials section at the end of this report. Supplemental A - Recipes for all solutions, supplemental B - Brain slice preparation. All procedures were approved by the University of Virginia Animal Care and Use Committee.
Equipment settings and preparation for recordings
We viewed GnRH neurons with an Olympus BX50WI upright fluorescent microscope equipped with infrared differential interference contrast (Opelco, Dulles,VA) using a 40x water immersion lens. GnRH neurons were identified by brief illumination (15-45 sec) at 470 nm to visualize the GFP signal. We tried to minimize cell exposure to fluorescence to avoid possible damage to the cell (11), although in our experience, exposure to fluorescence for up to 20 min (roughly the average duration of a whole-cell recording) does not affect cell physiology (DeFazio and Moenter, unpublished observations).
Recording pipettes were fabricated from capillary glass (type 7052, outer diameter/ inner diameter 1.65/1.1 mm, World Precision Instruments, Sarasota, FL) using a two-stage pipette puller (Narashige, Japan). We found pipettes with resistances of 1-3 MΩ when filled with normal HEPES pipette solution and held at 0 mV in the external solution were most suitable for these recording. When pipette resistance was > 3 MΩ, we found the diameters to be too small, and G-Ω seals often spontaneously formed. Conversely, pipettes < 1 MΩ were so large in diameter that cell morphology could be affected. That is, cells would sometimes be aspirated into the pipette barrel over the duration of the recording. We did not systematically change or study the shank or taper of our electrodes to test for effects on seal stability and longevity, as in our experience, electrode resistance was the primary determinant of recording success. Pipettes were filled with normal saline solution in early studies. Because this is a carbonate-buffered solution, we became concerned that pH could shift over time because internal solutions cannot be bubbled with carbon dioxide to maintain pH. We thus switched to normal HEPES solution for later studies. No significant difference was observed in firing patterns between these pipette solutions. We did find the longevity of recordings improved, but this could have been due to improved ability to acquire and maintain seals rather than a change in pipette solution.
Extracellular recordings were made using an EPC-7 or EPC-8 amplifier (HEKA, Germany) with Igor Pro software (Instrutech, Port Washington, NY) running on a G4 Macintosh computer (Apple Computers, Cupertino, CA) to acquire data. Recordings were made in voltage-clamp mode with a holding potential of 0 mV, initial gain of 0.5x, filtering at 10 kHz, and digitized with at ITC-18 acquisition interface (Instrutech). Using these settings, spontaneous electrical discharges (action currents) from cells could be observed. Action currents reflect very rapid and local changes in the electrochemical gradient.
We used the Pulse Control Event Tracker program operating within Igor Pro (Instrutech) to detect and record action currents (events), the membrane currents associated with action potential firing. Although the following details may not be of use to those without Igor Pro software (Instrutech), similar event detection software exists for pClamp (Axon Instruments). We used the following Event Tracker settings: 10 μsec sampling interval to ensure detection of action currents near their peaks, one point over threshold to trigger to ensure detection of all events above threshold, event polarity negative, and -50 to -150 pA threshold depending on action current amplitude. Noise typically ranged 20-25 pA peak-to-peak. For each detected event, the time of the event and 10 msec centered on the event were digitized and stored to a data file.
We based these settings on the amplitude and duration of spontaneous events observed in early recordings of GnRH neurons. By only digitizing the data surrounding the event itself, file sizes for 30-min records were typically 0.5 to 5 MB, depending on the number of events observed in that 30-min period. Files could easily exceed 100 MB if the baseline trace is continuously recorded. Note that in Igor Pro software (IGOR-PRO 3.16PPC), Event Tracker files cannot exceed ~37-min due to limitations in the way the program stores temporal information. Between the end of one 30-min file and the beginning of the next, we checked seal resistance and adjusted baseline to 0 pA if necessary.
Establishing a targeted extracellular recording
To approach the target cell, minimal positive pressure (forcing solution out of the pipette) was applied to the recording pipette, which was then lowered into the bath and moved to a position above the surface of the slice using an MP-285 micromanipulator (Sutter Instruments, Novato, CA). By focusing back and forth between the target cell and the pipette tip, the pipette was moved into position above the slice surface directly over the target cell. Readings of pipette resistance and pipette offset were taken at this time. The pipette was then moved downward through the slice in a zig-zag motion downward (z-axis) and forward (x-axis) then backward along the x-axis (Fig. 1).
