

Abstract
The use of capillary electrophoresis (CE) for the analysis, identification, and characterization of microorganisms has been gaining in popularity. The advantages of CE, such as small sample requirements, minimal sample preparation, rapid and simultaneous analysis, ease of quantitation and identification, and viability assessment, make it an attractive technique for the analysis of microbial analytes. As this instrumental method has evolved, higher peak efficiencies have been achieved by optimizing CE conditions, such as pH, ionic strength, and polymer additive concentration. Experimental improvements have allowed better quantitation and more accurate results. Many practical applications of this technique have been investigated. Viability and identification of microbes can be accomplished in a single analysis. This is useful for evaluation of microbial analytes in consumer products. Diagnosis of microbe-based diseases is now possible, in some cases, without the need for culture methods. Microbe-molecule, virus-antibody, or bacteria-antibiotic interactions can be monitored using CE, allowing for the screening of possible drug candidates. Fermentation can be monitored using this system. This instrumental approach can be adapted to many different applications, including assessing the viability of sperm cells. Progress has been made in the development of microelectrophoresis instrumentation. These advances will eventually allow the development of small, dedicated devices for the rapid, repetitive analyses of specific microbial samples. Although these methods may never fully replace traditional approaches, they are proving to be a valuable addition to the collection of techniques used to analyze, quantitate, and characterize microbes. This review outlines the recent developments in this rapidly growing field.
INTRODUCTION
Sophisticated instrumental techniques for the analysis and characterization of microorganisms are becoming more common. Although these newer, often experimental approaches will not replace traditional methods involving cultures, microscopy, and so forth in the immediate future, their development and use will continue to grow. In particular, methods based on capillary electrophoresis (CE) or microfluidic devices seem to be very promising. CE is well known to produce rapid, high-efficiency separations of biologically important molecules with minimum sample preparation and sample consumption. These advantages may also be realized for the analysis of microorganisms. The rapid and simultaneous analysis of several microorganisms in one sample, including their identification, quantitation, and viability evaluation, appears to be feasible. This review examines recent developments in this rapidly evolving field. The techniques outlined here will undoubtedly play a role in many future microbiological endeavors. They will also be utilized in the development of microchip-based microbiology laboratories of the future.
BACKGROUND
A variety of techniques exist for the analysis of microorganisms. Each of these methods may determine one or more aspects of a microbial system (e.g., identification, quantitation, or characterization). These techniques include differential staining, serological methods, flow cytometry, phage typing, protein analysis, and comparison of DNA nucleotide sequences, for example (9). Many of these procedures require the preparation of bacterial cultures, which dramatically lengthens the analysis time (63). For example, most flow cytometry protocols necessitate bacterial growth prior to analysis in order to increase the sensitivity of the assay (62). Also, in doing this procedure, it becomes impossible to determine the original concentration or population of the microorganism in the original sample (or the viability, i.e., the ratio of the percent live to percent dead in the original sample). Cell viability assessments can be achieved by using combinations of various fluorescent dyes (10, 29, 31, 36, 40).
Microbial phenotypic characteristics such as bacterial fermentation, parasitic morphology, and viral cytopathic effects are sometimes used for identification. However, techniques using these characteristics are not absolutely definitive for the identification of microbes (64). Many organisms have similar phenotypic characteristics, which make accurate identification very difficult. In addition, large concentrations of microbes are necessary for analysis. Recently, PCR techniques have become popular for this very reason. Amplification of DNA increases the sensitivity of the assay without the need for culture (7, 64). New methods have been developed to decrease the time required for PCR detection (7). However, sample purification and DNA isolation prior to PCR analysis prove to be both time-consuming and cumbersome.
Recently, the use of mass spectrometry in the identification of bacteria has become a growing field of interest as well. These methods primarily use the molecular components to identify a cell. The cell can be identified according to its characteristic fingerprint, a series of molecular mass/charge ratio intensities, recorded by the mass spectrometer (27, 28, 41, 54). Other techniques bind proteins to bacteria prior to ionization by matrix-assisted laser desorption/ionization mass spectrometry. This method allows the isolation of particular samples of interest due to preferential binding of the protein. Samples can therefore be concentrated, and better detection limits are achieved using this method (11).
As in the use of phenotypic characteristics, the use of molecular components or patterns of molecular components to identify cells can be highly problematic. Cells, in general, contain a large number and variety of different compounds. Many of these compounds are not restricted to one particular type of cell; most microorganisms are made up of very similar types of molecules. The amount of a particular component in a cell can also vary with its stage of development. Environmental factors can also influence the molecular contents in a cell (1). Methods that use only molecular components (other than DNA analysis) or pattern recognition of molecular components to identify a microorganism can yield false-positive and false-negative results.
Attempts have been made to use fluorescence spectrometry to identify microorganisms (49, 59). As with mass spectrometry analysis, the results are often less than definitive. Quantitation of microbes in a given sample can be achieved by using flow cytometry. In conjunction with other methods, flow cytometry can also be utilized to identify a microbe of interest (13, 33, 35).
Recently, microelectrophoresis methods have been developed that may allow the facile simultaneous separation, identification, quantitation, and characterization of intact microorganisms, alone or in mixtures. These methods rely on the fact that microbes are charged and move in a direct-current electric field.
BRIEF THEORY
Microbes as Colloidal Particles: Origin of Charge
A colloid is a particle or cluster of particles ranging in size from approximately 10−4 to a few micrometers. They are sufficiently small that they do not ordinarily settle out of solution solely under the influence of gravity (i.e., Brownian motion of the surrounding solution is sufficient to keep them suspended). Colloids can be solid, liquid, or gas. They are usually charged and have high surface-to-volume ratios. Therefore, based solely on their size, it is acceptable to characterize most microbes as colloidal particles. Colloids (including microorganisms) can obtain surface charge in at least two ways. The first occurs due to ionization of surface groups as a result of protonation or deprotonation of acidic or basic surface molecules. Surface charge can also be acquired by adsorption of ions from the surrounding solution (55). Indeed, there is almost always a differential adsorption of ions by colloidal particles. Both the type and amount of an ion adsorbed are controlled by the size and nature (surface chemistry) of the colloidal particle as well as by the properties of the surrounding solution (e.g., pH, ionic strength and ionic composition). The surface charge of microorganisms is usually due to a combination of these factors. The most common ionizable groups on the microbe's outer surface are carboxylic acids, organophosphates, amines, and sometimes sulfate moieties (17).
The chemistry of the outer cell wall determines not only surface charge but also (for bacteria) whether the organisms are gram-positive or gram-negative. The cell walls of gram-negative bacteria are composed of three layers, an inner bilayer phospholipid membrane, a thin peptidoglycan layer, and the outer bilayer membrane, coated with complex lipopolysaccharides. These groups give rise to surface charge on gram-negative bacteria. Gram-positive bacteria do not have the outer membrane, but they do have a much thicker peptidoglycan layer (which is their outer layer). Teichoic acids are prevalent in the cell walls of gram-positive bacteria giving them a negative charge at pHs ≥5. The phosphodiester bonds linking the polymers of the peptidoglycan layer also can acquire charge under the right conditions (17). Viruses have an outer protein coat in place of the membrane. The amino acid residues of the proteins enable charge formation. Membranes and cell walls of fungi contain proteins and lipid molecules, giving them a characteristic charge. This charge can be altered depending on pH, ionic strength, electrolyte composition, and temperature (1).
Electrical Double Layer and Electrophoretic Mobility
Charged colloidal particles in aqueous solutions will develop a solvation sphere, in which dipolar water molecules and oppositely charged ions acculmulate near the charged surface of colloids, forming the electrical double layer. The potential across the surface of shear of the double layer is referred to as the zeta potential (ξ). The entire diameter of the colloid is composed of the diameter of the particle itself as well as the thickness of the solvation sphere (55). Therefore, the thickness of the double layer also contributes to the electrophoretic mobility of a colloidal particle. The thickness of the double layer (1/κ) is given by
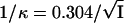 |
where I is the ionic strength of the solution.
Electrophoretic mobility (μ) is linked to ξ by
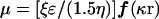 |
where ɛ is the dielectric constant of the solution, η is the solution viscosity, and r is the radius of the particle (55). The value of ƒ(κr) is dimensionless and varies between 1 and 1.5. If ƒ(κr) is large [ƒ(κr) = 1.5], the double layer is considered to be a flat plane. Therefore, the above relationship is simplified to (55)
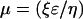 |
In this case, mobility is related to the ionic strength of the surrounding solution. This is also true in the case of microorganisms in a direct current electrical field. Their relatively large size in comparison to the thickness of the double layer makes ƒ(κr) very large. Therefore, it is possible to assume that the electrophoretic mobility of microbes, in a constant electrical field, is dependent primarily on the ionic strength of the surrounding solution. The protonation or deprotonation of the outer surface of biological entities is dictated by the pH of the surrounding solution. Therefore, altering the pH can also affect the electrophoretic mobility of microbes (55). If ƒ(κr) is small, the radius of a microbe also affects its mobility.
In CE, separation of analytes is based on their differential electrophoretic mobilities. Figure 1 shows an example of an electrophoretic separation. A quantity called resolution (Rs) determines how well two things have been separated. Two factors contribute to resolution: (i) peak-to-peak separation and (ii) efficiency (the narrowness of the peaks [the narrower the peaks, the better the efficiency]) (23). Dispersion of analyte bands usually occurs during the separation procedure and causes peak broadening, which leads to lower efficiencies. Based on peak width, efficiencies are calculated in terms of theoretical plates, N. The higher the plate count, the more efficient is the separation. The better the separation, the more reliable are the results of the analysis (23, 37).
FIG. 1.

