

Abstract
The v-SNARE proteins Snc1p and Snc2p are required for fusion of secretory vesicles with the plasma membrane in yeast. Mutation of a methionine-based sorting signal in the cytoplasmic domain of either Sncp inhibits Sncp endocytosis and prevents recycling of Sncp to the Golgi after exocytosis. snc1-M43A mutant yeast have reduced growth and secretion rates and accumulate post-Golgi secretory vesicles and fragmented vacuoles. However, cells continue to grow and secrete for several hours after de novo Snc2-M42A synthesis is repressed. DPL1, the structural gene for dihydrosphingosine phosphate lyase, was selected as a high copy number snc1-M43A suppressor. Because DPL1 also partially suppresses the growth and secretion phenotypes of a snc deletion, we propose that enhanced degradation of dihydrosphingosine-1-phosphate allows an alternative protein to replace Sncp as the secretory vesicle v-SNARE.
INTRODUCTION
The budding of a transport vesicle from a donor organelle followed by fusion of the vesicle with a target organelle allows the transfer of membrane constituents and soluble cargo between the organelles of the secretory and endocytic pathways. The fusion step requires assembly of a SNARE complex between a v-SNARE on the transport vesicle and t-SNAREs on the target organelle (Rothman and Warren, 1994; Nichols et al., 1997). The predominant structural feature of a SNARE complex is a parallel four helix-bundle that has transmembrane domains extending from its C terminus into both the vesicle and target membranes (Sutton et al., 1998). The hydrophobic interface between the amphipathic α-helicies that form the helical bundle is interrupted by a central hydrophilic layer comprising an arginine residue contributed by the v-SNARE and three glutamine residues contributed by the t-SNAREs (Sutton et al., 1998). There is structural homology between assembled SNARE complexes and the activated conformation of viral fusion proteins, suggesting that SNARE complexes directly catalyze intracellular membrane fusion (Skehel and Wiley, 1998). In fact, if v- and t-SNAREs are incorporated into separate populations of liposomes, these liposomes will fuse when mixed (Weber et al., 1998). Within the v- and t-SNARE protein families, there are many homologous proteins that are targeted to different transport vesicles and organelles within the cell. Thus, it has been proposed that specificity in the assembly or fusion capacity of SNARE complexes serves as a checkpoint to ensure that vesicles fuse only with appropriate target organelles (Rothman and Warren, 1994). Current evidence, however, suggests that v- and t-SNAREs bind promiscuously both in vivo and in vitro (Gotte and von Mollard, 1998; Grote and Novick, 1999; Yang et al., 1999).
After fusion, the v-SNARE is located in the target membrane where it remains associated with a t-SNARE in a cis-SNARE complex (Grote et al., 2000a). For a v-SNARE to be used in subsequent rounds of transport, it must be recycled from the target organelle back to the donor organelle by retrograde transport. The first step in recycling is disassembly of this postfusion cis-SNARE complex by NSF/Sec18p. Next, the v-SNARE must be sorted into a transport vesicle that buds from the acceptor organelle and is targeted back to the original donor organelle. A recycling v-SNARE may be passive cargo within the retrograde transport vesicle that is prevented from interfering with vesicle transport by binding to a chaperone (Edelmann et al., 1995; Pfeffer, 1996; Lustgarten and Gerst, 1999). Alternatively, it is possible that a single v-SNARE can be a component of distinct SNARE complexes that catalyze fusion of either anterograde or retrograde transport vesicles. If so, the other proteins on the transport vesicles must be necessary for targeting because a single v-SNARE cannot direct transport vesicles to two different targets (Lewis et al., 1997). Once the v-SNARE has recycled to the donor organelle, it can be sorted into a new vesicle for another round of transport.