Fig. 1.
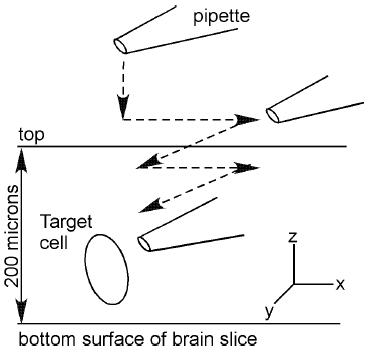
Recording pipette approaching a cell for targeted extracellular recording. Sketch showing the relationship between a brain slice containing the target cell and the recording pipette. The arrows illustrate the path the pipette should move to enter the slice and approach the cell. The sawing motion is critical for long-term stability as this avoids compression of tissue under the electrode.
Positive pressure was maintained by applying pressure gently but consistently through the mouth pipette during this time until the pipette tip was located at the same vertical level (z-axis) as the target cell (slightly above the target cell is also acceptable) and approximately 1/4 to 1/2 of a cell body in front of the target cell (so that the pipette tip could still be moved along the x-axis toward the cell). Positive pressure was then slowly released over 0.5-1 sec. The target cell and surrounding tissue typically moved slightly toward the pipette tip with the relief of positive pressure. We usually observed a gradual rise in pipette resistance from the values taken before entering the slice, often accompanied by the appearance of small capacitive transients in the trace. Within 5-min, seal resistances typically ranged from 5-20 MΩ and either remained stable or increased slowly over time up to as high as 50 MΩ. These seals typically formed spontaneously and did not require application of negative pressure (suction) by mouth. Only when no spontaneous activity and no change in pipette resistance were observed in the first 5-min did we apply negative pressure to improve seal resistance. On occasion, seal resistances continued to rise into the 100 MΩ to GΩ range. These recordings were aborted.
We next monitored the current trace for evidence of downward deflections indicative of action currents. Gain was increased to 5x for monitoring and recording action currents. Spontaneous action currents were sometimes very low amplitude initially. As the seal gradually increased in resistance over the first few minutes, this signal increased in amplitude. Before collecting data, we monitored the trace for 5-minutes to determine the initial amplitude of observed action currents, so that an appropriate threshold could be set for Event Tracker (-50 to -150 pA depending on event amplitude). We also checked for stability in the baseline of the trace during this time. We used the pipette offset dial to shift the baseline to 0 pA as needed to negate current due to a change in the junction potential. Typically, the trace would shift very little and usually very slowly once set to 0 pA. If baseline changes did occur during the course of a recording, we shifted the trace using the pipette offset to maintain 0 pA current injection. We rarely observed dramatic or abrupt shifts in baseline and aborted recordings if the baseline did not stabilize after a few minutes. If several consecutive recordings are marked by highly variable baselines, we suggest checking grounding wires, especially grounds to the bath solution. After monitoring the trace for 5-min, we took one more reading of the seal resistance, checked the baseline trace once more, and began data collection.
Maintaining a long-term recording
The best method for monitoring the quality of the recording while it is in progress is to note the amplitude of action currents periodically. Typically, action current amplitude rose over the first few minutes of a recording as the seal resistance rose. Action current amplitude then leveled off and could be maintained for many minutes to several hours. If amplitude decreased, there were several remedies that we used to extend the recording duration. First, we adjusted the location of the pipette with respect to the cell. Over long periods of time, cells within a slice or the slice itself can move slightly (micromanipulators can also drift). If the cell body appears to be far from the pipette, moving the pipette near the cell body often results in an increase in action current amplitude. Note that this technique requires a camera and monitor to visualize the cell and pipette. If the pipette and cell body do not appear to have shifted, we also checked the pipette resistance. If significantly lower than the previous reading, the seal had lost some of its integrity. We were often able to restore the seal by gently applying negative pressure. If action current amplitude remained low or continued to decrease to below threshold, we terminated the recording. We also found a recording could be re-established by applying very gentle positive pressure while moving away from the cell membrane and then targeting a different region of the soma and following the steps for establishing a recording. Making and breaking multiple loose-patch seals is an accepted and commonplace practice in many labs (1-2). We did not use this technique to maintain any recordings in our previous publication, however, because we did not know for certain if there are effects on long-term patterns due to repeated recordings. We have been able to confirm, however, that action currents are observable after making and breaking seals several times. Although this approach appears tenable, data must be interpreted with regard to possible effects on long-term rhythm generation.