Sample electropherogram showing the calculation of resolution, Rs, and efficiency in terms of theoretical plates, N. Resolution is essentially a measure of the quality of a separation. A resolution of 1.5 is considered a baseline separation of two peaks. The greater the resolution, the better the separation of the two peaks. Resolution is a combination of two factors. One factor is the selectivity, which takes into account the distance between the maxima of the two peaks. The second factor is the efficiency of the separation. Higher efficiency means that there are a greater number of theoretical plates, which, in turn, produces narrower peaks. Therefore, narrow peaks and large peak-to-peak separations produce large Rs values. The calculation of resolution uses the migration time of the two peaks (tm1, tm2) and the base width of the peaks (Wb1, Wb2). The greater the peak width at half the peak height, W1/2, the lower the value of N. Therefore, in this figure, peak 1 would have a higher efficiency than peak 2.
MICROBIAL ANALYSIS USING SEPARATION METHODS
Field Flow Fractionation and Free Flow Electrophoresis
There are many instrumental, separation-based methods for the analysis of biologically important molecules. There has been an effort to use these methods to separate and analyze colloid-size microorganisms. These efforts have been marginally successful. While gross separations of microorganisms and cells have sometimes been achieved, the selectivity, efficiency, robustness, and ease of the instrumental method are usually far lower than what is found for molecules. For example, a number of papers cite the use of field flow fractionation (FFF) techniques for the separation of bacteria and viruses (16, 30, 32, 58, 67). FFF is a fairly straightforward technique in which a field is applied perpendicular to fluid flow, causing particles, such as macromolecules and colloids, to migrate with different velocities. Fields of sedimentation, diffusion, and electrical diffusion are manipulated to optimize separations involving microbial analytes (67). Environmental samples can be analyzed using this technique. For instance, FFF has been used in the fractionation of three strains of bacteria found in dairy products (30). FFF, however, has poor efficiency, moderate to poor selectivity, and limited instrumentation is available (many instruments are home-made). In addition, microorganisms cannot be identified through FFF techniques alone.
Free-flow electrophoresis has long been used in the separation of biological analytes via their differential electrophoretic mobilities on the preparative scale (21, 52). Resnick et al. used stable-flow free-boundary (Staflo) electrophoresis to separate spores from diploid cells of yeast. This technique utilized the differential electrophoretic mobilities of spores and diploid cells to obtain a pure aqueous suspension of spores (52). It uses a density gradient for stabilization and is a variation of zone electrophoresis. Solutions of differential densities are layered, and then the sample is introduced. The sample forms an intermediate layer within the Staflo chamber. An electrical field is then applied, causing the spores to migrate into the aqueous layer for separation. The diploid cells have much lower electrophoretic mobilities than do the spores (52). Hansen-Hagge et al. used free-flow electrophoresis in the enrichment of mutants defective in lipid A synthesis. Mutants that were defective in lipid A synthesis had high electrophoretic mobilities under the influence of an electrical field. Fractions could then be collected at specific migration times (21). However, for both of these techniques, very low efficiencies were achieved. Separation was possible only if very different types of cells were used.
Early Attempts Using Capillary Electrophoresis for Microbial Separations
Hjertan et al. (24) described the first CE experiment involving microbes on an analytical scale. By using a fused silica capillary coated with methylcellulose to prevent adsorption on capillary walls, they analyzed Lactobacillus casei and tobacco mosaic virus (TMV) in separate experimental runs. TMV was detected using online UV-visible absorbance detection at 260 nm, while L. casei was detected at 220 nm. The resulting electropherograms showed fairly sharp peaks with migration times less than 4 min. However, there was no separation of the different microorganisms. They all moved together in the flow stream produced via the electro-osmotic flow (EOF). These experiments demonstrated the possibility of moving microbes through capillaries under applied electrical fields.
Subsequently, Zhu and Chen used capillary zone electrophoresis to analyze erythrocytes. Hydroxypropyl methylcellulose was used to coat the walls of the capillary in order to reduce electroosmotic flow. The migration times of the red blood cells for consecutive runs proved highly reproducible under specific controlled conditions, such as cell preparation and composition of electrolyte solutions. The reported migration time of the single red blood cells was ∼14 min. Zhu and Chen were the first to note that extraneous peaks in the electropherogram seemed to be the result of cell agglomeration. They also were able to establish a close correlation between peak height and number of cells injected (68).
Grossman and Soane found that the orientation of a microbe affects its electrophoretic mobility. The mobility of TMV, a rod-shaped virus, increased with higher applied voltages. It was thought that the orientation of the TMV within the electrical field was responsible for the observed differences. The increase in mobility with field strength was caused by a decrease in translational friction, resulting from the alignment of the virus with the field. The rod-shaped geometries were compared to spherical geometries, where no differences in mobility were observed (20).
Ebersole and McCormick reported the first separation of bacteria via capillary zone electrophoresis (12). They were able to separate or partially separate Enterococcus faecalis, Streptococcus pyogenes, Streptococcus agalactiae, Streptococcus pneumoniae, and Staphylococcus aureus into discrete bands. S. pyogenes and S. pneumoniae were separated as broad peaks in nearly 70 min. Ninety percent of the bacteria were thought to remain viable throughout the experiment. Injection of individual bacteria enabled identification of the peaks through mobility matching (12). Mobility matching has been used in traditional cell electrophoresis as well, in the characterization of bacteria (66). In order to give the bacteria sufficient time to move away from the EOF front, very long capillaries (250 cm) had to be used. One of the separations showed two peaks corresponding to E. faecalis. The first peak was thought to represent the bacteria alone, and the second peak was thought to be the result of agglomeration of the same bacteria. The difference in the surface charges of the bacteria and the bacterial chains was thought to cause the differences in migration times. Two of the peaks were well resolved, although the peaks were broad (low efficiency) and the shapes were poor. The broad bands and long retention times decreased the efficiency of this separation (12).
Four years later, Pfetsch and Welsch (50) also reported an analogous CE separation of three different bacteria, Pseudomonas putida, Pseudomonas species, and Alcaligenes eutrophus, using a method similar to that of Ebersole and McCormick (12). The analysis time was only slightly shorter than that reported by Ebersole and McCormick for their separations. The bandwidths were still relatively broad. Figure 2 shows a comparison of peak shapes for P. putida (a microbe) (Fig. 2a) and 2-nitrophenol (a molecule) (Fig. 2b) having the same retention time. The role played by analyte size in band broadening is apparent from the electropherograms (50).
FIG. 2.

