

Short abstract
Spin/Ssty genes might be important in the transition from sperm cells and oocytes to the early embryo. The discovery of a new protein motif of around 50 amino acids in length, the Spin/Ssty repeat is reported. Each repeat resides in its own exon, supporting the view that Spin/Ssty repeats are independent functional units.
Abstract
Background
The homologous genes Spin (spindlin) and Ssty were first identified as genes involved in gametogenesis and seem to occur in multiple copies in vertebrate genomes. The mouse spindlin (Spin) protein was reported to interact with the spindle apparatus during oogenesis and to be a target for cell-cycle-dependent phosphorylation. The transcript of the mouse Ssty gene is specific to sperm cells. In the chicken, spindlin was found to co-localize with SUMO-1 to nuclear dots during interphase in fibroblasts, but to co-localize with chromosomes during mitosis. Thus, Spin/Ssty genes might be important in the transition from sperm cells and oocytes to the early embryo, as well as in mitosis.
Results
Here we report the discovery of a new protein motif of around 50 amino acids in length, the Spin/Ssty repeat, in proteins of the Spin/Ssty (spindlin) family. We found that in one member of this family, the human SPIN gene, each repeat resides in its own exon, supporting our view that Spin/Ssty repeats are independent functional units. On the basis of different secondary-structure prediction methods, we propose a four-stranded β-structure for the Spin/Ssty repeat.
Conclusions
The discovery of the Spin/Ssty repeat might contribute to the further elucidation of the structure and function of spindlin-family proteins. We predict that the tertiary structure of spindlin-like proteins is composed of three modules of Spin/Ssty repeats.
Background
During early oocyte development, the transcription of maternal genes ceases with the onset of meiosis. After fertilization and zygote formation, transcription of the embryonic genome starts at the two-cell stage or later, depending on the organism [1,2,3]. Thus, the amount of maternal mRNAs must be sufficient to drive the gamete through meiosis, fertilization and through the first zygotic cell division - a time span of almost 2 days in mice [1]. During this period the activation of translation from many different deadenylated, and thus dormant, mRNAs is controlled by their cytoplasmic polyadenylation [1,4].
In these early phases of mouse development, one of the most frequent transcripts regulated in this manner is that of the spindlin (Spin) gene [1,5]. The protein encoded by Spin is a meiotic-spindle-associated protein specific to the oocyte [1,5], that is phosphorylated during meiosis [6,7]. Oh et al. showed that phosphorylation modulates the ability of the Spin protein to interact with the spindle apparatus during oogenesis [6]. Phosphorylation is dependent on the Mos/MAP kinase pathway, which is controlled by meiotic-checkpoint proteins cyclin B and Cdc2 in Xenopus laevis oocytes [6,8]. Sequence similarity and mRNA expression suggest that a complementary role in sperm development seems to be fulfilled by the gene Ssty (Y-linked spermiogenesis specific transcript), a multicopy testis-specific spermatogenesis gene on the mouse Y chromosome long arm [9]. In contrast to the oocyte-specific expression of Spin, the Ssty mRNA is specifically expressed in sperm cells [9]. Dosage reduction by partial deletion of Ssty genes was suggested to cause deformed sperm heads and infertility [10,11]. However, reports on Ssty expression on the protein level are still lacking. Recently, two Spin-type genes from the chicken, Gallus gallus, have been cloned - Spin-W and Spin-Z, located on the W and Z sex chromosomes, respectively [12]. They are nearly identical to each other in their coding regions, and both were reported to be expressed in early embryos, but Spin-Z is also expressed in various adult tissues. Transfection of fibroblasts with DNA expressing fluorescent protein-tagged chSpin-W and the small ubiquitin-related modifier SUMO-1 showed the co-localization of these proteins in nuclear dots during interphase. Localization was shown to depend on the carboxy-terminal 30 amino acids of chSpin-W, especially on the presence of two phenylalanines in positions 244 and 247. However, SUMO-1 and chSpin-W could not be shown to interact directly. In contrast to its interphase localization, the red fluorescent protein-chSpinW fusion associated with chromosomes during mitosis. Although experimental results indicate that the spindlin protein family includes important players in meiosis and early embryogenesis, as well as in mitosis, their biochemical function is largely unknown.