Data collection and analysis
Data collection using Event Tracker is fairly straightforward. By checking the box marked “event detection” in the set-up window, Event Tracker will digitize only the data surrounding a detected event, as mentioned above. To start a recording, check the storage box and click record in the main Event Tracker window. Note that Event Tracker will still detect and count events with the storage box unchecked, but the events will not be digitized, resulting in an empty file.
The output of an Event Tracker file consists of a series of detected events. Each event contains two important pieces of information: 1) 10 ms of digitized data surrounding the event which contains the waveform of the event and 2) the time when the event crossed threshold to 10 microsecond precision (based on above settings). The waveform is digitized so that the event can by verified as an action current or rejected as an artifact based on amplitude, duration and shape. To automate this process, we designed a program to detect spurious events using a minimum event width of 0.05 ms at half-peak amplitude and a minimum event amplitude. We do not use the waveform data in any other aspect of our analysis. In principle, however, the average of many action current waveforms during a control period could be compared to those under a test condition to look for qualitative changes in waveform shape. Because amplitude is dependent upon seal resistance and distance of the recording electrode to the cell body, we do not recommend any quantitative comparison of amplitude.
The key component of these data streams for purposes of pattern analysis is the time value associated with each event. Using custom programs we wrote for Igor Pro (Instrutech), verified action current events were counted and binned at 1-min intervals. These counts were then plotted across time to identify gradual changes in long-term firing patterns. We used the CLUSTER7 pulse detection algorithm (12), to identify peaks in firing rate using a series of pooled t-tests (7, 13). In later studies, we also binned activity at 1-sec intervals to characterize higher frequency rhythms using Fourier spectral analysis on firing patterns (14).
Results and Discussion
Loose vs. tight seals
We used the targeted extracellular recording technique primarily to record GnRH neurons that were identified by their GFP expression in coronal brain slices. We found this technique to be optimal for monitoring long-term firing patterns with minimal disturbance to the cell. We found the low resistance or “loose” seals of this technique to be specifically advantageous over the cell-attached technique in which tight seals (GΩ seals) are formed for several reasons. First, the signal amplitude was greater using the targeted extracellular approach with loose seals (Fig. 2).
Fig. 2.
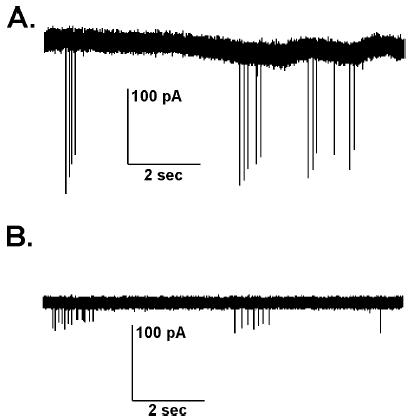
Differences between low-resistance (loose) and tight seal recordings. (A-B) a 10-sec digitized recording segment displaying both the baseline trace and downward vertical deflections indicating action currents (events). (A) With the loose seal (16 MΩ), event amplitude is ~200 pA and peak-to-peak noise ~25 pA, resulting is a signal-to-noise ratio of 8:1. In some low resistance seals, this ratio can exceed 50:1. (B) With the tight seal (GΩ, traditional cell-attached configuration), event amplitude is ~40 pA and peak-to-peak noise ~20 pA, resulting in a lower signal-to-noise ratio of 2:1. Note that A and B are recordings from two different cells, representative of the general finding using each technique.