Sorting signals in the cytoplasmic domains of membrane proteins are used to concentrate proteins into budding vesicles (Trowbridge et al., 1993). Among the most extensively characterized sorting signals are the tyrosine and dileucine signals for endocytosis via clathrin-coated pits. These signals bind to the μ subunit of the AP2 clathrin adaptor complex (Heilker et al., 1999). There are apparently several distinct binding sites for endocytosis signals because the transferrin receptor, the low density lipoprotein (LDL) receptor, and the epidermal growth factor (EGF) receptor all have tyrosine-based endocytosis signals but they do not compete with each other for internalization (Warren et al., 1998). In addition, monoubiquitination can also serve as a signal for endocytosis (Shih et al., 2000).
The synaptic vesicle v-SNARE vesicle-associated membrane protein 2 (VAMP2) has a novel sorting signal (Grote et al., 1995). VAMP2 recycling was studied in transfected PC12 cells by following the transport of an antibody bound to epitope-tagged VAMP2 from the plasma membrane to endosomes and then back to synaptic vesicles (Grote et al., 1995; de Wit et al., 1999). A targeting signal was identified in the cytoplasmic domain of VAMP2 that is required both for endocytosis and synaptic vesicle targeting. This signal, centered on methionine-46, is located on the hydrophobic face of the same amphipathic helix that binds to t-SNAREs, but the sequence requirements for targeting and t-SNARE binding are distinct (Grote et al., 1995). Synaptic vesicles recycle via clathrin-coated pits (Cremona and De Camilli, 1997). VAMP2 may either interact directly with a component of the coat, or be sorted to coated pits by a lateral interaction with another membrane protein. A direct interaction with the budding machinery is more likely because mutation of the endocytosis signal inhibits VAMP2 endocytosis in fibroblasts that do not express other synaptic vesicle membrane proteins (Grote and Kelly, 1996). VAMP must have an important role in synaptic vesicle recycling because proteolysis of VAMP by tetanus toxin prevents the budding of synaptic vesicles in vitro (Salem et al., 1998).
The Snc proteins are yeast orthologs of VAMP (Gerst et al., 1992). Two genes, SNC1 and SNC2, encode Snc proteins that are 98% identical in their α-helical t-SNARE-binding domains and 79% identical overall (Protopopov et al., 1993). Although the two Snc proteins are only 38% identical to VAMP within the t-SNARE-binding domain, the methionine and surrounding amino acids important for VAMP2 sorting are conserved. Sncp binds to the plasma membrane t-SNAREs Ssop and Sec9p to catalyze exocytic fusion (Protopopov et al., 1993). Sncp also binds to several other t-SNAREs, including Tlg1p, Tlg2p, and Pep12p, but there is no direct evidence that these interactions are functional (Abeliovich et al., 1998; Holthuis et al., 1998; Grote and Novick, 1999).
In this manuscript, we report on our studies of yeast expressing Sncp with a defective endocytosis signal. We tested the hypothesis that inhibiting Sncp recycling would prevent the biogenesis of fusogenic secretory vesicles. The results show an accumulation of secretory vesicles and a reduced rate of secretion in cells with a snc1-M43A mutation. In an attempt to identify a sorting receptor for the methionine-based endocytosis signal, we performed a high-copy suppressor screen. Overproduction of DPL1, the gene for dihydrosphingosine phosphate lyase, suppressed the snc endocytosis mutant. However, DPL1 overproduction also suppressed a snc deletion, so Dpl1p cannot be a sorting receptor for Sncp. We propose that alterations in sphingosine metabolism allow secretion to occur with an alternative SNARE in place of Sncp.