Results and discussion
Repeat identification and analysis
At the beginning of our analysis, pairwise sequence similarity among proteins of the spindlin family was already public knowledge, with the reported average sequence identity between members being approximately 70% (entry PF02513 (Spin/Ssty protein family) in the Pfam 6.2 protein database). When we tried to identify additional family members of this protein family by scanning the NCBI nonredundant protein database (nr) using BLASTP and the human Spin protein sequence (GenBank RefSeq identifier NP_006708) as a query, we noticed a second high-scoring segment pair in the hit of the human Spin sequence with itself. Therefore we scanned the human Spin sequence for internal repeats with the program dotter and found a triple repeat spanning nearly the complete protein sequence. We aligned the repeats using CLUSTALX and corrected the alignment manually for subsequent construction of a hidden Markov model (HMM). By scanning the nr database with this model we identified the repeat in open reading frames (ORFs) of other known members of the Spin/Ssty gene family with expectation (E) values below 1e-9. Among these, we detected three repeats of typical length of 53 amino acids in the ORF of mouse Ssty, encompassing the two smaller 71 base-pair (bp) repeats that were previously noticed at the cDNA level [9]. Spindlin-family protein sequences in the nr database are from human, mouse and chicken. Among the human and mouse sequences, many were hypothetical protein sequences translated from genomic or cDNA sequences. These sequences were too similar at the protein level to conclude that they derive from different genes. To determine the number of Spin/Ssty-like genes for Mus musculus and Homo sapiens, we decided to isolate an initial redundant set of possible transcripts on the basis of the human and mouse RefSeq and UniGene databases and the database of confirmed peptides of the Ensembl human genome annotation project (Version 1.1.3), and finally to reduce the redundancy of identified transcripts by thorough sequence comparison. We identified the initial set of Spin/Ssty-like transcripts in these databases by TBLASTN searches using known spindlin-family protein sequences as queries.
For H. sapiens, we detected four different genes of the Spin/Ssty family. According to Ensembl, the chromosomal region Xp11.1 contains two SPIN-like genes: one coding for a spindlin-like transcript (Ensembl: ENST00000218159; RefSeq: NM_019003.1; UniGene: Hs.2294334; GenBankClone: Z82211) and a second in close proximity, which was named spindlin-like 2 (Ensembl: ENST00000252781; GenBankClone: Z82211). These transcripts are 99.7% identical to each other at the nucleotide level in their protein-coding regions and were first described by Laval et al. as members of the human X-linked DXF34 sequence family [13]. Another SPIN-family gene resides on chromosome Xq12 (Ensembl: ENST00000253399). The best characterized family member, the human SPIN gene (Ensembl: ENST00000223559; RefSeq: NM_006717.1; UniGene: Hs.3335321; GenBankClone: AL353748) is located on chromosome 9q22.2 and comprises three exons.
For M. musculus, scanning the RefSeq and UniGene resources revealed three Spin/Ssty-like transcripts with complete coding regions. The known Spin gene (RefSeq: NM_011462.1; UniGene: Mm.S939555) and the Ssty gene (also called Smy; RefSeq: NM_009220.1; UniGene: Mm.S936711) are around 70% identical on the protein level. A novel 1,056 bp cDNA (RefSeq: NM_023546.1; UniGene: Mm.S1997937) seems to encode a complete spindlin family protein with around 80% protein sequence identity to Ssty. Other mouse transcripts that could potentially encode complete proteins of the spindlin family seem to exist, as there are 11 additional independent cDNA assemblies in UniGene (Mm.S1975038, Mm.S1922195, Mm.S499811, Mm.S227336, Mm.S1973836, Mm.S707442, Mm.S781768, Mm.S502745, Mm.S782972, Mm.S778767, Mm.S787945). Their ORFs are interrupted or incomplete, however. Increased expressed sequence tag (EST) coverage and quality of these assemblies might reveal more functional spindlin family members. The high number of SPIN-like transcripts in mice is in agreement with previous reports [11,13] that presented evidence for the existence of a multi-copy Ssty-like gene family on the mouse Y chromosome. As three of four human Spin/Ssty-like genes consist of a single exon, and alternative transcripts of the human triple-exon gene SPIN have not yet been reported, alternative splicing is unlikely to contribute to the diversity of Spin/Ssty transcripts in mouse.