Unlike an extracellular field recording, in which the electrode detects a small ion flux within a cell layer or region, we can visualize fibers and cells and target electrodes toward the membrane of a specific single cell, resulting in a large amplitude signal (Fig. 2A) without contamination of the signal from the activity of nearby cells. Note that with a cell-attached or tight seal approach (Fig. 2B), although noise is reduced, action current amplitude is reduced as well. Second, formation of a GΩ seal can cause significant local disruption to the cell membrane (15). The possible effects of this disruption on the ion channel properties and firing patterns are not known, but are of enough concern that we felt a low resistance seal would be preferable for studying long-term endogenous rhythms. Third, GΩ seals can rupture, thus puncturing and severely damaging the cell. The duration of the average recording using a tight seal in the cell-attached configuration ranged from 15 to 30 min, with an occasional recording lasting for as long as one hour before loss of seal integrity. Loose seals, as mentioned previously, do not compromise cell integrity and often can be maintained for much longer periods of time.
Long-term recordings
In many neuroendocrine systems, rhythms of activity are observed with periods on the order of many minutes to hours. We are now able to routinely make recordings of GnRH neurons for two or more hours using the targeted extracellular technique, enabling us to observe long-term changes in firing patterns. In Fig. 3, we show an example of one of the longest recordings thus far of a rhythmic GnRH neuron recorded from a mouse that was ovariectomized and implanted with an estradiol capsule to mimic estrogen feedback. This recording, made from a coronal brain slice, was over 3.5 hours in duration.
Fig. 3.
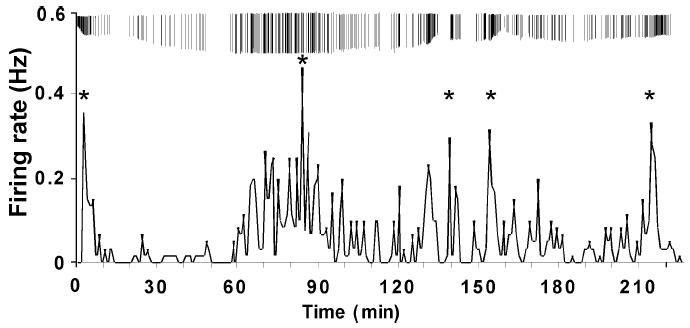
A long-term recording using the targeted extracellular technique. Firing rate of a GnRH neuron recorded in a coronal brain slice is displayed at 1-min intervals. Vertical bars above this graph indicate the time of occurrence for each action current composing the firing rate plot. * indicate episodes of significant changes in firing rate detected by CLUSTER7 pulse detection algorithm. We believe the changes in action current amplitude are due to shifts in the cell location with respect to the pipette tip and to changes in seal resistance. Data adapted from (7) with permission.
At several times during this recording, firing rate increased to produce a rhythmic pattern of firing activity, denoted by asterisks in Fig. 3 (mean interepisode interval for this recording: 53.0 +/- 13.2 min). Cells capable of firing action potentials with any long-term rhythmic components are thus amenable to this technique. Note that the firing rate between these episodes often approached zero. Although we do not require more than 2-3 hours of recording time to accurately assess rhythms in the firing patterns in GnRH neurons, we believe the maximum recording duration of a single cell could be between 6 and 8 hours based on our estimations of brain slice viability.
Because our cells were often quiescent for long durations, determining when to terminate a recording was a challenge when no action currents were observed for long periods of time. Because action current amplitude can sometimes diminish very rapidly when a seal becomes unstable (Fig. 4A), we needed to resolve whether a long period of quiescence was due to actual cell quiescence or to having lost the seal. To do this, we added 10-15 mM KCl to the bath whenever no action currents were observed for more than 30 minutes to induce firing (Fig. 4, see legend).
Fig. 4.