Comparison of peak widths between P. putida (a) and 2-nitrophenol (b). The buffer was Tris-boric acid-EDTA (pH 9.6); I = 0.5 mM; capillary length = 250 cm. Detection was by UV absorbance at 208 nm. Reprinted from reference 50 with permission from the publisher.
Figure 3 demonstrates the importance of ionic strength for peak width. As the ionic strength of the run buffer increases, the peaks become broader. The work also concluded that the optimal pH range for bacterial separations was between 7 and 10 when using a solution with an ionic strength of 1.5 mM. A steep increase in electrophoretic mobility was observed from pH 4 to 7. Between pH 7 and 10, the electrophoretic mobility of the bacteria changed very little (50). This was consistent with the theoretical model of microbe electrophoretic mobility presented earlier in this review (55).
FIG. 3.

Dependence of bandwidth of Rhodococcus erythropolis on the ionic strength of buffer. (a) I = 1.0 mM, (b) I = 2.0 mM, (c) I = 6 mM. The buffer was Tris-boric acid-EDTA (pH 9.6). Capillary length = 250 cm. Detection was by UV absorbance at 208 nm. Reprinted from reference 50 with permission from the publisher.
From these early CE studies (12, 50), it appears that (i) very long capillaries are needed for this procedure, (ii) the separation of different bacterial populations is possible only if there are sufficiently large differences in their electrophoretic mobilities, and (iii) compared to molecules, the CE peak capacity is small due to the limited migration window and large bandwidths. Despite the difficulties, this work provided a solid starting point for microbial analysis using CE. It was clear that the efficiencies and time of analysis must be substantially improved if CE was to become a dominant and highly useful method for microbial analysis. Such improvements could allow both identification and quantitation of microbes through analysis of peak position and peak area (as is done for molecules). Also, the technique(s) must be robust, reproducible, and accurate. Recent advances in the field indicate that these goals are within reach.
HIGH-EFFICIENCY SEPARATION OF INTACT MICROBES
Microbes versus Molecules
CE was originally developed for the analysis of molecules (and this is its predominant use today). Even the largest molecules are much smaller and less complex than any microorganism. Glucose, for example, is only 1.5 nm in length, whereas bacteria can be as large as 10 μm (9). Size is just one characteristic of microorganisms which can make separation via microanalytical methods challenging. Several other factors must be considered and controlled if a successful CE separation is to be accomplished (6). For example, microbes are amphoteric. At high pH, a microbe has a net negative charge, but at low pH, it has a net positive charge. Solutions of microbes can change with time depending on the age of the microbial population. Also, many bacteria can aggregrate to form long chains or clusters, which can alter the results. They can attach to other microbes and surfaces such as the walls of capillaries. There are few available microbial standards, making identification difficult. Microbes can also secrete substances that result in unwanted peaks, lysis, or altered mobilities. On a per-particle basis, microbial solutions are generally more dilute than solutions of molecules, which can lead to sampling problems. Microbes are highly sensitive to the environment (pH, ionic strength, and presence of O2, etc.) and can lyse under moderate to harsh conditions (6).
Surfactants have been used to alter EOF and eliminate the adsorption of molecules to capillary walls. These surfactants, however, are known to be causative agents for cell lysis. Hjertan and Kubo describe the use of hydrophilic polymers such as methylcellulose to eliminate EOF and wall adsorption (25). These types of polymers pose less of a danger to some highly sensitive microbes.
Sample preparation is critical for high-efficiency microbial separations. The preparation and separation of bacteria must be done under mild, carefully controlled conditions. Small changes in buffer concentration, pH, and environmental conditions can contribute to irreproducible results (6).
The first high-efficiency separation of a mixture of microbes was reported in late 1999 (6). In the first of a series of papers, two different CE methods were employed in the separation of three bacteria and Saccharomyces cerevisiae (baker's yeast). The first method used a dilute dissolved polymer, polyethylene oxide (PEO), in the run buffer. PEO has been used in CE as a nonbonded coating for the purpose of altering the EOF (51). Without PEO, the EOF was too fast, resulting in all the species eluting with the EOF marker. After addition of PEO (average Mn = 600,000), bacteria were focused into sharp, high-efficiency peaks and had differential migration times. The migrations times were also different from those of the EOF marker and could be altered by varying the concentration of PEO in the running buffer (as well as the pH and other parameters).
Another approach used capillary isoelectric focusing (CIEF) to separate bacteria. This method exploits the surface charge differences between the bacteria. Serratia rubidae, P. putida, and Escherichia coli were separated with very high efficiency (6).
Figure 4 shows the difference in electropherograms of lysed and intact P. fluorescens. The separation parameters were identical for both runs. After being warmed with sodium dodecyl sulfate (SDS), a commonly used EOF modifier, the bacteria were lysed. It is unclear whether the broad bands of Fig. 3 were a result of low efficiency or bacterial lysis or perhaps both (6).
FIG. 4.

Electropherograms of lysed (A) and intact (B) P. fluorescens. Separation conditions were identical. The buffer was 0.0125% PEO dissolved in Tris-boric acid-EDTA (pH 8.4). Capillary length = 50 cm. Bacteria were lysed by warming with 0.15% SDS prior to injection. Reprinted from reference 6 with permission from the publisher.
Figure 5 shows an electropherogram of three bacteria, P. fluorescens, Enterobacter aerogenes, and Micrococcus luteus, along with S. cerevisiae. The narrow, sharp peaks demonstrate the high efficiency of the separation (N ∼ 850,000 theoretical plates). All of the peaks are well resolved from one another and in an extremely short time (less than 10 min). The area of each peak is proportional to the number of bacteria detected (6).
FIG. 5.

Electropherogram showing the separation of S. cerevisiae and three bacteria, P. fluorescens, E. aerogenes, and M. luteus. PEO was added to the run buffer. The buffer was Tris-boric acid-EDTA (pH 8.4). Capillary length = 50 cm. The EOF marker was mesityl oxide. Reprinted from reference 6 with permission from the publisher.
Shintani et al. demonstrated a similar technique to optimize the peak efficiency of microbes by using a carbohydrate polymer additive that served the same function as PEO (61). Sharp peaks of Salmonella enterica serovar Enteritidis were obtained by adding 0.01% sodium alginate, a straight-chain, hydrophilic polyuronic acid, and 0.2% NaCl to the run buffer. These researchers theorized that the carbohydrate polymer might interact with a cell surface charged polymer such as carbohydrate chain, suppressing the characteristics of these surface polymers. They found that the additive improved the peak shape only for certain microbes. Therefore, they were unable to separate a mixture of nine different microbes including S. enteritidis by using this particular additive (61).
Armstrong and co-workers described a method for managing microbial aggregates by using CE (57). Aggregation can cause changes in surface charge as well as diffusional properties. It is therefore possible to have multiple peaks in electropherograms of a single species of microbe if different aggregates are present and are not dispersed before analysis. With analysis times of less than 10 min, efficiencies of 1,000,000 plates were achieved in the separation of certain bacteria and their aggregates. Figure 6 shows two electropherograms comparing the effect of aggregation on peak intensity. The first sample of M. luteus (Fig. 6A) contained a large number of aggregate chains, as can be seen by the accompanying photomicrograph, while the second sample (Fig. 6B), had fewer aggregate chains. Brief sonication (a few minutes at 43 kHz) of the samples disperses the microbial aggregates and produces a much better electropherogram. Association between the cells can be temporarily reversed by exposure to small amounts of ultrasonic energy. It is important to note that some microorganisms associate more strongly than others and may be more difficult to disperse by sonication alone. The original sample of M. luteus had a large number of aggregate clusters. After sonication, the sample has only one intense peak, representative of the single cells of bacteria, and few minor peaks, representing the remaining aggregate clusters (57). It is clear that in many cases, pretreating the sample by brief sonication or other means is necessary to eliminate ambiguities resulting from multiple aggregates of the cells. Obtaining one single aggregate, can, however, prove beneficial. This type of bacterial focusing is discussed later.
FIG. 6.