MATERIALS AND METHODS
SNC Plasmid and Strain Construction
The strains and plasmids used in this study are listed in Tables 1 and 2. A cluster of amino acids containing six lysine residues encoded by two complementary oligonucleotides was inserted into the XhoI site at the 3′ end of the hemagglutinin (HA)-tag in pNB841 (Abeliovich et al., 1998) to construct the pGal1-SNC2-HA-6K plasmid pNB1028. Starting with the third HA epitope, the C-terminal sequence of Snc2-HA-6K is YPYDVPDYATSLEKEKDKDSTKEKDKELEGGPGTQFAL. Methionine 42 of the SNC2 gene in this plasmid was mutated to alanine using the polymerase chain reaction (PCR) to construct pNB1029. The sequence of pNB1029, and all other plasmids in this study constructed via PCR, was verified by DNA sequencing. The SNC2 gene of pNB1028 was replaced with SNC1 and snc1-M43A to construct pNB1075 and pNB1076. pNB1028, pNB1029, pNB1075, and pNB1076 were integrated at the leu2 locus of the pep4Δ strain NY603 to minimize C-terminal proteolysis. A M43A mutation was integrated into the SNC1 gene of pADH-LSNC1 (Protopopov et al., 1993) to construct pNB1036.
Table 1.
Yeast strains used in this study
| Strain | Genotype |
|---|---|
| NY603 | MATa pep4∷URA3 leu2-3,112 ura3-52 |
| NY2202 | MATα leu2∷GAL1p-SNC2-HA-6K-LEU2 pep4∷URA3 ura3-52 |
| NY2203 | MATα leu2∷GAL1p-snc2-M42A-HA-6K-LEU2 pep4∷URA3 ura3-52 |
| NY2268 | MATα leu2∷GAL1p-SNC1-HA-6K-LEU2 pep4∷URA3 ura3-52 |
| NY2269 | MATα leu2∷GAL1p-SNC1-M43A-HA-6K-LEU2 pep4∷URA3 ura3-52 |
| SP1 | MATa ura3 ade8 leu2 trp1 his3 |
| SP1α | MATα ura3 ade8 leu2 trp1 his3 |
| JG8 | MATa snc1∷URA3 snc2∷ADE8 ura3 ade8 leu2 trp1 his (2μ GAL1p-SNC1-HA-TRP1) |
| NY2206 | MATa/α SNC1/snc1∷URA3 SNC2/snc2∷ADE8 ura3/ura3 ade8/ade8 leu2/leu2 trp1/trp1 his/his3 |
| NY2207 | MATa/α snc1-M43A/snc1∷URA3 SNC2/snc2∷ADE8 ura3/ura3 ade8/ade8 leu2/leu2 trp1/trp1 his/his3 |
| NY2264 | MAT? SNC1 snc2∷ADE8 ura3 ade8 leu2 trp1 his |
| NY2265 | MAT? snc1-M43A snc2∷ADE8 ura3 ade8 leu2 trp1 his |
| NY2204 | MATa leu2∷SNC2-LEU2 snc1∷URA3 snc2∷ADE8 ura3 ade8 trp1 his |
| NY2205 | MATa leu2∷snc2-M42A-LEU2 snc1∷URA3 snc2∷ADE8 ura3 ade8 trp1 his |
| NY2270 | MATa leu2∷GAL1p-SNC2-LEU2 snc1∷URA3 snc2∷ADE8 ura3 ade8 trp1 his |
| NY2271 | MATa leu2∷GAL1p-snc2-M42A-LEU2 snc1∷URA3 snc2∷ADE8 ura3 ade8 trp1 his |
| NY2258 | MATa/α snc1-M43A/snc1∷URA3 snc2∷ADE8/snc2∷ADE8 ura3/ura3 ade8/ade8 leu2/leu2 trp1/trp1 his3/his |
| NY2261 | MATα ura3-53 leu2-3,112 trp1 his3-Δ200 |
| NY2260 | MATα ura3-53 leu2-3,112 trp1 his3-Δ200 (pNB1039 Myc-DPL1) |
| NY2259 | MATa snc1∷URA3 snc2∷ADE8 ura3 ade8 leu2 trp1 his (2μ GAL1p-SNC1-HA-TRP1) (pNB1039 Myc-DPL1) |
| NY2262 | MATα DPL1∷LEU2 ura3-53 leu2-3,112 trp1 his3-Δ200 |
| NY2263 | MATα DPL1∷LEU2 ura3 ade8 leu2 trp1 his3 |
| NY1203 | MATa sec1-1 leu2-3,112 |
| NY775 | MATa sec4-8 leu2-3,112 |
| NY777 | MATa sec5-24 leu2-3,112 |
| NY1204 | MATa sec9-4 leu2-3,112 |
Table 2.