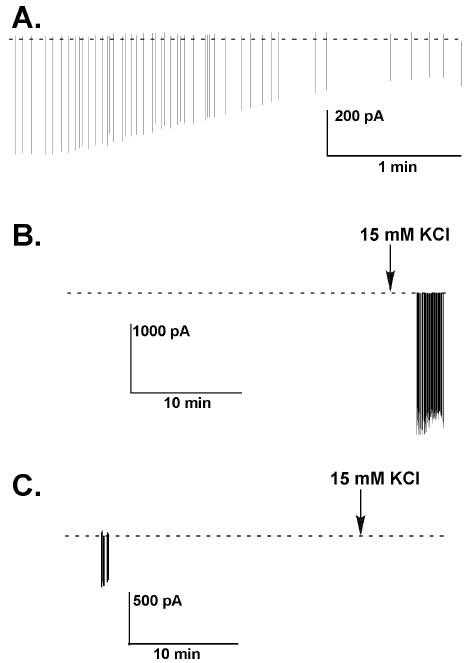
Examples of the termination of a recording. Recordings were made of a GFP-expressing GnRH neuron in a coronal brain slice. (A) An example of the decrease in amplitude that can occur when a low resistance seal deteriorates. (B) An example of a quiescent cell, in which no events were detected until stimulated after by bath application of 10-15 mM KCl. (C) An example of failed seal, in which bath application of 10-15 mM KCl did not evoke detectable events.
Although we always stopped data analysis after KCl treatment (because KCl would disrupt firing patterns), we could include the preceding 30-min of inactivity as data if the high potassium treatment successfully elicited firing. Approximately 20% of GnRH neurons were quiescent for 30 min or more, although this is a crude estimation because we have only examined two endocrine conditions and the duration of quiescence differs with respect to estrous cycle stage and steroid milieu (7). In about 50% of these cases, no action currents were observed during KCl treatment, indicating we were no longer able to detect action currents from the cell. In these cases, the last spontaneous action current marked the end of the data set. The inability to determine the fidelity of the recording without perturbing the recording is perhaps the greatest disadvantage to this technique. The advantages, nonetheless, far outweigh this disadvantage for use in our system of study.
Additional applications
This technique is not limited to study of labeled cells, such as GFP-expressing GnRH neurons. We and others (16-17) have also made long-term recordings of unidentified cells (cells that did not express GFP). We have also shown this technique can be adapted to acutely dissociated cells (data not shown), though it is much more difficult to avoid GΩ seals in dissociated cells without modifications to the pipette (see 1). Because low-resistance seals are easily obtainable, we also believe there are few technical limitations to recording several cells simultaneously with multiple electrodes using this technique in order to observe communication between cells or synchronization within networks of cells. For a detailed review of additional applications, see Roberts et al (1).
In conclusion, the targeted extracellular technique is particularly well suited to observing the natural activity of excitable cells. We have demonstrated the advantages of this technique and have reported methods to extend the duration of this type of recording to several hours. In addition, the ease with which a low resistance seal can be initiated using this sighted technique makes it an ideal tool for training students who are new to electrophysiology, enabling publishable quality recordings to be obtained in a relatively short time.
Acknowledgments
This publication makes use, with permission, of data and methodologies published in Nunemaker CS, DeFazio RA, Moenter SM. Estradiol-sensitive afferents modulate long-term episodic firing patterns of GnRH neurons. Endocrinology 2002; 143:2284-2292, Copyright 2002 by The Endocrine Society.
Supported by HD34860 and HD41469 (SMM), the National Institute of Child Health and Human Development/National Institute of Health through cooperative agreement U54HD28934 as part of the centers program in reproductive research, and the National Science Foundation Center for Biological Timing. We thank Drs. Xu-Zhi Xu and Glenn Harris for excellent technical assistance and Dr. Leslie Satin for editorial comments.
Appendix
Protocols
Appendix A - Solution recipes
Normal saline For 2 liters
| Component | MW | Stock conc M | Final conc mM | g added |
| NaCl | 58.44 | 130.5 | 15.25 | |
| NaHCO3 | 84.01 | 26.0 | 4.369 | |
| Na2HPO4-7H2O | 268.07 | 1.25 | 0.670 | |
| MgSO4-7H2O | 246.48 | 1.2 | 0.592 | |
| Glucose | 180.16 | 10 | 3.603 | |
| KCl | 1 | 3.5 | 7 ml in 2 liters |
Adjust osmolarity to 305 by adding NaCl or H2O. Sterile filter and aliquot at 400 ml. Store at 4 C until use.