Electropherograms of M. luteus before sonication (A) and after sonication for 3 min (B). The insets photomicrographs of the samples prior to injection. The peak at 6.8 min represents the single-cell bacteria. The buffer was 0.0125% PEO dissolved in Tris-boric acid-EDTA (pH 8.4). Capillary length = 27 cm. Reprinted from reference 57 with permission from the publisher.
Torimura et al. detected the separation of microbes by using differential light scattering. Electrophoretic mobilities measured by capillary zone electrophoresis were consistent with those from classical electrophoresis (65). Microscopic detection confirmed the presence of aggregates. Extraneous peaks corresponded to these aggregates. Peak broadening was noted to occur in part due to the nature of the microbes, i.e., size. It was found that enrichment of certain bacteria resulted in changes in electrophoretic mobility with respect to deficient samples. This would support a theory that enrichment of bacteria affects the structure and composition of the cell surface (65).
Many CE methods rely on mobility matching using pure laboratory-prepared samples. However, the scarcity of suitable microbial standards underlies the problem of peak identification for microbes (6). Okun et al. evaluated a different method for peak identification for human rhinovirus (HRV). The peaks were identified using various indirect methods involving (i) RNase treatment followed by enzymatic digestion and (ii) immunodepletion with monoclonal antibody binding (44). In a separate study, subviral particles of HRV were separated and identified using fast spectral scanning in conjunction with immunodepletion (46).
Mann et al. also overcame the problem of peak identification associated with PCR and biological activity assays (38). Using a polyvinyl alcohol-coated capillary, they monitored the mobility of adenovirus. The polyvinyl alcohol kept the virus from adsorbing onto the walls of the capillary. The applied voltage was 29.5 kV, and the detection wavelength was 214 nm. At a pH of 7.0 in 25 mM sodium phosphate buffer, the analysis time of adenovirus was ∼9 min. Observed minor peaks were thought to be a result of modifications in the surface charge of the virus (38).
Mann et al. found the best pH range for microbial separations to be between 7 and 10. Below pH 5.4, there was no detectable virus signal. At this pH, adenovirus is thought to spontaneously dissociate. The buffer concentration was also correlated with electrophoretic mobility. Relative intensities of major and minor peaks were stable throughout a wide range of conditions. Figure 7 shows a typical electropherogram of adenovirus (38).
FIG. 7.

Electropherogram of adenovirus. Polyvinyl alcohol was used to coat the capillary walls. The buffer was 25 mM sodium phosphate (pH 7.0). Capillary length = 57 cm. Reprinted from reference 38 with permission from the publisher.
Electrophoretic Mobility and pI Values
Glynn et al. compared measurements of electrophoretic mobilities of three bacterial strains by CE with those obtained by microelectrophoresis (19). They found that CE measurements of electrophoretic mobility were comparable to microelectrophoresis measurements. Figure 8 shows the electropherograms obtained for the various bacterial strains. These researchers also discovered, by using capillaries with larger internal diameters (75 μm) and by either modifying the buffer or coating the capillary walls, that they could reduce the analyte-wall interactions. Bacterial strains exhibited multiple electrophoretic mobilities. This was due in part to interpopulation heterogeneities resulting in differences in surface charge (19). This study recognized a potential imperfection with using mobility matching for identification.
FIG. 8.

Electropherograms for various strains of bacteria. (A) Neutral marker peak and two bacterial peaks for strain A1264. (B) Neutral marker peak and two bacterial peaks for CD1. (C) Neutral marker peak and a single bacterial peak for PL2W31. The buffer was 10 mM MOPS (pH 7.02). Capillary length = 57 cm. Reprinted from reference 19 with permission from the publisher.
It is evident that experimental conditions can have various effects on the electrophoretic mobility of microbes. A number of papers cite the use of pI value as a better method to establish identification (56, 60). Theoretically, pI values are independent of experimental conditions. Determination of pI values, however, can have significant uncertainties. Shen et al. used CIEF to determine pI values for yeast cells (60). The pI values were established using standard pI markers such as proteins. Capillaries were coated with hydroxypropyl methylcellulose. Formation of aggregates caused notable inaccuracies in pI determination. It was established that pI values also changed by up to 1.2 pH units depending on the stage of cell development. When cell concentrations were greater than 3 cells/μl, no signal was observed. At concentrations lower than 2.4 cells/μl, reproducible migration times were detected (Fig. 9) (60).
FIG. 9.

Yeast cell concentration range. At concentrations lower than 2.4 cells/μl but greater than 0.1 cells/μl, reproducible retention times were observed using CIEF. The catholyte was ammonium hydroxide (pH 10.7). The anolyte was acetic acid (pH 2.5). Capillaries were coated with hydroxypropyl methylcellulose. Capillary length = 65 cm. Detection was by UV absorbance at 280 nm. Reprinted from reference 60 with permission from the publisher.
Kenndler and co-workers determined the pI value of HRV to be 6.8 by CIEF (56). They accomplished this by applying a pressure to the cathodic end of the capillary to keep the zones focused through the detector. The voltage was kept constant to establish equilibrium between zones. Extrapolation of data allowed the calculation of the pI value for HRV (56).
Natural Samples and Consumer Products
A rapid CE-based assay for the identification of bacteria causing urinary tract infections (UTIs) was described recently (4). UTIs are typically caused by one of two types of bacterial pathogens, E. coli or Staphylococcus saprophyticus. UTIs are far more common in women than in men (39). The exact bacterium causing the problem is rarely identified, primarily due to a long analysis time, contamination, and high cost. CE proved to be an effective way to analyze spiked and authentic samples. Urine was directly injected into the CE unit. A blank urine sample is shown in Fig. 10 (A). The matrix (urea plus various salts) appears to elute near the EOF. Figure 10B shows a comparison between urine spiked with S. saprophyticus and urine spiked with E. coli. The bacteria clearly demonstrate two different electrophoretic mobilities by having unique migration times. With migration times shorter than than 10 min and sharp peak shapes, the efficiency of these separations neared ∼107 theoretical plates (4).
FIG. 10.

Electropherograms of a concentrated and diluted urine blank (A) and S. saprophyticus- and E. coli-spiked urine samples (B). The additive was PEO. The buffer was Tris-boric acid-EDTA (pH 9.0). Capillary length = 27 cm. Reprinted from reference 4 with permission from the publisher.
In addition to the UTI assay, a microbial assay for consumer products containing active bacteria was developed (5). Living microbes are used in many products alleged to benefit human health (42). For instance, many pills and powder-based products contain lactose-digesting bacteria as the active ingredient to treat lactose intolerance in people. Schiff tablets, containing Lactobacillus acidophilus, also help patients with lactose intolerance. Another product, BabyLife powder, contains Bifidobacterium infantis. It is used in conjunction with infant formulas to inoculate the gut of newborn babies with helpful bacteria. Both products were analyzed using CE to determine their bacterial content. Figure 11 shows electropherograms of cultured cells of B. infantis (Fig. 11A) and direct injection of dissolved BabyLife powder (Fig. 11B). The B. infantis peak in both samples has roughly the same migration time, confirming the identity of the bacteria. Note that both the microbial component and the molecular component (maltodextrin) of this material can be analyzed in the same run (Fig. 11B). Using the same experimental procedure, L. acidophilus could be analyzed in Schiff tablets (Fig. 12). (5).
FIG. 11.

Electropherograms of cultured cells of B. infantis from BabyLife powder (A) and direct injection of dissolved BabyLife powder (B). The buffer was 0.025% PEO dissolved in Tris-boric acid-EDTA (pH 8.4). Capillary length = 27 cm. Reprinted from reference 5 with permission from the publisher.
FIG. 12.

