

Abstract
Background
To demonstrate the involvement of tobacco smoking in the pathophysiology of lung disease, the responses of pulmonary epithelial cells to cigarette smoke condensate (CSC) — the particulate fraction of tobacco smoke — were examined.
Methods
The human alveolar epithelial cell line A549 and normal human bronchial epithelial cells (NHBEs) were exposed to 0.4 μg/ml CSC, a concentration that resulted in >90% cell survival and <5% apoptosis. Changes in gene expression and signaling responses were determined by RT-PCR, western blotting and immunocytofluorescence.
Results
NHBEs exposed to CSC showed increased expression of the inflammatory mediators sICAM-1, IL-1β, IL-8 and GM-CSF, as determined by RT-PCR. CSC-induced IL-1β expression was reduced by PD98059, a blocker of mitogen-actived protein kinase (MAPK) kinase (MEK), and by PDTC, a NFκB inhibitor. Analysis of intracellular signaling pathways, using antibodies specific for phosphorylated MAPKs (extracellular signal-regulated kinase [ERK]-1/2), demonstrated an increased level of phosphorylated ERK1/2 with increasing CSC concentration. Nuclear localization of phosphorylated ERK1/2 was seen within 30 min of CSC exposure and was inhibited by PD98059. Increased phosphorylation and nuclear translocation of IκB was also seen after CSC exposure. A549 cells transfected with a luciferase reporter plasmid containing a NFκB-inducible promoter sequence and exposed to CSC (0.4 μg/ml) or TNF-α (50 ng/ml) had an increased reporter activity of approximately 2-fold for CSC and 3.5-fold for TNF-α relative to untreated controls.
Conclusion
The acute phase response of NHBEs to cigarette smoke involves activation of both MAPK and NFκB.
Keywords: bronchial, cigarette, MAPK, NF-kappaB, signal transduction
Introduction
The association of inhaled particulate pollution and cigarette smoking with pulmonary disease, such as chronic obstructive pulmonary disease, is well documented [1], but the specific early responses of lung epithelial cells to toxic substances in particulates — that predispose the cells to disease — have not been elucidated. Cigarette smoke has also been considered a major player in the pathogenesis of asthma and as a trigger for acute symptoms [2]. Exposure to cigarette smoke activates an inflammatory cascade in the airway epithelium resulting in the production of a number of potent cytokines and chemokines, with accompanying damage to the lung epithelium, increased permeability, and recruitment of macrophages and neutrophils to the airway [3]. Even brief exposure to cigarette smoke has been shown to increase expression of IL-8 in primary cultures of human bronchial epithelial cells (HBEs) in the presence of dust mite allergen [4]. Increased release of the chemokine IL-8 has also been shown for cultured HBEs exposed to cigarette smoke extract (CSE) [5,6] and diesel exhaust particles [7]. However, the precise molecular events that bring about these intracellular changes and acute inflammation in lung epithelial cells are not well understood.
Upregulation of cytokine gene expression in epithelial cells has been linked to activation of specific signaling pathways. Involvement of the mitogen-activated protein kinase (MAPK) pathway, extracellular signal-regulated protein kinase (ERK)-1/2 (p44/p42), in cytokine regulation has been demonstrated in HBEs exposed to diesel exhaust particles [7] but not to cigarette smoke. NFκB is a ubiquitous transcription factor involved in regulating inflammatory responses, and its targets include several of the genes encoding cytokines and chemokines. The activation of NFκB by oxidative stress, inflammatory cytokines such as IL-1β and by TNF-α has been studied in depth [8], but much less is known about the effects of cigarette smoke on NFκB activation.