On day of use add 1 ml of 1 M CaCl2 per 400 ml (final concentration of CaCl2 2.5 mM).
Sucrose saline For 2 liters
| Component | MW | Stock conc M | Final conc mM | g added |
| Sucrose | 342.31 | 250 | 171.155 | |
| NaHCO3 | 84.01 | 26.0 | 4.369 | |
| Na2HPO4-7H2O | 268.07 | 1.25 | 0.670 | |
| MgSO4-7H2O | 246.48 | 1.2 | 0.592 | |
| Glucose | 180.16 | 10 | 3.603 | |
| MgCl2 | 1 | 2.5 | 5 ml in 2 liters | |
| KCl | 1 | 3.5 | 7 ml in 2 liters |
Sterile filter and aliquot at 400 ml. Store at 4 C until use.
Normal HEPES For 50 ml
| Component | MW | Final conc mM | g added |
| NaCl | 58.44 | 150 | 0.438 |
| HEPES | 238.31 | 10 | 0.119 |
| Glucose | 180.16 | 10 | 0.09 |
| Component | Stock conc M | Final conc mM | ml added |
| CaCl2 | 1 | 2.5 | .125 |
| MgCl2 | 1 | 1.3 | .065 |
| KCl | 1 | 3.5 | .175 |
Adjust pH to 7.4 by adding NaOH.
Adjust osmolarity to 310 by adding H2O or NaCl. Sterile filter and store at 4 C until use.
Appendix B - Brain Slice Preparation
Materials needed
Sucrose saline
Normal saline
Ice, ice bucket
Razor blade
Vibratome with tray
Large scissors
Bone scissors
Fine scissors
Plastic spoon
Blotting paper
Petri dish
Superglue
Bent weighing spatula
50 ml beaker
2- 400 ml beakers
2- slice chambers
2-lids from 50 ml conical tubes
100 ml graduated cylinder
water bath at 30-32 C
O2/CO2 tank 95%/5%
Fill vibratome with ice. Place vibratome tray in chuck. Break razor blade in half long ways and insert one half into vibratome so that blade protrudes as much as possible.
Place 150 ml sucrose saline, and empty 50 ml beaker on ice.
Place lids from 50 ml conical tubes upside-down in bottom of the 400 ml beakers and place slice chambers on top of these. (We make slice chambers by drilling out the bottom of a portion of a 24-well tissue culture plate and supergluing fine nylon mesh on the bottom). Fill one with normal saline until near top of slice chamber (~80 ml) and place in water bath. Mix 40 ml sucrose saline with 40 ml normal saline in the other 400 ml beaker and keep at room temperature.
Bubble all three solutions (sucrose saline, 50:50 mix and normal saline) with 5% CO2/95% O2 for 15 minutes before use. Sucrose saline may be bubbled as vigorously as you wish; bubbling in other two solutions should be moderate to maintain pH.
Fill Petri dish with ice and keep on ice. Place blotting paper on top of Petri dish.
Humanely decapitate mouse and remove scalp. Use bone scissors to carefully remove top of skull, working around foramen magnum to first remove rear of skull, then cutting up sides until it is possible to lift up skull and snap off at front of head.
Once skull is removed, pour sucrose saline over brain into 50 ml beaker on ice to chill brain. Time elapsed from decapitation to this point should be under 2 minutes.
Cut olfactory bulbs with fine scissors. Then use fine scissors inserted gently into rostral cortex to lever brain out of skull until optic tracts are exposed. Cut these with fine scissors and remove brain to 50 ml beaker on ice. Add more sucrose saline and let brain rest here 30-60 seconds. Moisten blotting paper with cold sucrose saline.
Remove brain with plastic spoon and place ventral side down on moist blotting paper. Use second half of razor blade to block the brain as appropriate for your area of interest.
Place small drop of glue on vibratome tray and spread to roughly size of brain.
Pick up brain using bent spatula-cut side down-use blade to gently nudge brain onto spatula.
Carefully blot excess moisture off of brain on dry blotting paper.
Place brain on glue in vibratome tray to obtain the orientation of slices you desire.
Fill tray with sucrose saline-replace after every second slice with fresh sucrose saline.