Electropherograms of cultured cells of L. acidophilus from Schiff tablets (A) and direct injection of dissolved Schiff tablets (B). The buffer was 0.025% PEO dissolved in Tris-boric acid-EDTA (pH 8.4). Capillary length = 27 cm. Reprinted from reference 5 with permission from the publisher.
Viability
Bacteria found in consumer products are helpful only if they are alive. Indeed, the determination of cell viability can be important in many other studies as well. CE can be used to assess the viability of cells in a number of ways. One method is to monitor microbial activity on-line. Torimura et al. monitored the signal obtained from E. coli treated with various chemicals (65). The colistin-treated samples showed similar electropherograms to untreated samples. Therefore, it was assumed that the outer surface of the cell remained unchanged after treatment. KCN-treated E. coli exhibited similar behavior. However, ofloxacin-treated bacteria produced a significantly different electropherogram, most probably due to bacteriolysis. These experiments were able to monitor cell lysis due to chemical treatment. Indeed, using the high-efficiency CE approach, it is quite easy to tell the difference between intact and lysed cells (Fig. 4). Cell metabolism is a direct indication of cell viability; therefore, monitoring metabolism through on-line activity measurements would undoubtedly assess cell viability (65).
It has already been noted that fluorescent dyes have long been used to evaluate the viability of microbes (10, 29, 31, 36, 40). It was determined that the use of fluorescent dyes and CE coupled with laser-induced fluorescence (LIF) detection would provide a rapid determination of cell viability (5). SYTO-9 stains all bacteria, while propidium iodide stains only bacteria with damaged membranes (i.e., dead bacteria). Using both dyes in conjunction, it was found that living bacteria fluoresce green whereas dead bacteria fluoresce red. CE separation, using a LIF detector with a 520-nm bandpass filter and a 663-nm longpass filter, was used to simultaneously monitor both dyes in the bacteria. By comparing the ratio of the two peaks, it was determined that this sample of Schiff tablets contained only 60% viable cells (5).
The analytical figures of merit for CE cell viability determinations were explored in detail recently (1). It was established that CE coupled with LIF detection offers the advantages of speed, sensitivity, efficiency, automation, and overall effectiveness for assessing cell viability. It was also found that the CE-LIF viability numbers were comparable, within experimental error, to those obtained using flow cytometry. In this study, a number of dye types and concentrations were used to determine the optimum conditions for viability analysis. FUN-1, an asymmetrical cyanine dye, has extremely efficient membrane permeability, making it useful in the staining of yeast and fungi. This dye diffuses into the cytoplasm of the cell and produces green fluorescence. Living cells will process this dye, producing red fluorescence. For bacterial analysis, the SYTO-9/propidium iodide dye system works best. With this combination of dyes, Fig. 13 shows the simultaneous separation of B. infantis, L. acidophilus, and S. cerevisiae with the detection of live (green-fluorescent spectra) and dead (red-fluorescent spectra) cells. Using a standard curve (Fig. 14) and the ratio of green to red light intensity (from Fig. 13), the viability is easily determined (1). In CE-LIF, the total luminescence measured is proportional to the number of cells present. The linear dynamic range for this specific procedure was determined to be from 0 to 2.4 × 108 cells/ml. This method unequivocally demonstrates that it is now possible to separate, identify, quantitate, and determine viability in a single run (1).
FIG. 13.

Simultaneous separation of B. infantis, L. acidophilus, and S. cerevisiae. (A) Detection of the live cells; (B) detection of the dead cells. Total luminescence is proportional to the total number of cells present. The buffer was 0.025% PEO dissolved in Tris-boric acid-EDTA (pH 8.4). Capillary length = 30 cm. Reprinted from reference 1 with permission from the publisher.
FIG. 14.

Standard viability curves for B. infantis (▪), L. acidophilus (•), and S. cerevisiae (▴). Reprinted from reference 1 with permission from the publisher.
Shintani et al (61) used a CE-LIF method for the sensitive detection of S. enterica serovar Enteritidis, the most dominant species of human Salmonella infections. Using SYTO-9 fluorescent dye, they were able to detect as few as three cells per injection from a pure culture. Cells were counted via microscopic observation. This sensitivity would be high enough to detect Salmonella directly from food and other clinical samples. In addition to the SYTO-9 bacterial staining method, they used a fluorescent-labeled polyclonal antibody for the selective identification of targeted serovar Enteritidis by CE-LIF detection. This enabled the detection of small quantities of Salmonella within 10 min. proving CE-LIF to be a powerful tool for the identification of microbes (61).
Evaluation of Microbe-Molecule Binding
Kenndler and co-workers recently reported the monitoring of complex formations of antibodies and viruses by affinity CE (22, 45, 47, 53). Figure 15 shows the formation of complexes as a function of added antibody. The decrease in peak absorption and shifted migration times were a result of increasing antibody concentration. When the amount of antibody added reached saturation, two peaks were observed (45). This group also has shown that receptor fragments of differing lengths will bind to HRV. At low molar ratios of receptor to virus, peaks for the complex were rather broad. This was thought to result from inhomogeneities in receptor sites. Sharper complex peaks were representative of saturated binding sites (47).
FIG. 15.