The purpose of this study was to gain insight into the mechanism of inflammation induced by cigarette smoke, specifically the acute response to smoke. The human alveolar epithelial cell line A549 and normal human bronchial epithelial cells (NHBEs) were exposed to the particulate fraction of cigarette smoke (cigarette smoke condensate; CSC) and examined for the production of proinflammatory cytokines and activation of MAPK and NFκB. The results demonstrate that the acute phase response of pulmonary epithelial cells to cigarette smoke involves activation of ERK1/2 and NFκB
Materials and methods
Reagents
RPMI 1640, penicillin, streptomycin, fungizone, and trypsin/EDTA were obtained from Cellgro (Herndon, VA, USA). Fetal bovine serum was from Atlanta Biochemicals (Atlanta, GA, USA). The culture medium (BEGM), growth factors, trypsin, trypsin neutralizing solution, and Hanks' balanced salt solution for growing and treating primary human epithelial cells were purchased from Clonetics (San Diego, CA, USA. CSC was purchased from Murty Pharmaceuticals (Lexington, KY, USA) and was prepared using a Phipps-Bird 20-channel smoking machine designed for FTC testing. The particulate matter from Kentucky standard cigarettes (1R3F; University of Kentucky, KY, USA) was collected on Cambridge glass fibre filters and the amount obtained determined by weight increase of the filter. CSC was prepared by dissolving the collected smoke particulates in dimethyl sulfoxide (DMSO) to yield a 4% solution (w/v). The average yield of CSC was 26.1 mg/cigarette. The CSC was diluted into DMSO and aliquots were kept at -80°C. Oligonucleotide primers for PCR were synthesized by Operon Technologies (Alameda, CA, USA). Taq polymerase, deoxynucleotide triphosphates and Taq buffer were from Promega (Madison, WI, USA). Anti-ERK1/2 (p44/p42) polyclonal antibody, anti-phosphorylated ERK1/2 polyclonal antibody, and horseradish peroxidase (HRP)-conjugated goat anti-rabbit IgG were purchased from New England Biolabs (Beverly, MA, USA). Anti-phosphorylated IκB-α polyclonal antibody, IκB monoclonal antibody and chemiluminescent detection reagents were from Cell Signaling Technology (Beverly, MA, USA).
Cell culture
The human alveolar epithelial cell line A549 (ATCC, Rockville, MD, USA) was grown in RPMI plus 10% fetal bovine serum, 100 U/ml penicillin, 100 U/ml streptomycin, and 250 ng/ml amphotericin-B in an atmosphere of 5% carbon dioxide and 95% air at 37°C. Primary cultures of NHBEs (Clonetics, San Diego, CA, USA) were obtained at second passage and cultured according to the manufacturer's instructions using the media and growth factors supplied with the cells. The cells were seeded into 12-well culture plates at 105 cells per well and used between passages three and seven.
Exposure of epithelial cells to cigarette smoke condensate
Cultures of A549 cells or NHBEs were grown to 50–75% confluence, then incubated with serum-deficient media for 4 hours before exposure to CSC. Treatment with CSC, at a final concentration of 0.4 μg/ml in 0.5% DMSO, was carried out in modified Eagle's medium (MEM) containing Earle's salts at 37°C for 30 min to 5 hours. Control cells were treated with the same concentration of DMSO without CSC. For CSC dose-response experiments, cells were incubated for 30 min at 37°C with concentrations of CSC ranging from 0.004 to 20 μg/ml in 0.5% DMSO. Cell viability after CSC treatment was determined using the trypan blue dye exclusion test and was compared to viability of untreated control cells.
TUNEL assay
The induction of apoptosis was determined by means of the TUNEL (deoxynucleotidyl transferase-mediated dUTP nick end labeling) assay using a kit supplied by Promega (Madison, WI, USA). Cultures of A549 cells or NHBEs were grown on 8-well slides and treated with CSC as described above, fixed with 4% paraformaldehyde for 15 min at room temperature, then examined by fluorescent microscopy. Nuclei were counterstained by the addition of 4',6-diamidino-2-phenylindole dihydrochloride (DAPI; Molecular Probes, Eugene, OR, USA) to the mounting medium. The percentage of apoptotic cells was determined by counting green fluorescent nuclei relative to the total number of DAPI-positive nuclei.
Reverse-transcriptase PCR
A549 cells or NHBEs were grown to approximately 75% confluence in 12-well plates, treated with various concentrations of CSC in 0.5% DMSO, and total RNA was isolated using RNeasy method (Qiagen, Valencia, CA, USA). RNA (1 μg) was converted to cDNA in a 50 μl reaction containing 1 U of avian myeloblastosis virus reverse transcriptase (New England Biolabs, Beverly, MA, USA), 1 mM dNTP's, and 10 μg/ml random hexamer primers in 1× reverse transcriptase buffer at 42°C for 50 min; the reaction was terminated by heating to 90°C for 10 min. The resulting cDNA was used as template for PCR amplification using the primer pairs (Operon Technologies, Alameda, CA, USA) listed in Table 1. As a loading control parallel PCR reactions were carried out using a primer pair for human GAPDH (see Table 1). PCR products were separated on agarose gels, the ethidium bromide-stained bands were digitally photographed using a ChemImager 4400 (Alpha Inotech, San Leandro, CA, USA) and scanned using the AphaEase version 5.5 densitometry program. The experimental values were normalized to the corresponding GAPDH value and expressed as relative intensity.
Table 1.