To identify Spin/Ssty-family genes from other organisms, we scanned the dbEST database using the TBLASTN program and known spindlin-family proteins as queries. We found additional ESTs in several organisms. We assembled ESTs from Bos taurus (GenBank AV588979, AV588980, BE667003, BF045945), determined the full coding region by alignment with the human Spin protein sequence and added the Spin/Ssty repeat regions to the repeat alignment (Figure 1). Furthermore, we detected Spin/Ssty repeats in several single ESTs that represent fragments of putative Spin/Ssty-family genes. However, we were not able to obtain full coding regions by assembling these ESTs. Among them were several ESTs from Rattus norvegicus, an EST from X. laevis (GenBank BG018656), two ESTs from Oryzias latipes (GenBank AU169984, AU178597) and one EST from Danio rerio (GenBank AW077586), indicating the existence of Spin/Ssty repeats in fish and frog proteins. We did not detect Spin/Ssty repeats in the proteomes of Drosophila melanogaster or Caenorhabditis elegans. Thus, Spin/Ssty repeats are currently restricted to vertebrate proteins.
Figure 1.

Alignment, consensus and secondary structure of Spin/Ssty repeats. The upper part shows the alignment of Spin/Ssty repeats. A two-letter organism-specific code (mm, Mus musculus; hs, Homo sapiens; ol, Oryzias latipes; bt, Bos taurus; gg, Gallus gallus) appears on the far left of each line, followed by a protein identifier, the repeat subtype (type A, amino-terminal; type B, central; type C, carboxy-terminal), the database identifier, the start and end residue of the Spin/Ssty repeat in the protein and the protein sequence. Amino acids are colored according to an 80% consensus. h, hydrophobic (ACFGILMVWP, white letters on dark blue); l, aliphatic (ILV, cyan); p, polar (NQSTY, yellow); a, aromatic (FHWY, purple); -, acidic (ED, green); +, basic (HKR, red letters on yellow); t, tiny (GAS, gray). The secondary-structure predictions of various programs run by the Jpred2 server and the Jpred2 consensus prediction are presented below. E, β strand; H, α helix.
The subsequent analysis of Spin/Ssty repeats is exclusively based on repeats from known proteins or complete ORFs, in order to exclude low-quality sequences from the analysis. To include Spin/Ssty repeats from a fish protein, an exception is made for the O. latipes EST AU169984, which contains an incomplete ORF comprising two complete Spin/Ssty repeats without interruption by frameshift errors.
Using our initial HMM we identified three repeats per protein (two for the incomplete O. latipes protein) with E values below 1e-15. We aligned the repeats (Figure 1) and constructed three HMMs: two by using only repeats with less than 75 and 90% pairwise sequence identity, another by using all repeats in the seed alignment. All HMMs re-identified the repeats with E values below 1e-22. However, scanning the nr database with these new models did not identify further Spin/Ssty repeats. We submitted a description and an alignment of the Spin/Ssty repeat to Pfam (Pfam 6.6: PF02513), which replaced the previous Spin/Ssty protein family entry.
For single combinations of Spin/Ssty repeats, the pairwise sequence identity drops below 15%. To test the significance of the similarity among the repeat subtypes (amino-terminal, central, carboxy-terminal) and to exclude HMM training artifacts, we carried out a cross-validation test. We constructed HMMs for each repeat subtype and tried to detect the repeats of the remaining subtypes. For this approach we used five nonredundant proteins (gg_SPINZ, bt_SPINH, hs_SPINX2, mm_SSTY, mm_SPINL; Figure 1). We could identify the complete set of repeats from the five proteins with E values below 5e-3 and thus confirmed that the subgroups are evolutionarily related.
Phylogenetic analysis of the Spin/Ssty repeats from the five nonredundant proteins with the neighbor-joining method after removal of gapped alignment columns confirmed the existence of three subtypes of repeats, the amino-terminal, central and carboxy-terminal subtype (Figure 2). In the genomic structure of the human SPIN gene on chromosome 9 each Spin/Ssty repeat resides in its own exon, supporting our view that the Spin/Ssty repeats represent structural or functional units. In summary, the phylogenetic analysis and the gene structure of the SPIN gene suggest that the first spindlin-family protein evolved by subsequent duplications of an ancient exon and that these duplications preceded the speciation events leading to birds and mammals.
Figure 2.