Formation of virus-antibody complexes. IS, internal standard. The buffer was 100 mM borate-boric acid with 0.26% SDS (pH 8.3). Capillary length = 60 cm. mAb, monoclonal antibody. Reprinted from reference 45 with permission from the publisher.
In another study, the stoichiometry of binding between monoclonal antibodies and receptor sites on HRV was examined (48). The equilibrium concentration of the antibody was determined by the initial concentration of the virus and the thermodynamic equilibrium constant. These factors determine the shape of the calibration curve used to determine the stoichiometry of high-affinity complexes. Intermediate-affinity complexes could also use the calibration curve for stoichiometric determination but with lower accuracy. The stoichiometry of low-affinity complexes could not be determined (48).
In a recent study, CE was used to distinguish between high-binding and nonbinding behavior between a wide range of ligands and receptors including microbes (8). Determinations of binding constants for cells were more complex than for molecules. Cell death can produce chemical changes in membrane permeability or leakage of cell material, which can cause changes in the molecular binding constant. These changes can affect the reproducibility of measurements. The number of molecules of a DNA stain contained in an individual cell could be calculated using CE methods (8). Generally, it was found that evaluation of binding behavior was easier for viruses than for bacteria, primarily due to the relative simplicity and smaller size of viruses (8).
Affinity complex studies provide important information for understanding how certain antibodies bind with particular viruses. These methods are applicable as rapid screening techniques for investigating antibody-virus interactions.
PROSPECTS
Focusing Microbes into Discrete Bands
In a recent publication, apparent efficiencies up to 109 theoretical plates were achieved using CE (3). Efficiencies of this magnitude are generally a result of artifacts such as gas bubbles and precipitates. Using a charge-coupled device camera, the movement of S. cerevisiae through the capillary was monitored in real time. It was observed that shortly after the voltage was applied, the broad sample slug was compacted into a very narrow band of microbes. The high efficiencies achieved in this study were a result of the tendency of microbes to form noncovalent attachments to one another. The focused microbes will travel through the capillary as a single aggregate cluster. The ability to control the focusing and aggregation of cells is vital for the separation of microbial analytes (3).
It was found that three experimental conditions are necessary for the focusing to occur: (i) a dilute polymer must be added to the run buffer, (ii) a direct-current electrical field must be present, and (iii) EOF must be in a direction countercurrent to the charged microbe (microbes generally have a negative surface charge pulling them toward the anode). If any of these conditions are negated, then focusing does not occur (3).
Three theories have been proposed to explain the mechanism behind the focusing (3). The first theory, the field aggregation model, simply describes how colloidal particles will form disk-like aggregates, which line up perpendicular to the applied electrical field. This mechanism depends on the polarizability of the microbe. Microbes, in contrast to other colloidal particles, are polarizable and deformable, allowing for band compression. If was found, however, that polarizability is not the only factor controlling their focusing behavior (3).
The second theory, the hairy-particle model, takes into account the role of PEO on the focusing of the microbe as well as the countercurrent flow of the analyte and run buffer. It is thought that the PEO reversibly attaches to the surface of the microbe, altering the microbe's mobility. This “hairy-particle” layer decreases the mobility of small ions and therefore the conductivity in the vicinity of the microbe. This could also enhance the electrical field in the vicinity of the injection plug. Cations are concentrated on the cathodic side of the injection plug, and anions are concentrated on the anodic side. The combination of the countercurrent flow of the microbes and the run buffer and the altered field due to the “hairy” particle focus the sample zone before it goes through the detector as a single band (3).
The third theory, the shape-induced differential-mobility model, is perhaps the easiest mechanism to visualize. Simply stated, nonspherical microbes and small aggregates have different mobilities depending on their orientation in the capillary. Since the orientation of the particles constantly changes, so do their individual mobilities. Because of the differential and constantly fluctuating mobilities, there is a higher probability that the particles will collide with one another forming aggregates. Finally, all the individual microbes in the injection slug form noncovalent attachments to each other, forming a single aggregate. Addition of PEO to the run buffer slows the EOF, enabling enough time for this aggregate to form (3).
Miniaturization
In many areas of science and technology, miniaturization of instrumentation has become increasingly important. Li and Harrison published a paper describing the transport of biological cells on a microchip device by using electrokinetic methods. The microfluidic system was fabricated on a silicon substrate using a modified chemical etching technique. S. cerevisiae, E. coli, and canine erythrocytes were transported through the channels on the chip. At a T-junction within the chip, SDS was added to lyse the canine erythrocytes in order to demonstrate the capability for reactions on-chip (34).
Fu et al. have illustrated the use of microfabrication in cell sorting (15). EOF was initiated and maintained by three platinum electrodes placed at the input and output wells. The micromachined chip was silicon based, similar to the chip proposed by Li and Harrison (34). Using EOF, fluorescent and nonfluorescent E. coli cells were separated using the fluorescence-activated cell sorter. Cells were viable after sorting, allowing for the possibility of collection. The direction of flow could be manipulated, permitting the cells to pass through the detection window numerous times, thus increasing sensitivity (15).
Fiedler et al. also developed a system for microscale cell sorting (14). Using high-frequency alternating-current fields under conditions of negative dielectrophoresis, they sorted mammalian cells. A series of electrodes was used to concentrate cells into a single band, break up cell aggregates, trap particular cells, and then direct the cells into one of two output channels. This method, in conjunction with high-performance optical detection, would make possible microscale flow cytometry (14).
A number of papers have been published regarding real-time microchip-based PCR detection of microbes (26, 43). The papers describe the development of a miniature analytical thermal cycling instrument (MATCI) to amplify and detect DNA via PCR. MATCI uses thermal cycling and CE on-chip for analysis. The instrument, including accessory components, is the size of a briefcase (25). The miniaturized devices have cut costs tremendously, and their portability allows for on-site testing of various samples (43). Miniature CE instruments, in-house, would allow doctors to diagnose infections with ease. The future of microbial analysis seems to be poised toward miniaturization.
Fertility Studies
Using the previously described method for viability determination, boar sperm cells were analyzed by LIF-CE (21a). Boar sperm removed from its gel matrix was obtained from Iowa State University College of Veterinary Medicine. Extender was added to keep the sperm cells viable. The extender was a mixture of glucose, sodium carbonate, EDTA, trisodium citrate, and some antioxidants. A 50-μl aliquot of sample was diluted with an additional 2 ml of extender. After the dilution, the samples were stained with SYBR-14 and propidium iodide. Similar to the SYTO-9/propidium iodide system, viable cells fluoresced green and dead cells fluoresced red. The stained sample was then directly injected into the CE system. The CE run buffer contained 1 mM Tris, 0.33 mM citric acid, 1% fructose, and 0.025% PEO (600 kDa). LIF detection using two channels monitored the green and red fluorescence. A single peak was obtained reproducibly with retention times around 15 min. The ratio of green to red fluorescence was determined to be the live-to-dead ratio of the sperm cells (Armstrong and He, unpublished).