Vibratome at speed ~1, amplitude 8 (approximate and will vary). Begin to cut off sections at 500 μm. Before retracting blade, raise it 100 µ to avoid dragging blade over brain. If sections bunch up, slow speed to ~0 (blade will idle forward extremely slowly at this setting).
Begin collecting sections (200-300 μm) when desired brain region is reached.
Place slices in the 50:50 mix at room temperature in individual chambers. Keep slices in this for 15 minutes then transfer to normal saline in water bath.
Keep slices in normal saline for at least 90 minutes for recovery before initiating recording.
Dispose of slices 8 hours after their initial preparation.
Appendix C - Targeted extracellular recording method
A non-invasive way to monitor the firing pattern of a cell and provide information on how individual components you might measure in an animal model (e.g., synaptic input, specific currents) are integrated into overall output of the cell.
Set up
Global controls-similar to “bath settings” except amplifier gain set at 5 rather than 1.
Event tracker
Sampling interval 10 µsec
NO external trigger
YES event detection
Recording channel 0
Event polarity-negative
Threshold, typically 100 pA. May need to decrease or increase depending on signal to noise ratio. Range typically 50-200 pA. Noise is typically 20 pA peak to peak, maxing out around 40 pA. If you set the threshold too high and the signal diminishes, you loose the recording more quickly. Note if you end up in the cell-attached configuration, both signal and noise will plummet as seal resistance increases.
Number of points below threshold-1
Sample time before-5 msec
Sample time after-5 msec (could probably get away with 2 ms for each of these)
Max record size-1 sec
Graphing time base-50 sec (although can see smaller events if you reduce this parameter to 1 sec, it is hard to see patterning with short time bases)
Recording
Use larger electrode (1-3 MΩ).
Fill electrode with filtered HEPES buffered extracellular solution so you don’t have to worry about pH.
Approach cell as you would for patch-clamp, but not quite as close. “Seal” will generally be 5-30 MΩ.
Unlike in traditional patch-clamp, you want to adjust the pipette offset to keep the baseline at zero. Adjust before approaching cell and once at cell using Scope Window, then switch to Event Tracker.
Start Event Tracker and set so that you are viewing ADC channel 0. There are three lines on the Event Tracker (ET) screen.
An almost solid line with a 0 on it (this is zero)
A dashed line (this is the baseline you accept-see below)
A dotted line (this is your threshold setting)
Adjust the Vp offset knob so that the data trace is over the zero line. Click the baseline button and then click accept. The data baseline should now be near the zero line-repeat until this is true.
After accepting the baseline, use Vp offset knob to adjust data trace so that it stays over the accepted baseline. You shouldn’t need to do this more than every couple of minutes. If the trace remains unstable for more than 5 minutes at the start of an experiment, you should strongly consider abandoning the recording.
Check to make sure the threshold you have set makes sense relative to the size of the events you are recording and the noise level.
Click the storage box at the top of the ET screen and this will begin data collection. Record for 30 min max (at 37 min, the time buffer is exceeded). Then check the “seal” in the scope window, return to Event Tracker and begin again with a new baseline. The gaps in the data for this switch will be less than a minute so shouldn’t impact on your overall pattern.
Each 30 min data window will generate an ET file. These appear in the IGOR Pro folder and are named by date with an extension to indicate the order in which they were generated. (So if you are the second person that day to record ET files, make a note of where your data begins-you can do this by noting the time on the computer when you start the ET file). Each ET file will have several records in it-one for every time threshold is crossed.
References
- Roberts WM, Almers W. Patch voltage clamping with low-resistance seals: loose patch clamp. Methods Enzymol. 1992;207:155–176. doi: 10.1016/0076-6879(92)07011-c. [DOI] [PubMed] [Google Scholar]
- Stuhmer W, Roberts WM, Almers W. The loose patch clamp. In Sakmann B, Neher (eds): “Single-Channel Recording.” 1983 New York: Plenum Press, 2nd ed. pp. 123-132.