Primer pairs used for PCR of cDNA from CSC-treated NHBEs and A549 cells
| Name | Forward primer sequence (5'→3') | Reverse primer sequence (5'→3') |
| SICAM-1 | Atggctcccagcagcccc | cacctggcagcgtagggt |
| Il-1b | Gacctggacctctgccctctg | aggtattttgtcattactttc |
| RANTES | tgaaggtctccgcggcacgcct | ctagctcatctccaaagagttg |
| IL-6 | Aactccttctccacaagcg | tggactgcaggaactcctt |
| IL-8 | Atttctgcagctctgtgtaa | tcctgtggcatccacgaaact |
| GM-CSF | Atgtggctgcagagcctgc | tcactcctggactggctcc |
| GAPDH | atggggaaggtgaaggtcgga | gagatgatgacccttttggc |
Western blotting and immunocytochemistry
A549 cells or NHBEs were treated with CSC as described above and whole-cell protein extracts were prepared by scraping cells into lysis buffer containing 20 mM Tris-HCl (pH 8.0), 1 mM EDTA, 1 mM sodium orthovanadate, 1 mM sodium fluoride, 1 μg/ml each of leupeptin and pepstatin, and 1 μM PMSF. Cell lysis was achieved by sonicating the suspension on ice with three 10-second bursts at a power setting of 2 mW (Sonic Dismembrator, Fisher Scientific Co., Atlanta, GA, USA). Lysates were clarified by centrifugation at 10,000 × g for 30 min at 4°C and the protein concentration of the whole-cell extracts was determined by the bicinchoninic protein assay (Pierce Biochemical Co., Rockford, IL, USA). Aliquots of 10 μg of protein were separated by SDS-PAGE and electroblotted to nitrocellulose. The filters were blocked with 2% nonfat dry milk in 10 mM Tris-HCl (pH 8.0), 0.15 M sodium chloride, 0.2% Tween-20 (TBST) then incubated with primary antibody (1:1000 dilution) for 2 hours at room temperature. After washing in Tris-buffered saline with tween 20 (TBST), blots were incubated for one hour at room temperature with HRP-conjugated secondary antibody, washed again and detected by chemiluminescence. The protein bands were quantified by densitometry as described above. For immunocytochemistry, NHBEs grown on 8-well chamber slides were exposed to CSC at a concentration of 0.4 μg/ml for 30 min. Cells were fixed with 4% paraformaldehyde, blocked with BSA, immunostained using anti-phosphorylated ERK1/2, and visualized using HRP-conjugated secondary antibody and bisbenzamidine substrate.
Transfection with NFκB-luciferase reporter constructs and assay for gene expression
A549 cells were grown to 50–80% confluence on 12-well plates and transfected with the Mercury pathway profiling system vector, pNFκB-TA-Luc (Clontech, Palo Alto, CA, USA), using lipofectamine (Gibco/BRL, Rockville, MD, USA) and used according to the manufacturer's recommendations. Cells were transfected using lipofectamine from Bibco/BRL (Rockville, MD). The plasmid, pVAX-LacZ, (Clontech, Palo Alto, CA, USA) was used as transfection control.
DNA/lipofectamine complexes were allowed to form for 30 min at room temperature in RPMI (without antibiotics) containing 1 μg of reporter plasmid, 20 ng of pVAX-LacZ for monitoring transfection efficiency, and 10 μg of lipofectamine. Cells were transfected for 18 h. Prior to CSC challenge, the transfected cells were preincubated for 4 hours in minimal Eagle's medium with Earle's salts to remove serum factors. Assays were done in triplicate and consisted of untreated controls, treatment with 50 ng/ml TNF-α and treatment with 0.4 μg/ml CSC. After treatments, the cells were lysed by addition of cell lysis buffer as supplied in the luciferase assay kit from Clontech, scraped from the wells and centrifuged 1 min at 12,000 × g. Aliquots of 20 μl of each lysate were transferred to flat-bottomed, white microtiter plates (Dynex, Chantilly, VA, USA) and read on a Dynex MLX plate reader in flash mode. The transfection efficiency was normalized by determining β-galactosidase activity in the lysates from the cotransfected pLacZ plasmid.
Statistical Analysis
All values are expressed as mean ± standard error of the mean (SEM). Comparisons of experimental values for CSC-treated cells to untreated controls were done by analysis of variance (ANOVA). Probability values of < 0.05 were considered significant.