Clearly, the aforementioned results demonstrate the highly efficient, rapid, and sensitive nature of CE for characterization and identification of microbes. The future of this technique appears to be quite promising. From analysis of fermentation to diagnosis of disease to defense against bioterrorism, the applications of such a system are numerous. With automation, miniaturization, and high-throughput analysis, the benefits of this method are immeasurable.
CONCLUSIONS
Diagnostic microbial separations have come a long way in last few years. The efficiency of CE separations of these microbes has been greatly improved by optimizing the pH and ionic strength and using dilute run buffer modifiers such as PEO. Ebersole and McCormick reported one of the first separations of microbes with efficiencies around 580 theoretical plates (12). More recently, apparent efficiencies of over 109 theoretical plates have been achieved (3).
Recent research has demonstrated very practical uses for microbial separations. Assays to determine viability and to identify microbes in products are likely to become common regulatory procedures. Rapid diagnosis of microbe-based diseases without the need to isolate pure cultures is of obvious importance. There is the added benefit that only small volumes of sample are needed for the analysis. Also, the ability to simultaneously monitor several microorganisms in a complex mixture in real time is becoming a real possibility. Binding experiments provide quick screening of antibody-virus interactions, bacterium-antibiotic interactions, or virtually any molecule-microbe interactions. Indeed the miniaturized, high-throughput analysis of microbes and microbe-molecule interactions may soon become commonplace. Clearly, applications of microbial separations are extensive. The impact of these high-efficiency separations on modern microbiology will continue to grow.
Acknowledgments
We thank the National Institutes of Health (NIH RO1 GM53825-07) for support of this work.
REFERENCES
- 1.Armstrong, D. W., and L. He. 2001. Rapid determination of cell viability in single or mixed samples using capillary electrophoresis LIF microfluidic systems. Anal. Chem. 73:4551-4557. [DOI] [PubMed] [Google Scholar]
- 2.Reference deleted.
- 3.Armstrong, D. W., M. Girod, L. He, M. A. Rodriguez, E. S. Yeung, W. Wei, and J. Zheng. 2002. Mechanistic aspects in the generation of apparent ultra-high efficiencies for colloidal (microbial) electrokinetic separations. Anal. Chem. 74:5523-5530. [DOI] [PubMed] [Google Scholar]
- 4.Armstrong, D. W., and J. M. Schneiderheinze. 2000. Rapid identification of the bacterial pathogens responsible for urinary tract infections using direct injection CE. Anal. Chem. 72:4474-4476. [DOI] [PubMed] [Google Scholar]
- 5.Armstrong, D. W., J. M. Schneiderheinze, J. P. Kullman, and L. He. 2001. Rapid CE microbial assays for consumer products that contain active bacteria. FEMS Microbiol. Lett. 194:33-37. [DOI] [PubMed] [Google Scholar]
- 6.Armstrong, D. W., G. Schulte, J. M. Schneiderheinze, and D. J. Westenberg. 1999. Separating microbes in the manner of molecules. 1. Capillary electrokinetic approaches. Anal. Chem. 71:5465-5469. [DOI] [PubMed] [Google Scholar]
- 7.Belgrader, P., W. Benett, D. Hadley, J. Richards, P. Stratton, R. Mariella, and F. Milanovich. 1999. PCR detection of bacteria in seven minutes. Science 284:449-450. [DOI] [PubMed] [Google Scholar]
- 8.Berthod, A., M. Rodriguez, and D. W. Armstrong. 2002. Evaluation of molecule-microbe interactions with capillary electrophoresis: procedures, utility, and restrictions. Electrophoresis 23:847-857. [DOI] [PubMed] [Google Scholar]
- 9.Black, J. G. 1996. Microbiology principles and applications, 3rd ed., p. 227-228, 235-240, 249-250. Prentice-Hall, Upper Saddle River, N.J.
- 10.Breeuwer, P., and T. Abee. 2000. Assessment of viability of microorganisms employing fluorescence techniques. Int. J. Food Microbiol. 55:193-200. [DOI] [PubMed] [Google Scholar]
- 11.Bundy, J., and C. Fenselau. 1999. Lectin-based affinity capture for MALDI-MS analysis of bacteria. Anal. Chem. 71:1460-1463. [DOI] [PubMed] [Google Scholar]
- 12.Ebersole, R. C., and R. M. McCormick. 1993. Separation and isolation of viable bacteria by capillary zone electrophoresis. Bio/technology 11:1278-1282. [DOI] [PubMed] [Google Scholar]
- 13.Ferrari, B. C., G. Vesey, K. A. Davis, M. Gauci, and D. Veal. 2000. A novel two-color flow cytometric assay for the detection of Cryptosporidium in environmental water samples. Cytometry 41:216-222. [PubMed] [Google Scholar]
- 14.Fiedler, S., S. G. Shirley, T. Schnelle, and G. Fuhr. 1998. Dielectrophoretic sorting of particles and cells in a microsystem. Anal. Chem. 70:1909-1915. [DOI] [PubMed] [Google Scholar]
- 15.Fu, A. Y., C. Spence, A. Scherer, F. H. Arnold, and S. R. Quake. 1999. A microfabricated fluorescence-activated cell sorter. Nat. Biotechnol. 17:1109-1111. [DOI] [PubMed] [Google Scholar]
- 16.Gao, Y. S., S. C. Lorbach, and R. Blake. 1997. Separation of bacteria by sedimentaion field-flow fractionation. J. Microcol. Separ. 9:497-501. [Google Scholar]
- 17.Garrett, R. H., and C. M. Grisham. 1999. Biochemistry, 2nd ed., p. 279-282. Saunders College Publishers, Fort Worth, Tex.
- 18.Girod, M., and D. W. Armstrong. 2002. Monitoring the migration behavior of living microorganisms in capillary electrophoresis using laser-induced fluorescence detection with a charge-coupled device imaging system. Electrophoresis 23:2048-2056. [DOI] [PubMed] [Google Scholar]
- 19.Glynn, J. R., B. M. Belongia, R. G. Arnold, K. L. Ogden, and J. C. Baygents. 1998. Capillary electrophoresis measurements of electrophoretic mobility for colloidal particles of biological interest. Appl. Environ. Microbiol. 64:2572-2577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Grossman, P. D., and D. S. Soane. 1990. Orientation effects on the electrophoretic mobility of rod-shaped molecules in free solution. Anal. Chem. 62:1592-1596. [DOI] [PubMed] [Google Scholar]
- 21.Hansen-Hagge, T., V. Lehmann, and O. Luderitz. 1985. Free flow electrophoresis as a tool for enrichment of mutants with temperature-dependent lethal mutations in lipid A synthesis. Eur. J. Biochem. 148:21-27. [DOI] [PubMed] [Google Scholar]
- 21a.He, L., R. J. Jepsen, L. E. Evans, and D. W. Armstrong. 2003. Electrophoretic behavior and potency assessment of boar sperm using a capillary electrophoresis-laser induced fluorescence system. Anal. Chem. 75:825-834. [DOI] [PubMed] [Google Scholar]
- 22.Hewat, E., and D. Blaas. 1996. Structure of neutralizing antibody bound bivalently to human rhinovirus 2. EMBO J. 15:1515-1523. [PMC free article] [PubMed] [Google Scholar]
- 23.Hjertan, S. 1990. Zone broadening in electrophoresis with special reference to high performance electrophoresis in capillaries: an interplay between theory and practice. Electrophoresis 11:665-690. [DOI] [PubMed] [Google Scholar]
- 24.Hjertan, S., K. Elenbring, F. Kilar, J. L. Liao, A. J. Chen, C. J. Siebert, and M. D. Zhu. 1987. Carrier-free zone electrophoresis, displacement electrophoresis and isoelectric focusing in a high-performance electrophoresis apparatus. J. Chromatogr. 403:47-61. [DOI] [PubMed] [Google Scholar]
- 25.Hjertan, S., and K. Kubo. 1993. A new type of pH- and detergent-stable coating for elimination of electroendosmosis and adsorption in (capillary) electrophoresis. Electrophoresis 14:390-395. [DOI] [PubMed] [Google Scholar]
- 26.Ibrahim, M. S., R. S. Lofts, P. B., Jahrling, E. A. Henchal, V. W. Weedn, M. A. Nothrup, and P. Belgrader. 1998. Real-time microchip PCR detecting single-base differences in viral and human DNA. Anal. Chem. 70:2013-2017. [DOI] [PubMed] [Google Scholar]
- 27.Jarman, K. H., D. S. Daly, C. E. Peterson, A. Saenz, N. B. Valentine, M. T. Kingsley, and K. L. Wahl. 1999. Extracting and visualizing matrix-assisted laser desorption/ionization time of flight mass spectral fingerprints. Rapid Commun. Mass Spectrom. 13:1586-1594. [DOI] [PubMed] [Google Scholar]
- 28.Jarman, K. H., S. T. Cebula, A. Saenz, C. E. Peterson, N. B. Valentine, M. T. Kingsley, and K. L. Wahl. 2000. An algorithm for automated bacterial identification using matrix-assisted laser desorption/ionization mass spectrometry. Anal. Chem. 72:1217-1223. [DOI] [PubMed] [Google Scholar]
- 29.Jones, K. H., and J. A. Senft. 1985. An improved method to determine cell viability by simultaneous staining with fluorescein diacetate-propidium iodide. J. Histochem. Cytochem. 33:77-79. [DOI] [PubMed] [Google Scholar]