- Anson BD, Roberts WM. A novel voltage clamp technique for mapping ionic currents form cultured skeletal myotubes. Biophysical Journal. 1998;74:2963–2972. doi: 10.1016/S0006-3495(98)78003-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke IJ, Cummins JT. The temporal relationship between gonadotropin-releasing hormone (GnRH) and luteinizing hormone (LH) secretion in ovariectomized ewes. Endocrinology. 1982;111:1737–1739. doi: 10.1210/endo-111-5-1737. [DOI] [PubMed] [Google Scholar]
- Levine JE, Ramirez VD. Luteinizing hormone-releasing hormone release during the rat estrous cycle and after ovariectomy, as estimated with push-pull cannulae. Endocrinology. 1982;111:1439–1448. doi: 10.1210/endo-111-5-1439. [DOI] [PubMed] [Google Scholar]
- Moenter SM, Caraty A, Locatelli A, Karsch FJ. Pattern of gonadotropin-releasing hormone (GnRH) secretion leading up to ovulation in the ewe: existence of a preovulatory GnRH surge. Endocrinology. 1991;129:1175–1182. doi: 10.1210/endo-129-3-1175. [DOI] [PubMed] [Google Scholar]
- Nunemaker CS, DeFazio RA, Moenter SM. Estradiol-sensitive afferents modulate long-term episodic firing patterns of GnRH neurons. Endocrinology. 2002;143:2284–2292. doi: 10.1210/endo.143.6.8869. [DOI] [PubMed] [Google Scholar]
- Hille B. Hille B. Ion channels of excitable membranes. 3rd ed. Sunderland, MA: Sinauer Associates, Inc.; 2001.
- Sherman-Gold R (ed.) The axon guide for electrophysiology and biophysics laboratory techniques. Axon Instruments, 1993. No longer available in print, but is available on-line at http://www.axon.com.
- Suter KJ, Song WJ, Sampson TL, Wuarin JP, Saunders JT, Dudek FE, Moenter SM. Genetic targeting of green fluorescent protein to gonadotropin-releasing hormone neurons: characterization of whole-cell electrophysiological properties and morphology. Endocrinology. 2000;141:412–419. doi: 10.1210/endo.141.1.7279. [DOI] [PubMed] [Google Scholar]
- Liu HS, Jan MS, Chou CK, Chen PH, Ke NJ. Is green fluorescent protein toxic to the living cells? Biochem Biophys Res Comm. 1999;260:712–717. doi: 10.1006/bbrc.1999.0954. [DOI] [PubMed] [Google Scholar]
- Velhuis JD, Johnson ML. Cluster analysis: a simple versatile, and robust algorithm for endocrine pulse detection. Am J Physiol Endocrinol Metab. 1986;250:E486–E493. doi: 10.1152/ajpendo.1986.250.4.E486. [DOI] [PubMed] [Google Scholar]
- Nunemaker CS, DeFazio RA, Geusz ME, Herzog ED, Pitts GR, Moenter SM. Long-term recordings of networks of immortalized GnRH neurons reveal episodic patterns of electrical activity. J Neurophysiol. 2001;86:86–93. doi: 10.1152/jn.2001.86.1.86. [DOI] [PubMed] [Google Scholar]
- Nunemaker CS, Straume M, DeFazio RA, Moenter SM. Gonadotropin-releasing hormone neurons generate interacting rhythms in multiple time domains. Endocrinology March 2003;144 (in press). [DOI] [PubMed]
- Milton RL, Caldwell JH. How do patch clamp seals form? A lipid bleb model. Pflugers Arch Eur J Phys. 1990;416:758–762. doi: 10.1007/BF00370626. [DOI] [PubMed] [Google Scholar]
- Reuss S, Vollrath L. Electrophysiological properties of rat pinealocytes: evidence for circadian and ultradian rhythms. Exp Brain Res. 1984;55:455–461. doi: 10.1007/BF00235276. [DOI] [PubMed] [Google Scholar]
- Schenda J, Vollrath L. Single-cell recordings from chick pineal glands in vitro reveal ultradian and circadian oscillations. Cell Mol Life Sci. 2000;57:1785–1792. doi: 10.1007/PL00000658. [DOI] [PMC free article] [PubMed] [Google Scholar]