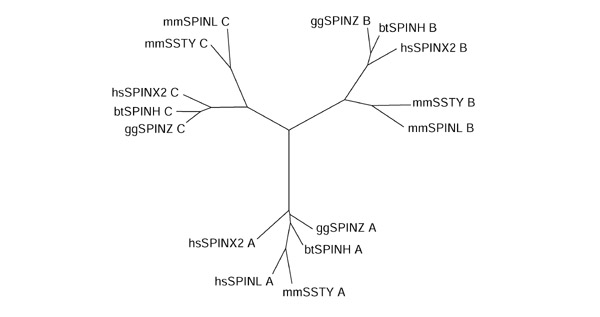
Phylogenetic tree of Spin/Ssty repeats. The tree was built from 15 repeats of five sequences. The labels stand for the repeats in the five proteins and consist of three fields: a two-letter code for the organism, an identifier for the protein sequence and the repeat subtype (see Figure 1 for terminology). Note the three groups of repeats: the amino-terminal repeats form one subtree, the central repeats form a second, and the carboxy-terminal repeats form a third. Thus, the phylogenetic classification of repeats matches the classification of the repeats by their positions in the proteins.
Structure prediction
We made secondary-structure predictions using several programs via the Jpred2 server with the alignment of the whole family and the alignments of each of the amino-terminal, central and carboxy-terminal repeat subfamilies as a query. The consensus prediction for the whole alignment suggests four β strands for the Spin/Ssty repeat. Although the isolated central Spin/Ssty repeat is predicted to form an α helix in exchange for the second β strand, the single predictions for the amino- and carboxy-terminal repeat subtypes are in agreement with the prediction based on the whole family. Because in most cases the accuracy of secondary-structure predictions is higher when alignments of more diverse protein-family members are used, we believe that the predictions based on the whole family are the most reliable, and we suggest an all-β structure with four β strands for all Spin/Ssty repeats. Attempts to assign a known protein fold to the Spin/Ssty repeat using different fold-prediction methods via the Structure Prediction Meta Server did not lead to significant predictions.
Conclusions
Our findings might serve as a basis for future work on this new class of repeats. The Spin/Ssty repeat alignment will assist in detecting further family members in other species and in the search for an evolutionary origin of the spindlin family of proteins. The detection of Spin/Ssty repeats in proteins with other domain architectures might provide a clue to the function of the spindlin family. Knowledge of the repeat structure of spindlin-like proteins can support further experimental work. Once interaction partners or biochemical functions are identified for the spindlin-like proteins, hypotheses based on the repeat architecture can be generated for further experiments: site-directed mutagenesis studies, that are targeted on conserved residues, are most likely to disrupt the structure or destroy the function of a protein; attempts to delete certain regions of spindlin-family proteins or to swap regions between family members in order to explore their function, can now be guided by the repeat architecture in these sequences to choose more reasonable borders. Finally, we hope that our findings will support the exploration of the tertiary structure of spindlin-like protein, as the Spin/Ssty sequence repeat is probably reflected by a repeated structural element with four β strands, which currently cannot be assigned to a known type of protein fold.
Materials and methods
Searching sequence databases
We scanned several databases to identify ESTs, ORFs, known protein sequences or gene structures of the Spin/Ssty gene family. We used the following databases, which can all be downloaded from the NCBI ftp server [14] (database filenames are given in brackets) or the ENSEMBL ftp server [15]: the Non-redundant Protein Sequence Database (nr.Z), dbEST (est.Z), the mouse and human RefSeq mRNA and peptide sequences (hs.fna.gz, hs.faa.gz, mouse.fna.gz, mouse.faa.gz), the mouse and human UniGene databases (Hs.seq.uniq, Build #141; Mm.seq.uniq, Build #95) and the ENSEMBL set of confirmed human peptides and corresponding transcripts (ensembl.pep.gz, ensembl.cdna.gz, Ver.1.1.3). Pairwise sequence-similarity searches in these databases were carried out using the gapped versions of the programs of the BLAST program package version 2.1.2 with default scoring schemes [16].
Repeat analysis
The aim of the program dotter [17] is to visualize local sequence similarity between two sequences by allowing the user to view the dot matrix of the sequence comparisons and the alignment of the sequences in parallel. Here, dotter was used to compare sequences with themselves to examine them for repeats. Finally, it was used to refine the borders of repeat regions before their selection for the alignment.
Multiple alignment and phylogenetic tree construction
Multiple alignments were carried out with CLUSTALX version 1.8.1 [18] using the BLOSUM62 substitution matrix. The neighbor-joining algorithm [19] of CLUSTALX was used to build phylogenetic trees after gaps were removed from the alignments. The drawtree program of the PHYLIP package version 3.5 was used to visualize the tree [20].
Protein-sequence profile searches
For sensitive detection of repeats we built profile HMMs from the diverse alignments using the HMMER program suite [21] with default options for model building with hmmbuild (hmmls/domain alignment) and calibration with hmmcalibrate (sampled sequences: 5,000; mean length 350). Protein database searches with these HMMs were carried out using the hmmsearch program.
