

Abstract
To assess how automatic function assignment will contribute to genome annotation in the next five years, we have performed an analysis of 31 available genome sequences. An emerging pattern is that function can be predicted for almost two-thirds of the 73,500 genes that were analyzed. Despite progress in computational biology, there will always be a great need for large-scale experimental determination of protein function.
Letter
The completion of a first draft for the human genome sequence represents an outstanding scientific achievement of our times. Despite the unprecedented hype in the popular media, it is evident that the completion of this phase is just the beginning of a long march towards an understanding of the structure and function of our genetic blueprint [1]. What is to be expected from the analysis of the human genome five years from now? One guide in answering this question is a thorough assessment of our capability to analyze the genomes that have already been sequenced in the past five years, starting in 1995 with the genome of Haemophilus influenzae [2,3]. Since then, 31 entire genome sequences have been made available to the public domain (as of September 2000) [4]. To obtain a snapshot of the maximum possible set of database-driven sequence annotations across species, we have used GeneQuiz, a system for automated large-scale sequence analysis [3,5].
Automated genome sequence analysis and annotation has the advantage that the analysis strategy is uniformly applied to all genome sequences against the same database, rendering results comparable [5]. The updated annotations also form a resource for the scientific community for further analysis and assessment. The set of annotations we obtained contains an additional 5,534 novel protein function assignments, a 15% increase over the original genome sequence publications (Figure 1a). Evaluation of the results has shown that the agreement of automated function prediction reaches 95%, when compared to expert manual analysis [5,6]. Through this procedure, patterns that are not discernible from individual genomes become apparent, with interesting implications for the future.
Figure 1.
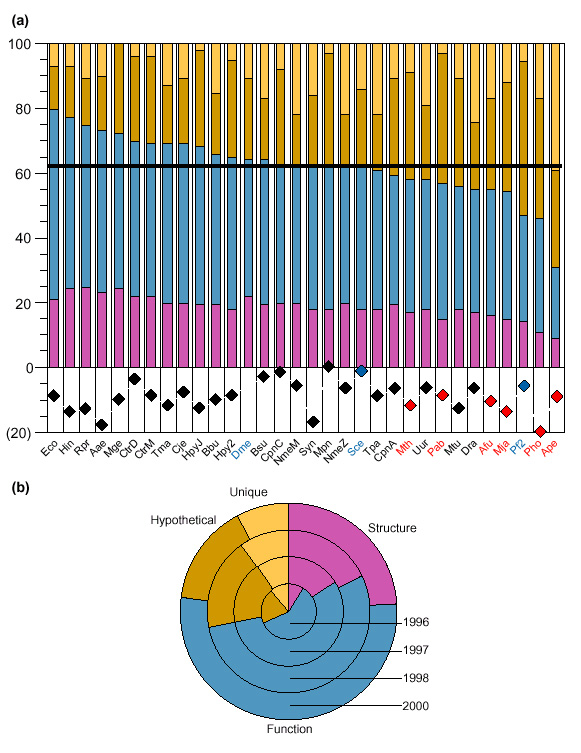
A summary of the annotation levels for 31 genomes. Annotations for all genomes (for 73,500 unique genes, 134,000 annotations in total - approximately a twofold annotation coverage) are available on the world wide web at the European Bioinformatics Institute Computational Genomics Group Services page [15] - then point and click at 'GeneQuiz'. Total computation required 2,400 CPU-hrs on a 16-CPU SGI Power Challenge and 68GB of storage. Results for other genomes will be made available at the same URL as they are completed. (a) Information snapshot for 31 entire genomes and a eukaryotic chromosome (Plasmodium falciparum, chromosome 2). For species (and strain) name abbreviations, please refer to the website [15]. Bacteria are shown in black, Archaea in red and Eukarya in blue. Percentages for proteins with homologs of known structure (pink) or function (blue), hypothetical proteins (dark brown) and unique proteins (light brown) are shown. Species are sorted according to the sum of structure and function information; the horizontal line represents the average of known/predicted functions across species. Diamonds (bottom panel) represent the percentage increase in new findings over the original (or public database) annotations (except Drosophila melanogaster, for which such comparison is not currently possible). This percentage range, ranging from 0 to 20, is indicated in brackets. (b) An 'information clock' for the genome of Haemophilus influenzae, showing the relative levels of annotation over time, reflecting a general increase of information in the public databases. Colours are used as in (a).