Plasmids used in this study
| Plasmid | Description |
|---|---|
| pNB529 | GAL1 promotor and ADH1 terminator in pRS305 |
| pNB841 | SNC2-HA in pNB529 |
| pNB1028 | SNC2-HA-6K in pNB529 (from pNB841) |
| pNB1029 | snc2-M42A-HA-6K in pNB529 (from pNB841) |
| pNB1080 | SNC2 in pNB529 |
| pNB1077 | snc2-M42A in pNB529 |
| pNB1075 | SNC1-HA-6K in pNB529 |
| pNB1076 | snc1-M43A-HA-6K in pNB529 |
| Ycp50-HO | from Ira Herskowitz |
| pNB1030 | SNC2 in pRS305 |
| pNB1031 | snc2-M42A in pRS305 |
| pNB1040 | DPL1, SSD1, and YDR925C in YEplac181 (10.7 kb genomic DNA insert) |
| pNB1041 | DPL1 and YDR925C in YEplac181 (Xba1 dropout from pNB1040) |
| pNB1042 | DPL1 and SSD1 in YEplac181 (SacI-NaeI dropout from pNB1040) |
| pNB1037 | DPL1 in pBluescript KS- |
| pNB1048 | DPL1 in pRS425 |
| pNB1049 | dpl1-K370R in pRS425 |
| pNB1050 | dpl1-K370N in pRS425 |
| pNB1047 | DPL1 in pRS315 |
| pNB1038 | dpl1∷LEU2 in pBluescript KS- (from pNB1037) |
| pNB1039 | Myc-DPL1 in pRS425 (from pNB1048) |
The mating type of the SNC1 SNC2 host strain SP1 (Protopopov et al., 1993) was changed by first transforming with the plasmid Ycp50-HO and then selecting against the plasmid on plates containing 5-fluoroorotic acid to construct SP1 α. The snc1-m43a mutation was constructed in SP1α by pop-in/pop-out gene replacement (Guthrie and Fink, 1991; page 297). SP1α and snc1-M43A mutant SP1α were mated with the snc1Δ snc2Δ strain JG8 (Protopopov et al., 1993). The diploid strains NY2206 and NY2207 were identified by screening for diploid colonies derived from these crosses that had lost the SNC1 TRP1 balancer plasmid from JG8 and were thus unable to grow without tryptophan. To test for growth defects associated with snc mutations, NY2206 and NY2207 were sporulated, dissected onto YPD plates, and grown for 3 d at 25°C or for 2 d at 34°C. The genotype of each colony was determined by following the URA3 and ADE8 disruption markers. If 2:2 segregation of markers was assumed, all of the colonies that failed to grow had a deletion of both SNC genes. The surface area of the remaining colonies was measured from a scanned image using NIH image software and categorized by genotype. NY2264 and NY2265 are sporulation products of NY2206 and NY2207 that were dissected onto synthetic complete plates. These strains were maintained on synthetic media to reduce the opportunity to accumulate or elo3 mutations (David et al., 1998). NY2258 was constructed by crossing JG8 to a MATα snc1-M43A strain created by sporulating the diploid strain NY2207.
A 2.7-kb genomic DNA fragment containing the SNC2 gene and 5′ and 3′ regulatory sequences was amplified by PCR and inserted between the BamHI and HindIII sites of pRS305 to construct pNB1030. A M42A mutation in the SNC2 gene of pNB1030 was introduced by PCR to construct pNB1031. The SNC2 and snc2-M42A open reading frames were amplified by PCR and subcloned into pNB529 to construct pNB1080 and pNB1077. pNB1030, pNB1031, pNB1080, and pNB1077 were integrated at the LEU2 locus of JG8 to construct NY2204, NY2205, NY2270, and NY2271.