Results
Effects of cigarette smoke condensate on cell viability and apoptosis
To determine the effects of CSC on cell viability, A549 cells and NHBEs were exposed to increasing concentrations of CSC for 30 min then examined using the trypan blue dye-exclusion test (Fig. 1A). Maximum toxicity for A549 at the highest concentration of CSC (20 μg/ml) was 15%. At lower CSC concentrations (<0.1 μg/ml), survival was greater than 90% for both A549 cells and NHBEs. Induction of apoptosis by CSC was determined in A549 cells and NHBEs by using the TUNEL assay and confirmed by DNA fragmentation analysis (data not shown). NHBEs showed 5 to 10% apoptosis at moderate doses of CSC; A549 cells showed less than 5% apoptosis (Fig. 1B). However, with increasing concentration of CSC, NHBEs showed as high as 20% apoptosis, while A549 cells remained unchanged.
Figure 1.
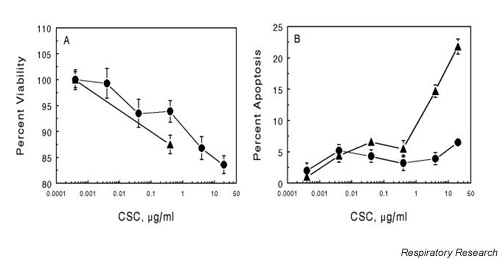
Effect of CSC on viability and apoptosis in A549 cells and NHBEs. Cells were grown to about 70% confluence in 12-well plates (Panel A, viability) or 8-well chamber slides (Panel B, apoptosis). Cells were exposed to the indicated concentration of CSC for 30 min then rinsed with PBS and processed for each assay. Viability was determined by trypan blue vital staining and expressed as percent of total cells. Apoptosis was measured by fluorescent TUNEL assay and is expressed as the percent of cells exhibiting apoptosis relative to the total number of cells. The values are the mean ± SEM for n = 3. (--●-- A549; --▲-- NHBE) CSC = cigarette smoke condensate; NBHE = normal human bronchial epithelial cell; PBS = phosphate-buffered saline; SEM = standard error of the mean; TUNEL = deoxynucleotidyl transferase-mediated dUTP nick end labeling
Changes in expression of specific cytokines and chemokines after exposure to cigarette smoke condensate
To characterize the response of epithelial cells to CSC we determined the changes in message levels for a group of cytokines, chemokines and cell-adhesion molecules. Total RNA isolated from NHBEs following exposure to CSC was subjected to RT-PCR, and the results are shown in Fig. 2. IL-1β was induced to a level 9.5 times that of untreated controls, while the expression of IL-8 and GM-CSF was increased 2.7-fold and 2.2-fold, respectively. Expression of the adhesion molecule ICAM-1 was found to be approximately 1.6 times the level of control. IL-6 and the chemokine RANTES were elevated slightly (Fig. 2). This pattern of enhancement of proinflammatory mediator expression implies that specific effector compounds are present in CSC.
Figure 2.
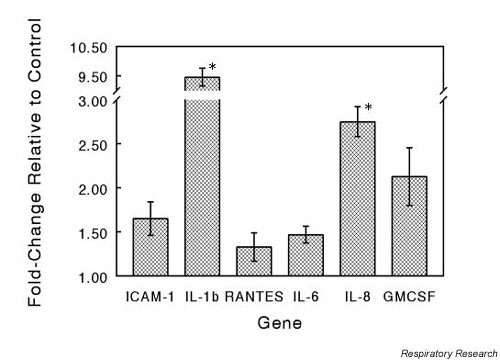
Effect of CSC treatment on proinflammatory gene expression. NHBEs were exposed to CSC at a concentration of 0.4 μg/ml for 30 min at 37°C. Total RNA was isolated and specific gene expression measured by RT-PCR assay. Target gene expression levels were normalized to the level of GAPDH expression in each reaction and expressed as fold change relative to control. The values are means ± SEM for n = 3. Significant differences at P < 0.05 are indicated by *. CSC = cigarette smoke condensate; NBHE = normal human bronchial epithelial cell; RT-PCR = reverse transcriptase-polymerase chain reaction; SEM = standard error of the mean
Involvement of the MAPK pathway in response to cigarette smoke condensate
To examine if CSC-upregulation of cytokine production involves activation of MAPKs we examined CSC-treated NHBEs for specific phosphorylated species. Fig. 3A shows the results of a western blot of whole-cell extracts from NHBEs exposed to increasing concentrations of CSC and probed for phosphorylated ERK1/2, the activated form of ERK1/2. Band densities on the film were measured and the protein loading variations were adjusted by normalizing to the levels of non-phosphorylated ERK1/2. The level of ERK1/2 phosphorylation increased rapidly with increasing exposure to CSC, to a maximum of nearly twice the control. To confirm the results of immunoblot detection of phosphorylated ERK1/2, NHBEs were exposed to 0.4 μg/ml CSC for 30 min, then fixed and stained for phosphorylated ERK1/2. Exposure to CSC caused a marked increase in nuclear staining of cells with phosphorylated ERK1/2 antibodies (Fig 3B). This nuclear localization and phosphorylated ERK1/2 staining was largely blocked by pretreating with the MEK inhibitor PD98059. The CSC-induced increase in IL-1β expression was also reduced by treatment of the cells with PD98059 or the NFκB inhibitor, PDTC (Fig. 3C). These results suggest that CSC activates ERK1/2 in these cells within 30 min.