- 30.Jussila, M. A., G. Yohannes, and M. L. Riekkola. 1997. Flow field-flow fractionation in the study of dairy products. J. Microcol. Separ. 9:601-609. [Google Scholar]
- 31.Kaneshiro, E. S., M. A. Wyder, Y. P. Wu, and M. T. Cushion. 1993. Reliability of calcein acetoxy methyl ester and ethidium homodimer or propidium iodide for viability assessment of microbes. J. Microbiol. Methods 17:1-16. [Google Scholar]
- 32.Khoshmanesh, A., R. Sharma, and R. Beckett. 2001. Biomass of sediment bacteria by sedimentation field-flow fractionation. J. Environ. Eng. 127:19-25. [Google Scholar]
- 33.Legendre, L., C. Courties, and M. Troussellier. 2001. Flow cytometry in oceanography 1989-1999: environmental challenges and research trends. Cytometry 44:164-172. [DOI] [PubMed] [Google Scholar]
- 34.Li, P. C. H., and D. J. Harrison. 1997. Transport, manipulation, and reaction of biological cells on-chip using electrokinetic effects. Anal Chem. 69:1564-1568. [DOI] [PubMed] [Google Scholar]
- 35.Li, W. K. W., and P. M. Dickie. 2001. Monitoring phytoplankton, bacterioplankton, and virioplankton in a coastal inlet (Bedford Basin) by flow cytometry. Cytometry 44:236-246. [DOI] [PubMed] [Google Scholar]
- 36.Lloyd, D., and A. J. Hayes. 1995. Vigour, vitality, and viability of microbes. FEMS Microbiol. Lett. 133:1-7. [Google Scholar]
- 37.Lukacs, K. D., and J. W. Jorgenson. 1985. Capillary zone electrophoresis: effect of physical parameters on separation efficiency and quantitation. J. High Res. Chromatogr. 8:407-411. [Google Scholar]
- 38.Mann, B., J. A. Traina, C. Soderblom, P. K. Murakami, E. Lehmberg, D. Lee, J. Irving, E. Nestaas, and E. Pungor. 2000. Capillary zone electrophoresis of a recombinant adenovirus. J. Chromatogr. Ser. A 895:329-337. [DOI] [PubMed] [Google Scholar]
- 39.McCarty, J. M., G. Richard, W. Huck, R. M. Tucker, R. L. Tosiello, M. Shan, A. Heyd, and R. M. Echols. 1999. A randomized trial of short-course ciprofloxacin, ofloxacin, or trimethoprim/sulfamethoxazole for the treatment of acute urinary tract infection in women. Am. J. Med. 106:292-299. [DOI] [PubMed] [Google Scholar]
- 40.McFeters, G. A., F. P. Yu, B. H. Pyle, and P. S. Stewart. 1995. Physiological assessment of bacteria using fluorochromes. J. Microbiol. Methods 21:1-13. [DOI] [PubMed] [Google Scholar]
- 41.Meuzelaar, H. L. C., and P. G. Kistemaker. 1973. A technique for fast and reproducible fingerprinting of bacteria by pyrolysis mass spectrometry. Anal. Chem. 45:587-590. [DOI] [PubMed] [Google Scholar]
- 42.Mustapha, A., T. Jiong, and D. A. Savaiano. 1997. Improvement of lactose digestion by humans following ingestion of unfermented acidophilus milk: influence of bile sensitivity, lactose transport, and acid tolerance of Lactobacillus acidophilus. J. Dairy Sci. 80:1537-1545. [DOI] [PubMed] [Google Scholar]
- 43.Northrup, M. A. 1998. A miniature analytical instrument for nucleic acids based on micromachined silicon reaction chambers. Anal. Chem. 70:918-922. [DOI] [PubMed] [Google Scholar]
- 44.Okun, V. M., B. Ronacher, D. Blaas, and E. Kenndler. 1999. Analysis of common cold virus (human rhinovirus serotype 2) by capillary zone electrophoresis: the problem of peak identification. Anal. Chem. 71:2028-2032. [DOI] [PubMed] [Google Scholar]
- 45.Okun, V. M., B. Ronacher, D. Blaas, and E. Kenndler. 2000. Affinity capillary electrophoresis for the assessment of complex formation between viruses and monoclonal antibodies. Anal. Chem. 72:4634-4639. [DOI] [PubMed] [Google Scholar]
- 46.Okun, V. M., D. Blaas, and E. Kenndler. 1999. Separation and biospecific identification of subviral particles of human rhinovirus serotype 2 by capillary zone electrophoresis. Anal. Chem. 71:4480-4485. [DOI] [PubMed] [Google Scholar]
- 47.Okun, V. M., R. Moser, B. Ronacher, D. Blaas, and E. Kenndler. 2001. VLDL receptor fragments of different lengths bind to human rhinovirus HRV2 with different stoichiometry. J. Biol. Chem. 276:1057-1062. [DOI] [PubMed] [Google Scholar]
- 48.Okun, V. M., R. Moser, D. Blaas, and E. Kenndler. 2001. Complexes between monoclonal antibodies and receptor fragments with a common cold virus: determination of stoichiometry by capillary electrophoresis. Anal. Chem. 73:3900-3906. [DOI] [PubMed] [Google Scholar]
- 49.Pau, C. P., G. Patonay, C. W. Moss, G. M. Carlone, T. M. Rossi, and I. M. Warner. 1986. A rapid enzymatic procedure for “fingerprinting” bacteria by using pattern recognition two-dimensional fluorescence data. Clin. Chem. 32:987-991. [PubMed] [Google Scholar]
- 50.Pfetsch, A., and T. Welsch. 1997. Determination of the electrophoretic mobility of bacteria and their separation by capillary zone electrophoresis. Fresenius J. Anal. Chem. 359:198-201. [Google Scholar]
- 51.Preisler, J., and E. S. Yeung. 1996. Characterization of nonbonded poly(ethylene oxide) coating for capillary electrophoresis via continuous monitoring of electroosmotic flow. Anal. Chem. 68:2885-2889. [DOI] [PubMed] [Google Scholar]
- 52.Resnick, M. A., R. D. Tippets, and R. K. Mortimer. 1967. Separation of spores from diploid cells of yeast by stable-flow free-boundary electrophoresis. Science 158:803-804. [DOI] [PubMed] [Google Scholar]
- 53.Ronacher, B., T. C. Marlovits, R. Moser, and D. Blaas. 2000. Expression and folding of human very-low density lipo-protein receptor fragments: neutralization capacity toward human rhinovirus HRV2. Virology 278:541-550. [DOI] [PubMed] [Google Scholar]
- 54.Saenz, A., C. E. Peterson, N. B. Valentine, S. L. Gantt, K. H. Jarman, M. T. Kingsley, and K. L. Wahl. 1999. Reproducibility of matrix-assisted laser desorption/ionization time of flight mass spectrometry for replicate bacterial culture analysis. Rapid Commun. Mass Spectrom. 13:1580-1585. [DOI] [PubMed] [Google Scholar]
- 55.Schnabel, U., C. H. Fischer, and E. Kenndler. 1997. Characterization of colloidal gold nanoparticles according to size by capillary zone electrophoresis. J. Microcol. Separ. 9:529-534. [Google Scholar]
- 56.Schnabel, U., F. Groiss, D. Blaas, and E. Kenndler. 1996. Determination of the pI of human rhinovirus serotype 2 by capillary isoelectric focusing. Anal. Chem. 68:4300-4303. [DOI] [PubMed] [Google Scholar]
- 57.Schneiderheinze, J. M., D. W. Armstrong, G. Schulte, and D. J. Westenberg. 2000. High efficiency separation of microbial aggregates using capillary electrophoresis. FEMS Microbiol. Lett. 189:39-44. [DOI] [PubMed] [Google Scholar]
- 58.Sharma, R., R. T. Edwards, and R. Beckett. 1998. Analysis of bacteria in aquatic environments using sedimentation field-flow fractionation: (II) physical characterization of cells. Water Res. 32:1508-1514. [Google Scholar]
- 59.Shelly, D. C., J. M. Quarkes, and I. M. Warner. 1980. Identification of fluorescent Pseudomonas species. Clin. Chem. 26:1127-1132. [PubMed] [Google Scholar]
- 60.Shen, Y., S. J. Berger, and R. D. Smith. 2000. Capillary isoelectric focusing of yeast cells. Anal. Chem. 72:4603-4607. [DOI] [PubMed] [Google Scholar]
- 61.Shintani, T., K. Yamada, M. Torimora. 2002. Optimization of a rapid sensitive identification system for Salmonella enteritidis by capillary electrophoresis with laser-induced fluorescence. FEMS Microbiol. Lett. 210:245-249. [DOI] [PubMed] [Google Scholar]
- 62.Shvalov, A. N., J. T. Soini, I. V. Surovtsev., G. V. Kochneva, G. F. Sivolobova., A. K. Petrov., and V. P. Maltsev. 2000. Individual Escherichia coli cells studied from light scattering with the scanning flow cytometer. Cytometry 41:41-45. [DOI] [PubMed] [Google Scholar]
- 63.Stinson, S. C. 1999. Identifying bacteria: looking for a fast track. Chem. Eng. News 77:36-38. [Google Scholar]
- 64.Tang, Y. W., G. W. Procop, and D. H. Persing. 1997. Molecular diagnosis of infectious diseases. Clin. Chem. 43:2021-2038. [PubMed] [Google Scholar]
- 65.Torimura, M., S. Ito, K. Kano, T. Ikeda, Y. Esaka, and T. Ueda. 1999. Surface characterization and on-line activity measurements of microorganisms by capillary zone electrophoresis. J. Chromatogr. Ser. B 721:31-37. [DOI] [PubMed] [Google Scholar]
- 66.Uhlenbruck, G. A. Froml, R. Lutticken, and K. Hannig. 1988. Cell electrophoresis of group B Streptococci: separation of types Ia, Ib/c, II, III, and IV before and after neuraminidase treatment. Zentiol. Bakteriol. Mikrobiol. Hyg. Ser. A 270:28-34. [DOI] [PubMed] [Google Scholar]
- 67.Yager, P., M. A. Afromowitz, D. Bell, F. K. Forster, J. P. Holl, A. Kamholz, and B. Weigla. 1988. Design of microfluidic sample preconditioning systems for detection of biological agents in environmental samples. Proc. SPIE-Int. Soc. Opt. Eng. 3515:252-259. [Google Scholar]
- 68.Zhu, A., and Y. Chen. 1989. High-voltage capillary zone electrophoresis of red blood cells. J. Chromatogr. 470:251-260. [DOI] [PubMed] [Google Scholar]