Endocytosis of Snc2-HA-6K
Forty A600 units of yeast were washed twice with phosphate-buffered saline (PBS) and then incubated in 100 mM NaCO3, pH 9.4, for 10 min at room temperature to loosen the cell wall. The cells were then chilled to 4°C and incubated with 3.0 mg/ml NHS-SS-biotin in 400 μl of PBS twice for 20 min. Surface biotinylated cells were washed twice with ice-cold PBS and then incubated (twice for 5 min) in PBS + 50 mM glycine to quench unreacted NHS-SS-biotin. Cells were incubated for the indicated times in 1 ml of YPD prewarmed to 30°C and then transferred to ice cold PBS and washed twice with ice-cold PBS/1% bovine serum albumin (BSA). Where indicated, biotin was stripped from the cell surface by two 20-min incubations in reducing solution (50 mM glutathione, 75 mM NaCl, 150 mM NaOH, 10% fetal bovine serum). Stripped cells were washed twice with PBS/BSA, and then incubated twice for 15 min with 5 mg/ml iodoacetamide in PBS/BSA. All samples were then lysed in HKNE buffer (20 mM HEPES, pH 7.4, 150 mM KCl, 0.5% NP-40, 1 mM EDTA) supplemented with 1 mM phenylmethylsulfonyl fluoride and Sncp was collected by immunoprecipitation by using anti-Sncp antiserum. Two aliquots of each washed immunoprecipitate were run on 12% acrylamide gels under nonreducing conditions, transferred to nitrocellulose, and probed for biotinylated Snc-HA-6Kp with streptavidin-horseradish peroxidase (HRP) and for total Snc-HA-6Kp with anti-Sncp antibodies. The amount of total Snc-HA-6Kp immunoreactivity was identical for all samples.
Growth Rate Curves and Sncp Expression Assays
Cells were grown overnight to early log phase in SC galactose media without methionine at 30°C. At t = 0, cells were collected by centrifugation and resuspended in SC glucose media without methionine at concentrations ranging from A600 0.1 to 0.001 by 3-fold serial dilution. At the indicated times, A600 readings were taken from cultures in the linear range of the spectrophotometer to measure the growth rate. A600 readings were corrected for dilution of the cultures and averaged to prepare growth curves. Two A600 unit aliquots were collected by centrifugation, washed at 4°C with TAF buffer (20 mM Tris, pH 7.5, 20 mM NaN3, 20 mM NaF), and frozen in a dry ice/ethanol bath for subsequent analysis of Sncp expression. The frozen pellets were thawed, suspended in 150 μl of TAF buffer at 4°C, and lysed by homogenization with glass beads. Sncp expression was quantified by densitometry of immunoblots with reference to a standard curve prepared by serial dilutions of the NY2268, t = 0 sample as previously described (Grote and Novick, 1999). Expression of Ssop, which is not affected by the shift to galactose media, was measured as a loading control.
Electron Microscopy
Cells grown to early log phase in synthetic media at 25°C were shifted to 37°C for 20 min and then prepared for electron microscopy as previously described (Salminen and Novick, 1987).
Membrane Trafficking Assays
To measure secretion of 35S-labeled proteins, 1.5 A600 units of cells grown to early log phase in methionine-free synthetic media at 25°C were pelleted and resuspended in 350 μl of media supplemented with 150 μCi 35S-ProMix (Amersham, Arlington Heights, IL), 0.06 mg/ml BSA, and 1 mM phenylmethylsulfonyl fluoride. After incubating for 15 min at 37°C, the cells were pelleted by a 5-s microfuge spin and 300 μl of media was transferred to a chilled tube containing 20 μl of 200 mM NaN3, 200 mM NaF. Stray cells were removed by a 1-min microfuge spin and 300 μl of the supernatant was transferred to a tube containing 20 μl of 100% trichloroacetic acid (TCA), 10 mg/ml deoxycholate. TCA precipitates were collected by centrifugation, washed twice with acetone at −20°C, and air dried. The cell pellets were homogenized by vortexing with glass beads in 300 μl of Lamelli sample buffer. The secreted proteins were resuspended in 30 μl of sample buffer. The samples were boiled for 5 min, and 10 μl of each sample was separated on an 8% SDS-polyacrylamide gel. The gels were dried, and 35S-labeled proteins were detected by using a STORM phosphor imaging system. The assay was performed in triplicate for each strain.