Figure 3.
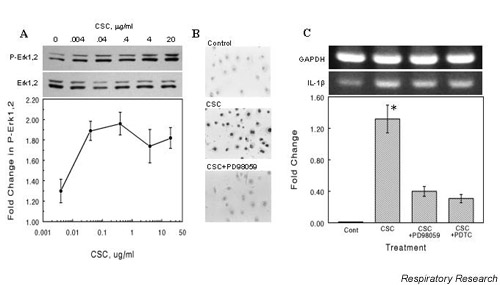
CSC-induced phosphorylation of ERK1/2 and IL-1β expression. (A) CSC induces ERK1/2 phosphorylation. NHBEs were incubated with the indicated concentrations of CSC for 30 min at 37°C. Whole-cell protein extracts were prepared from cell lysates, aliquots of 10 μg of protein were separated by SDS-PAGE, transferred to nitrocellulose and probed with an antibody to phosphorylated ERK1/2. Blots were then stripped and reprobed for ERK1/2. The band intensities were determined by densitometry and the fold-change relative to control was plotted as means ± SEM. (B) Nuclear localization of phosphorylated ERK1/2 in NHBEs. Cells were incubated with 0.4 μg/ml CSC for 30 min at 37°C, then fixed with paraformaldehyde and probed with antibody to phosphorylated ERK1/2. Visualization was achieved by incubating the cells with HRP-conjugated secondary antibody and bisbenzamidine substrate. Cells were treated with vehicle alone (Control), with CSC (CSC), or with CSC and the MEK inhibitor, PD98059 (20 μM) (CSC + PD98059). (C) Inhibition of ERK1/2 or IκB phosphorylation attenuates IL-1β expression in A549. Cells were treated with 50 μg/ml PD98059 or 1 mM pyrrolidine dithiocarbamate (PDTC) for 30 min then exposed to 0.4 μg/ml CSC for 60 min. RT-PCR was performed on RNA extracted from these cells using IL-1β-specific primers. Band densities were normalized to GAPDH levels, expressed as fold change relative to untreated controls, and plotted as mean ± SEM for n = 3. The * indicates significance at P < 0.05. CSC = cigarette smoke condensate; ERK = extracellular signal-regulated kinase; IκB = inhibitor of NFκB; MEK = MAPK kinase; NBHE = normal human bronchial epithelial cell.
Cigarette smoke condensate treatment induces rapid activation of the NFκB pathway
To determine whether upregulation of cytokine production by CSC involved activation of NFκB, we examined CSC-treated epithelial cells for phosphorylation of the inhibitor of NFκB (IκB). NHBEs were treated with increasing concentrations of CSC and whole-cell extracts were immunoblotted for phosphorylated IκB. Activation of NFκB involves the increased phosphorylation and proteolytic degradation of IκB. Treatment of cells with increasing concentrations of CSC resulted in an initial rise in phosphorylated IκB levels followed by a decrease (Fig. 4A). Protein bands were quantitated by densitometry on the film and the phosphorylated IκB densities were normalized to the levels of IκB. NHBEs exposed to CSC also showed an increase in nuclear staining of phosphorylated IκB as detected by immunocytochemical staining using an antibody to phosphorylated IκB (Fig. 4B).
Figure 4.