First, the detection of proteins of known structure and/or function has increased over time (Figure 1b). This is especially true for homologs of known structure, a trend that should be further enhanced during the next five years as a result of the recent initiatives in structural genomics [7,8]. The total number of homologs of known function has also increased, but at a slower pace (Figure 1b). This may be attributable to two factors: either high-throughput experimental characterization has not yet provided genome-wide functional information or that this type of functional annotation has not yet been adequately transferred into the public sequence databases (Figure 1b) [9]. It is imperative that provisions be made to start recording this type of functional information for the human genome [10].
Second, there is some degree of variability for the 31 genomes that we have analyzed. On average, function is known or can be predicted for 62% of the proteins encoded in each genome (Figure 1a). Of these, a significant proportion has homologs in the structure database (19% on average) and the remaining majority (43% on average) have homologs of known function. The total fraction of proteins of predicted structure/function varies significantly across species, ranging from 31% for Aeropyrum pernix (9% structure and 22% function) to 80% for Escherichia coli (21% and 59%, respectively; see Figure 1a). It remains to be seen how the human genome ranks within this range of functional assignments.
Third, there is a substantial number of uncharacterized proteins, including hypothetical proteins (with homologs of unknown function; 26% on average) or 'unique' proteins (without homologs; 12% on average). Hypothetical proteins range from 13% for E. coli and 14% for Rickettsia prowazekii to 40% for Pyrococcus abyssii and 47% for Plasmodium falciparum (chromosome 2). These protein families represent excellent candidate targets for functional genomics. Species with high percentages of hypothetical proteins demarcate taxa whose properties generally remain unknown. This group includes Archaea (34% on average), Mycobacterium tuberculosis (33%), Helicobacter pylori and Chlamydia pneumoniae strains (all at 30%) and yeast (24%). At the other end of the spectrum, certain species are sufficiently covered by their relatives (e.g. H. influenzae at 14%).
Finally, the percentage of 'unique' proteins delineates the number of uncharacterized protein families. This number varies widely, ranging from 0% for Mycoplasma genitalium and 4% for two Chlamydia trachomatis strains to 39% for A. pernix. A few of these genes may encode taxon-specific proteins or may represent false gene predictions. It is interesting that species with few unique proteins are bacterial (Figure 1a), an indication that many protein families in their phylogenetic neighbourhood have been detected. At the other extreme, species with many unique sequences also include Treponema pallidum, the two Neisseria meningitidis strains (all at 22%) and Deinococcus radiodurans (24%). It is expected that the human genome will also contain a high number of unique proteins, because it currently represents the only vertebrate genome that has been fully sequenced.
The annotation level for each species crucially depends on the existence of homologs of the proteins that are potentially encoded in the genome sequence. Automatic function assignment also depends on the quality of the underlying databases and the availability of proteins of known structure or function. Another factor that significantly influences annotation quality is the definition of gene structure, an exceedingly difficult task for eukaryotic genomes [11]. The difficulties of eukaryotic genome annotation have been recently reviewed [12], with specific details on the Ensembl and GadFly projects for the human and the fruitfly genomes, respectively.
The present study suggests that a great deal should be expected from the analysis of the human genome. On the one hand, using up-to-date databases and automatic approaches, a considerable proportion of the human genome will be annotated, to guide further experimentation and discovery. On the other hand, no matter how much the database increases over time, there will still be a great need for functional experiments, to detect the cellular roles of novel proteins. In spite of progress in the field of bioinformatics [13], filling this information gap is going to be the next major challenge for genomics research, where large-scale experimentation will lead the way [14].
Acknowledgments
Acknowledgements
The authors thank members of the Systems Group at the European Bioinformatics Institute for help. This work was fully supported by the TMR Programme of the European Commission (DG-XII - Science, Research and Development).