To stain vacuoles, five A600 units of cells grown to early log phase in synthetic media were pelleted and resuspended in 500 μl of media containing 50 mM FM4-64. The cells were incubated for 15 min at 37°C, washed, and chased for an additional 45 min at 37°C. Stained cells were chilled to 4°C in TAF (20 mM Tris, pH 7.4, 20 mM NaN3, 20 mM NaF) and visualized using a digital imaging microscope.
Multicopy snc1-M43A Suppressor Screen
NY2258 cells grown in SC galactose media were transformed with a yeast genomic library contained in the 2 μ LEU2 vector YEplac181. The transformed yeast were plated on SC dextrose−leucine plates and grown for 5 d at 38°C. Plasmids were recovered into Escherichia coli and tested for the presence of the SNC1 or SNC2 genes by diagnostic PCR. Plasmids not containing SNC genes were retransformed into NY2258 to test for plasmid-dependent suppression. DNA sequencing with primers complementary to sites flanking the insertion site in YEplac181 revealed that the three plasmids with suppression activity that were isolated in this screen were identical. This original suppressing plasmid was named pNB1040.
Construction of DPL1 Plasmids and Strains
pNB1040 derivatives lacking a 1384 bp XbaI fragment within YDR925C (pNB1041) or a 4541 bp SacI-NaeI fragment within SSD1 (pNB1042) retained snc1-M43A suppression activity. A 5076-bp EcoRI fragment from pE4-7 containing the DPL1 gene flanked by 1499 bp of 5′ and 1908 bp of 3′ untranslated sequences was subcloned into pBluescript(KS−) to construct pNB1037. Directed mutagenesis by PCR was performed on a 722-bp PmlI-BglII fragment of DPL1 to construct the dpl1-K370R and dpl1-K370N mutations. BamHI-HindIII fragments containing the DPL1 wild-type (pNB1048) and mutant (pNB1049 and pNB1050) inserts were subcloned from pBluescript into pRS425 (2 μ LEU2). Wild-type DPL1 was also subcloned into pRS315 (CEN LEU) to construct pNB1047.
To disrupt DPL1, a 3.5-kb HindIII-BamHI fragment from pJA51 containing the LEU2 and KnR markers was subcloned between the HindIII and BglII sites of pBluescript-DPL1, resulting in the deletion of 196 bp of 5′ untranslated DNA and 1769 of 1965 bp of coding sequence. The DPL1::LEU2 disruption construct was transformed into NY2261 and SP1α to construct NY2262 and NY2263. To test for genetic interactions, NY2262 was crossed with NY1203, NY775, NY777, and NY1204, and NY2263 was crossed to JG8.
A Myc epitope was appended to the amino-terminus of the DPL1 gene in pNB1048 by replacing an NheI-SphI fragment encompasing the 5′ end of the gene with a PCR product amplified from two complementary oligonucleotides to construct pNB1039. The nucleotide sequence GAGCAGAAGCTTATCTCGGAGGAAGATCTG coding for the epitope tag sequence EQKLISEEDL was inserted between the first and second codons of DPL1. pNB1039 was transformed into wild-type (NY2260) and sncΔ (JG8) strains to construct NY2260 and NY2259.