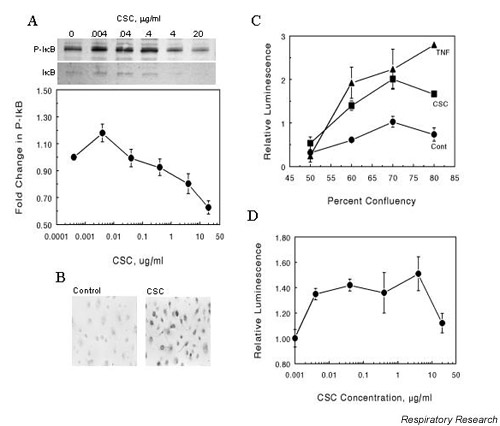
Phosphorylation of IκB and expression of NFκB-luciferase reporter gene in cells exposed to CSC. (A) CSC exposure results in phosphorylation of IκB in NHBEs. Cells were grown to approximately 80% confluency then treated with the indicated concentrations of CSC for 30 min at 37°C. Whole-cell protein extracts were prepared and 10 μg aliquots were separated by SDS-PAGE, blotted and probed with an antibody to phosphorylated IκB. The blot was then stripped and reprobed for IκB. The band intensities for phosphorylated IκB were determined by densitometric scanning and normalized to the levels of IκB in each lane. Values are means ± SEM for n = 3. (B) CSC-induced immunofluorescent staining for phosphorylated IκB in NHBEs. Cells were treated with vehicle (Control) or 0.04 μg/ml CSC (CSC) for 30 min at 37°C. Cells were fixed with paraformaldehyde and probed with antibody to phosphorylated IκB. (C) and (D) Effect of CSC on expression of an NFκB-regulated luciferase reporter gene in A549 cells. Cells were transfected with a plasmid containing an NFκB binding site upstream of a luciferase reporter sequence. The transfected cells were exposed to 0.4 μg/ml CSC for 30 min at 37°C (C) or increasing doses of CSC (D) and lysates were assayed for luciferase activity using a luminometer. Differences in transfection efficiency were normalized by cotransfection with a LacZ-containing plasmid. The treatments were performed in triplicate and expressed as mean ± SEM for n = 3. CSC = cigarette smoke condensate; IκB = inhibitor of NFκB; SEM = standard error of the mean; NFκB = nuclear factor-kappa B; NHBE = normal human bronchial epithelial cell.
Effect of cigarette smoke condensate on transcriptional activation of luciferase reporter construct
CSC appears to activate NFκB, which should result in transcriptional activation of NFκB-inducible genes. Therefore, A549 cells were transfected with a luciferase reporter plasmid containing a promoter sequence that bound NFκB and luciferase activity was measured after challenge with CSC. Treatment with 0.4 μg/ml CSC for 30 min resulted in significant enhancement of luciferase reporter expression compared to control (Fig. 4C), indicating activation of NFκB. TNF-α used as a positive control also activated luciferase gene expression with the same plasmid system. Exposure of cells transfected with the reporter plasmid to increasing doses of CSC showed NFκB expression peaking at a CSC concentration between 1 and 4 μg/ml (Fig. 4D).
Discussion
The complex changes in lung function, morphology, and gene expression caused by compounds in cigarette smoke involve a combination of direct and indirect effects on cells, but principally center around an increase in airway inflammation as a result of cigarette smoking. The results of our studies demonstrate that the mechanism underlying these acute inflammatory events involves activation of specific signaling pathways such as MAPK (ERK1/2) and NFκB, leading to the release of proinflammatory cytokines.
One of the important observations of this study is the effect of CSC on NHBEs. Even at a concentration of 0.4 μg/ml, which does not induce significant changes in the viability of these cells, CSC induced increases in expression of several cytokines at the mRNA level. IL-1β, which was significantly upregulated in NHBEs following CSC exposure, has been shown to be important for activation of IL-8 [6] and ICAM-1 [4]. This is also consistent with the report that epithelial cells regulate inflammatory events by secreting cytokines and chemokines such as IL-1β, IL-8, GM-CSF, TNF-α, and sICAM-1 [2]. These paracrine effectors can then produce further primary local inflammation or amplify the effects of previously activated macrophages, eosinophils, mast cells, or lymphocytes. A notable finding from our study is that exposure of NHBEs to CSC elicits the synthesis and release of a pattern of mediators similar to that seen in A549 cells in response to CSC. These results imply that neoplastic cells and normal cells respond to CSC in a similar manner.