EST assembly
Having identified ESTs of a putative novel SPIN-family gene, we used the program Gap version 4.4 [22] for their assembly to derive a consensus representation of the complete mRNA sequence.
Secondary-structure prediction
Secondary-structure predictions were performed with the consensus method of the Jpred2 server [23]. This method is built on several other well-known secondary-structure prediction algorithms such as DSC [24], Jnet [25], NNSSP [26], PHD [27] and Zpred [28]. According to the authors, the Jpred2 consensus method reaches a level of 75% accuracy in secondary-structure prediction and outperforms the single methods.
Tertiary-structure prediction
We tried to assign known protein folds to the identified repeats by four widely used methods: 3D-PSSM [29], FUGUE [30], SUPERFAMILY [31] and SAM-T99 [32].
References
- Oh B, Hwang S, McLaughlin J, Solter D, Knowles BB. Timely translation during the mouse oocyte-to-embryo transition. Development. 2000;127:3795–3803. doi: 10.1242/dev.127.17.3795. [DOI] [PubMed] [Google Scholar]
- Schultz RM. Regulation of zygotic gene activation in the mouse. BioEssays. 1993;15:531–538. doi: 10.1002/bies.950150806. [DOI] [PubMed] [Google Scholar]
- Telford NA, Watson AJ, Schultz GA. Transition from maternal to embryonic control in early mammalian development: a comparison of several species. Mol Reprod Dev. 1990;26:90–100. doi: 10.1002/mrd.1080260113. [DOI] [PubMed] [Google Scholar]
- Huarte J, Stutz A, O'Connell ML, Gubler P, Belin D, Darrow AL, Strickland S, Vassalli JD. Transient translational silencing by reversible mRNA deadenylation. Cell. 1992;69:1021–1030. doi: 10.1016/0092-8674(92)90620-r. [DOI] [PubMed] [Google Scholar]
- Oh B, Hwang SY, Solter D, Knowles BB. Spindlin, a major maternal transcript expressed in the mouse during the transition from oocyte to embryo. Development. 1997;124:493–503. doi: 10.1242/dev.124.2.493. [DOI] [PubMed] [Google Scholar]
- Oh B, Hampl A, Eppig JJ, Solter D, Knowles BB. SPIN, a substrate in the MAP kinase pathway in mouse oocytes. Mol Reprod Dev. 1998;50:240–249. doi: 10.1002/(SICI)1098-2795(199806)50:2<240::AID-MRD15>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- Howlett SK. A set of proteins showing cell cycle dependent modification in the early embryo. Cell. 1986;45:387–396. doi: 10.1016/0092-8674(86)90324-7. [DOI] [PubMed] [Google Scholar]
- Frank-Vaillant M, Haccard O, Ozon R, Jessus C. Interplay between Cdc2 kinase and the c-Mos/MAPK pathway between metaphase I and metaphase II in Xenopus oocytes. Dev Biol. 2001;231:279–288. doi: 10.1006/dbio.2000.0142. [DOI] [PubMed] [Google Scholar]
- Bishop CE, Hatat D. Molecular cloning and sequence analysis of a mouse Y chromosome RNA transcript expressed in the testis. Nucleic Acids Res. 1987;15:2959–2969. doi: 10.1093/nar/15.7.2959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgoyne PS, Mahadevaiah SK, Sutcliffe MJ, Palmer SJ. Fertility in mice requires X-Y pairing and a Y-chromosomal "spermiogenesis" gene mapping to the long arm. Cell. 1992;71:391–398. doi: 10.1016/0092-8674(92)90509-b. [DOI] [PubMed] [Google Scholar]
- Conway SJ, Mahadevaiah SK, Darling SM, Capel B, Rattigan AM, Burgoyne PS. Y353/B: a candidate multiple-copy spermiogenesis gene on the mouse Y chromosome. Mamm Genome. 1994;5:203–210. doi: 10.1007/BF00360546. [DOI] [PubMed] [Google Scholar]
- Itoh Y, Hori T, Saitoh H, Mizuno S. Chicken spindlin genes on W and Z chromosomes: transcriptional expression of both genes and dynamic behavior of spindlin in interphase and mitotic cells. Chromosome Res. 2001;9:283–299. doi: 10.1023/a:1016694513051. [DOI] [PubMed] [Google Scholar]