Myc-DPL1p Fractionation
Twenty A600 units of cells expressing Myc-DPL1 were pelleted, resuspended in HKDE (20 mM HEPES, pH 7.4, 150 mM KCl, 1 mM dithiothreitol, 1 mM EDTA) buffer containing protease inhibitors at 4°C, and lysed by homogenization with glass beads. Four fractions were prepared from the lysate by differential centrifugation: P1 is the pellet of a 1000 × g spin for 1 min, P10 is the pellet of a 10,000 × g spin for 10 min, P100 is the pellet of a 100,000 × g spin for 20 min, and S100 is the supernatant from the 100,000 × g spin. The pellet and supernatant fractions were suspended in equal volumes of sample buffer, separated on a 10% polyacrylamide gel, transferred to nitrocellulose, probed with a monoclonal anti-Myc antibody (9E10), and detected by chemiluminescence.
Immunofluorescent Staining of MycDpl1 and Kar2
Three A600 units of Myc-DPL1 transformed cells grown to early log phase in SC−uracil media were fixed with 3.7% formaldehyde in PBS, 2% glucose, 20 mM EDTA for 4 h at room temperature. For cell wall removal, fixed cells were washed twice in KPi/sorbitol (100 mM KPO4 at pH 7.4, 1.2 M sorbitol), resuspended in 0.5 ml KPi/sorbitol containing 25 mM 2-mercaptoethanol and 40 μg/ml zymo-lyase 100-T, and incubated for 30 min at 37°C. Cells were then washed once in ice-cold PBS and resuspended in 100 μl of PBS. Twenty-five microliters of this cell suspension was applied to eight-well slides coated with polylysine (1 mg/ml). Cells were then permeabilized with 0.5% SDS in PBS, 1 mg/ml BSA for 5 min at room temperature and washed 10 times with PBS/BSA. Anti-Kar2 antiserum (1/5000) and affinity purified 9E10 (anti-Myc) monoclonal antibody (1/1000) were diluted in PBS/BSA and incubated with the cells for 1 h at room temperature. The cells were washed 10 times with PBS/BSA and then incubated with dichlorotriazinyl amino fluorescein (DTAF)-conjugated donkey anti-rabbit and Texas Red-conjugated goat anti-mouse antibodies (Jackson ImmunoResearch, West Grove, PA) diluted 1/250 in PBS/BSA for 30 min. The cells were washed as described above, mounted in antifade solution (90% glycerol, 1 mg/ml p-phenylenediamine), and sealed with nail polish. Cells were observed with a Zeiss Axiophot epifluorescent microscope with a 100× objective. Control experiments established that the secondary antibodies were specific.
RESULTS
Reduced Endocytosis of Snc2-M42Ap
The effect of a methionine 42 to alanine mutation on Snc2p endocytosis was measured by using a cell-surface biotinylation assay. To facilitate surface labeling of Sncp, an extension containing three HA epitope tags and six lysines was appended to the C-terminal, extracytoplasmic domain of Snc2p to construct Snc2-HA-6Kp (Figure 1A). The extension was expected to be exposed either within the lumen of intracellular vesicles and organelles or on the extracellular surface of the plasma membrane. The tagged Snc protein is functional because it can support growth of a snc1Δ snc2Δ strain. The membrane impermeant biotinylation reagent NHS-SS-biotin was added to cells to selectively biotinylate the tertiary amines of accessible proteins present either on the extracellular surface of the plasma membrane or in the cell wall. Sncp was collected from a total cell lysate by immunoprecipitation, and the pool of Snc2-HA-6Kp on the plasma membrane was detected on a Western blot probed with streptavidin-HRP. The SS-biotin adduct can be stripped off proteins by reduction of the disulfide linkage. When surface-biotinylated yeast cells were maintained on ice and then stripped with the membrane impermeant reducing agent glutathione, biotin was completely cleaved off the Snc2-HA-6Kp that remained on the plasma membrane. However, when the biotinylated cells were warmed to 30°C to allow membrane trafficking to resume for a short period of time before stripping, a portion of the surface-labeled Snc2-HA-6Kp was internalized to a glutathione-inaccessible compartment by endocytosis (Figure 1B, upper panel). To determine the effect of a M42A mutation on Snc2p internalization, an identical assay was performed on cells expressing Snc2-M42A-HA-6Kp (Figure 1B, lower panel). At least 90% less of the total surface-labeled pool of Snc2-M42A-HA-6Kp was internalized to a glutathione-inaccessible compartment after 10 min at 30°C compared with wild-type Snc2-HA-6Kp. Thus, the M42A mutation ablates a signal for Snc2p endocytosis. Endocytosis of wild-type and M43A mutant Snc1p was also measured using the surface biotinylation assay (Figure 1C). As with Snc2p, the M43A mutation inhibited endocytosis of Snc1-HA-6Kp by >90% after 10 min at 30°C. Thus, mutation of a critical methionine conserved between Snc1p, Snc2p, and the synaptic vesicle v-SNARE VAMP2 ablates an evolutionarily conserved endocytosis signal.