Cigarette smoke has been shown by others to induce a synthesis of glutathione and gamma-glutamylcysteine that was associated with transcription factor AP-1 or an AP-1-like response element [9]. Although MAPKs and NFκB have been implicated in the upregulation of proinflammatory cytokines, the role of these pathways in normal human pulmonary epithelial cells in response to CSC has not been investigated. The results of our study implicate the ERK1/2 (p42/44) MAPK pathway as a key element in this inflammatory response, an association that has not heretofore been reported in normal human pulmonary epithelial cells. A significant finding of this study is that MAPKs such as ERK1/2 (p44/p42) are consistently activated in NHBEs. However, upon exposure to CSC, the magnitude of phosphorylation increases. The finding that IL-1β expression and MAPK activation are concomitantly increased by CSC exposure and inhibited by PD98059 is consistent with the report that MAPKs activate expression of IL-1β genes [10]. Although the control of cytok–80% confluence on 12-well plates and transfected with the Mercury pathway profiling system vector, pNFκB-TA-Luc (Clontech, Palo Alto, CA, USA), using lipofectamine (Gibco/BRL, Rockville, MD, USA) and used according to the manufacturer's recommendations. Cells were transfected using lipofectamine from Bibco/BRL (Rockville, MD). The plasmid, pVAX-LacZ, (Clontech, Palo Alto, CA, USA) was used as transfection control.
DNA/lipofectamine complexes were allowed to form for 30 min at room temperature in RPMI (without antibiotics) containing 1 μg of reporter plasmid, 20 ng of pVAX-LacZ for monitoring transfection efficiency, and 10 μg of lipofectamine. Cells were transfected for 18 h. Prior to CSC challenge, the transfected cells were preincubated for 4 hours in minimal Eagle's medium with Earle's salts to remove serum factors. Assays were done in triplicate and consisted of untreated controls, treatment with 50 ng/ml TNF-α and treatment with 0.4 μg/ml CSC. After treatments, the cells were lysed by addition of cell lysis buffer as supplied in the luciferase assay kit from Clontech, scraped from the wells and centrifuged 1 min at 12,000 × g. Aliquots of 20 μl of each lysate were transferred to flat-bottomed, white microtiter plates (Dynex, Chantilly, VA, USA) and read on a Dynex MLX plate reader in flash mode. The transfection efficiency was normalized by determining β-galactosidase activity in the lysates from the cotransfected pLacZ plasmid.
Conclusion
In essence, these results demonstrate that NHBEs respond to moderate doses of CSC by activating both the ERK1/2 and the NFκB pathways, and by upregulating the expression of several genes encoding proinflammatory molecules.
Abbreviations
BSA = Bovine serum albumin; CSC = cigarette smoke condensate; ERK = extracellular signal-regulated kinase; GM-CSF = granulocyte-macrophage colony-stimulating factor; HBE = human bronchial epithelial cell; IL = interleukin; IκB = inhibitor of NFκB; MAPK = mitogen-activated protein kinase; MEK-1 = MAPK kinase; NFκB = nuclear factor-kappa B; NHBE = normal human bronchial epithelial cell; PBS = phosphate-buffered saline; PDTC = pyrrolidine dithiocarbamate; RT-PCR = reverse transcriptase-polymerase chain reaction; sICAM = soluble intercellular adhesion molecule; TNF = tumour necrosis factor; TUNEL = deoxynucleotidyl transferase-mediated dUTP nick end labeling
Acknowledgments
Acknowledgements
This study was supported by grants from the VA Merit Review Award and the American Heart Association, Florida Affiliate award and the generous support from the Joy McCann Culverhouse endowment to the Division of Allergy and Immunology.
Contributor Information
Gary R Hellermann, Email: ghellerm@hsc.usf.edu.
Szilvia B Nagy, Email: bsnagy@hotmail.com.
Xiaoyuan Kong, Email: xkong@hsc.usf.edu.
Richard F Lockey, Email: rflockey@hsc.usf.edu.
Shyam S Mohapatra, Email: smohapat@hsc.usf.edu.