- Laval SH, Reed V, Blair HJ, Boyd Y. The structure of DXF34, a human X-linked sequence family with homology to a transcribed mouse Y-linked repeat. Mamm Genome. 1997;8:689–691. doi: 10.1007/s003359900538. [DOI] [PubMed] [Google Scholar]
- National Center for Biotechnology Information ftp server ftp://ncbi.nlm.nih.gov
- ENSEMBL ftp server ftp://ftp.ensembl.org
- Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonnhammer ELL, Durbin R. A dot-matrix program with dynamic threshold control suited for genomic DNA and protein sequence analysis. Gene. 1995;167:GC1–GC10. doi: 10.1016/0378-1119(95)00714-8. [DOI] [PubMed] [Google Scholar]
- Jeanmougin F, Thompson JD, Gouy M, Higgins DG, Gibson TJ. Multiple sequence alignment with CLUSTALX. Trends Biochem Sci. 1998;23:403–405. doi: 10.1016/s0968-0004(98)01285-7. [DOI] [PubMed] [Google Scholar]
- Saitou N, Nei M. The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol. 1987;4:406–425. doi: 10.1093/oxfordjournals.molbev.a040454. [DOI] [PubMed] [Google Scholar]
- Felsenstein J. PHYLIP - Phylogeny Inference Package (Version 3.2). Cladistics. 1989;5:164–166. [Google Scholar]
- Eddy SR. Profile hidden Markov models. Bioinformatics. 1998;14:755–763. doi: 10.1093/bioinformatics/14.9.755. [DOI] [PubMed] [Google Scholar]
- Bonfield JK, Smith KF, Staden R. A new DNA sequence assembly program. Nucleic Acids Res. 1995;23:4992–4999. doi: 10.1093/nar/23.24.4992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuff JA, Clamp ME, Siddiqui AS, Finlay M, Barton GJ. Jpred: A consensus secondary structure prediction server. Bioinformatics. 1998;14:892–893. doi: 10.1093/bioinformatics/14.10.892. [DOI] [PubMed] [Google Scholar]
- King RD, Sternberg MJE. Machine learning approach for the prediction of secondary structure. J Mol Biol. 1990;216:441–457. doi: 10.1016/S0022-2836(05)80333-X. [DOI] [PubMed] [Google Scholar]
- Cuff JA, Barton GJ. Application of multiple sequence alignment profiles to improve protein secondary structure prediction. Proteins. 2000;40:502–511. doi: 10.1002/1097-0134(20000815)40:3<502::AID-PROT170>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- Salamov AA, Solovyev VV. Prediction of protein secondary structure by combining nearest-neighbor algorithms and multiple sequence alignments. J Mol Biol. 1995;247:11–15. doi: 10.1006/jmbi.1994.0116. [DOI] [PubMed] [Google Scholar]
- Rost B, Sander C. Combining evolutionary information and neural networks to predict protein secondary structure. Proteins. 1994;19:55–72. doi: 10.1002/prot.340190108. [DOI] [PubMed] [Google Scholar]
- Zvelebil MJJM, Barton GJ, Taylor WR, Sternberg MJE. Prediction of protein secondary structure and active sites using the alignment of homologous sequences. J Mol Biol. 1987;195:957–961. doi: 10.1016/0022-2836(87)90501-8. [DOI] [PubMed] [Google Scholar]
- Kelley LA, MacCallum RM, Sternberg MJE. Enhanced genome annotation using structural profiles in the program 3D-PSSM. J Mol Biol. 2000;299:501–522. doi: 10.1006/jmbi.2000.3741. [DOI] [PubMed] [Google Scholar]
- Shi J, Blundell TL, Mizuguchi K. FUGUE: sequence-structure homology recognition using environment-specific substitution tables and structure-dependent gap penalties. J Mol Biol. 2001;310:243–257. doi: 10.1006/jmbi.2001.4762. [DOI] [PubMed] [Google Scholar]
- Gough J, Karplus K, Hughey R, Chothia C. Assignment of homology to genome sequences using a library of hidden Markov models that represent all proteins of known structure. J Mol Biol. 2001;313:903–919. doi: 10.1006/jmbi.2001.5080. [DOI] [PubMed] [Google Scholar]
- Karplus K, Barrett C, Hughey R. Hidden Markov models for detecting remote protein homologies. Bioinformatics. 1998;14:846–856. doi: 10.1093/bioinformatics/14.10.846. [DOI] [PubMed] [Google Scholar]