References
- Butler D, Smaglik P. Draft data leave geneticists with a mountain still to climb. Nature. 2000;405:984–985. doi: 10.1038/35016703. [DOI] [PubMed] [Google Scholar]
- Fleischmann RD, Adams MD, White O, Clayton RA, Kirkness EF, Kerlavage AR, Bult CJ, Tomb JF, Dougherty BA, Merrick JM, et al. Whole-genome random sequencing and assembly of Haemophilus influenzae Rd. Science. 1995;269:496–512. doi: 10.1126/science.7542800. [DOI] [PubMed] [Google Scholar]
- Casari G, Andrade MA, Bork P, Boyle J, Daruvar A, Ouzounis C, Schneider R, Tamames J, Valencia A, Sander C. Challenging times for bioinformatics. Nature. 1995;376:647–648. doi: 10.1038/376647a0. [DOI] [PubMed] [Google Scholar]
- Kyrpides NC. Genomes OnLine Database (GOLD 1.0): a monitor of complete and ongoing genome projects worldwide. Bioinformatics. 1999;15:773–774. doi: 10.1093/bioinformatics/15.9.773. [DOI] [PubMed] [Google Scholar]
- Andrade MA, Brown NP, Leroy C, Hoersch S, de Daruvar A, Reich C, Franchini A, Tamames J, Valencia A, Ouzounis C, Sander C. Automated genome sequence analysis and annotation. Bioinformatics. 1999;15:391–412. doi: 10.1093/bioinformatics/15.5.391. [DOI] [PubMed] [Google Scholar]
- Andrade M, Casari G, de Daruvar A, Sander C, Schneider R, Tamames J, Valencia A, Ouzounis C. Sequence analysis of the Methanococcus jannaschii genome and the prediction of protein function. Comput Appl Biosci. 1997;13:481–483. doi: 10.1093/bioinformatics/13.4.481. [DOI] [PubMed] [Google Scholar]
- Burley SK, Almo SC, Bonanno JB, Capel M, Chance MR, Gaasterland T, Lin D, Sali A, Studier FW, Swaminathan S. Structural genomics: beyond the human genome project. Nat Genet. 1999;23:151–157. doi: 10.1038/13783. [DOI] [PubMed] [Google Scholar]
- Skolnick J, Fetrow JS, Kolinski A. Structural genomics and its importance for gene function analysis. Nat Biotechnol. 2000;18:283–287. doi: 10.1038/73723. [DOI] [PubMed] [Google Scholar]
- Tsoka S, Ouzounis CA. Recent developments and future directions in computational genomics. FEBS Lett. 2000;480:42–48. doi: 10.1016/s0014-5793(00)01776-2. [DOI] [PubMed] [Google Scholar]
- Brazma A, Robinson A, Cameron G, Ashburner M. One-stop shop for microarray data. Nature. 2000;403:699–700. doi: 10.1038/35001676. [DOI] [PubMed] [Google Scholar]
- Stormo GD. Gene-finding approaches for eukaryotes. Genome Res. 2000;10:394–397. doi: 10.1101/gr.10.4.394. [DOI] [PubMed] [Google Scholar]
- Lewis S, Ashburner M, Reese MG. Annotating eukaryotic genomes. Curr Opin Struct Biol. 2000;10:349–354. doi: 10.1016/s0959-440x(00)00095-6. [DOI] [PubMed] [Google Scholar]
- Eisenberg D, Marcotte EM, Xenarios I, Yeates TO. Protein function in the post-genomic era. Nature. 2000;405:823–826. doi: 10.1038/35015694. [DOI] [PubMed] [Google Scholar]
- Lockhart DJ, Winzeler EA. Genomics, gene expression and DNA arrays. Nature. 2000;405:827–836. doi: 10.1038/35015701. [DOI] [PubMed] [Google Scholar]
- The European Bioinformatics Institute Computational Genomics Group - Services. http://www.ebi.ac.uk/research/cgg/services/ http://www.genomes.org.