References
- Office of Environmental Health Hazard Assessment: Health Effects of Exposure to Environmental Tobacco Smoke. California Environmental Protection Agency; 1997. [Google Scholar]
- Floreani AA, Rennard SI. The role of cigarette smoke in the pathogenesis of asthma and as a trigger for acute symptoms. Curr Opin Pulm Med. 1999;5:38–46. doi: 10.1097/00063198-199901000-00007. [DOI] [PubMed] [Google Scholar]
- Adler KB, Fischer BM, Wright DT, Cohen LA, Becker S. Interactions between respiratory epithelial cells and cytokines: relationships to lung inflammation. Ann NY Acad Sci. 1994;725:128–145. doi: 10.1111/j.1749-6632.1994.tb00275.x. [DOI] [PubMed] [Google Scholar]
- Rusznak C, Sapsford RJ, Devalia JL, Shah SS, Hewitt EL, Lamont AG, Davies RJ, Lozewicz S. Interaction of cigarette smoke and house dust mite allergens on inflammatory mediator release from primary cultures of human bronchial epithelial cells. Clin Exp Allergy. 2001;31:226–38. doi: 10.1046/j.1365-2222.2001.01000.x. [DOI] [PubMed] [Google Scholar]
- Mio T, Romberger DJ, Thompson AB, Robbins RA, Heires A, Rennard SI. Cigarette smoke induces interleukin-8 release from human bronchial epithelial cells. Am J Respir Crit Care Med. 1997;155:1770–6. doi: 10.1164/ajrccm.155.5.9154890. [DOI] [PubMed] [Google Scholar]
- Nakamura H, Yoshimura K, Jaffe HA, Crystal RG. Interleukin-8 gene expression in human bronchial epithelial cells. J Biol Chem. 1991;266:19611–7. [PubMed] [Google Scholar]
- Kawasaki S, Takizawa H, Takami K, Desaki M, Okazaki H, Kasama T, Kobayashi K, Yamamoto K, Nakahara K, Tanaka M, Sagai M, Ohtoshi T. Benzene-extracted components are important for the major activity of diesel exhaust particles: effect on interleukin-8 gene expression in human bronchial epithelial cells. Am J Respir Cell Mol Biol. 2001;24:419–26. doi: 10.1165/ajrcmb.24.4.4085. [DOI] [PubMed] [Google Scholar]
- Ghosh S. Regulation of inducible gene expression by the transcription factor NFκB. Immunol Rev. 1999;19:183–189. doi: 10.1007/BF02786486. [DOI] [PubMed] [Google Scholar]
- Rahman I, Smith CA, Lawson MF, Harrison DJ, MacNee W. Induction of gamma-glutamylcysteine synthetase by cigarette smoke is associated with AP-1 in human alveolar epithelial cells. FEBS Lett. 1996;396:21–5. doi: 10.1016/0014-5793(96)01027-7. [DOI] [PubMed] [Google Scholar]
- Eberhardt W, Huwiler A, Beck KF, Walpen S, Pfeilschifter J. Amplification of IL-1 beta-induced matrix metalloproteinase-9 expression by superoxide in rat glomerular mesangial cells is mediated by increased activities of NF-kappa B and activating protein-1 and involves activation of the mitogen-activated protein kinase pathways. J Immunol. 2000;165:5788–97. doi: 10.4049/jimmunol.165.10.5788. [DOI] [PubMed] [Google Scholar]
- Wang H, Ye Y, Zhu M, Cho C. Increased interleukin-8 expression by cigarette smoke extract in endothelial cells. Environ Toxicol Pharmacol. 2000;9:19–23. doi: 10.1016/S1382-6689(00)00056-9. [DOI] [PubMed] [Google Scholar]
- Means TK, Pavlovich RP, Roca D, Vermeulen MW, Fenton MJ. Activation of TNF-alpha transcription utilizes distinct MAP kinase pathways in different macrophage populations. J Leukoc Biol. 2000;67:885–93. doi: 10.1002/jlb.67.6.885. [DOI] [PubMed] [Google Scholar]
- Modur V, Zimmerman GA, Prescott SM, McIntyre TM. Endothelial cell inflammatory responses to tumor necrosis factor alpha. Ceramide-dependent and -independent mitogen-activated protein kinase cascades. J Biol Chem. 1996;271:13094–102. doi: 10.1074/jbc.271.22.13094. [DOI] [PubMed] [Google Scholar]
- Mochida-Nishimura K, Surewicz K, Cross JV, Hejal R, Templeton D, Rich EA, Toossi Z. Differential activation of MAP kinase signaling pathways and nuclear factor-kappaB in bronchoalveolar cells of smokers and nonsmokers. Mol Med. 2001;7:177–85. [PMC free article] [PubMed] [Google Scholar]
- Bowie A, O'Neill LA. Oxidative stress and nuclear factor-kappaB activation: a reassessment of the evidence in the light of recent discoveries. Biochem Pharmacol. 2000;59:13–23. doi: 10.1016/S0006-2952(99)00296-8. [DOI] [PubMed] [Google Scholar]
- Quay JL, Reed W, Samet J, Devlin RB. Air pollution particles induce IL-6 gene expression in human airway epithelial cells via NF-kappaB activation. Am J Respir Cell Mol Biol. 1998;19:98–106. doi: 10.1165/ajrcmb.19.1.3132. [DOI] [PubMed] [Google Scholar]
- Carter JD, Ghio AJ, Samet JM, Devlin RB. Cytokine production by human airway epithelial cells after exposure to an air pollution particle is metal-dependent. Toxicol Appl Pharmacol. 1997;146:180–8. doi: 10.1006/taap.1997.8254. [DOI] [PubMed] [Google Scholar]