Figure 1.

Reduced endocytosis of Snc2-M42A-HA-6Kp. (A) Snc2-M42A-HA-6Kp in a budding endosomal vesicle. Snc2p has a t-SNARE-binding α-helical domain adjacent to a C-terminal transmembrane anchor. Snc2-HA-6Kp was constructed by appending three HA epitope tags (HA) and six lysines (6K) to the C terminus of Snc2p. The extension is accessible from outside of the cell when Snc2-HA-6Kp is on the plasma membrane, but is protected after internalization of Snc2-HA-6Kp to endosomal vesicles. The M42A mutation is located within the α-helical domain. (B) Endocytosis of wild-type and M42A mutant Snc2-HA-6Kp. Cells expressing wild-type (NY2202) or M42A mutant (NY2203) Snc2-HA-6Kp were surface labeled with NHS-SS-biotin at 4°C. Aliquots of cells were incubated at 30°C for the indicated times (in minutes). Biotin was stripped off proteins remaining at the cell surface by reduction with glutathione. One aliquot of cells (total) was not warmed to 30°C or stripped. The cells were lysed and Snc proteins were collected by immunoprecipitation with anti-Sncp antiserum. A Western blot was probed for biotinylated Sncp with streptavidin-HRP. (C) Endocytosis of wild-type and M43A mutant Snc1-HA-6Kp. Endocytosis was measured in cells expressing Snc1-HA-6K (wt, NY2268) and Snc1-M43A-HA-6K (A, NY2269) as described above.
Growth Defect of Cells Expressing Snc1-M43Ap
To determine whether Sncp endocytosis is important for its functions in membrane traffic, we began by measuring the effect of the endocytosis mutations on growth of cells expressing only mutant Snc proteins. In a preliminary experiment, Snc1-M43Ap was expressed at high levels in a snc1Δ snc2Δ strain by using a multicopy plasmid with the ADH promotor. Cells expressing high levels of Snc1-M43Ap grew as well as cells expressing high levels of wild-type Snc1p. One interpretation of this result is that Sncp recycling is not required if sufficient Sncp is delivered to secretory vesicles by the biosynthetic pathway.
A M43A mutation was incorporated into the genomic copy of the SNC1 gene to observe the phenotypes of reduced Sncp endocytosis under conditions where Sncp is not overproduced. The masking effect of the wild-type SNC2 gene was removed by crossing snc1-M43A SNC2 cells to snc1Δ snc2Δ cells (see MATERIALS AND METHODS), and growth of the resulting tetrads was observed on YPD plates. snc1-M43A snc2Δ colonies were smaller than wild-type colonies on both rich and synthetic media at all temperatures tested and failed to grow at 38°C (Figure 2). Unexpectedly, SNC1 snc2Δ colonies also grew more slowly than wild-type colonies at 25°C. To quantify the difference between growth rates, measurements were made of the surface area of colonies grown for 3 d at 25°C after a tetrad dissection. SNC1 snc2Δ colonies were 68% smaller (SD = 7%, n = 7) than wild-type colonies. Nevertheless, snc1-M43A snc2Δ colonies were 62% smaller than SNC1 snc2Δ colonies and 88% smaller than wild-type colonies (SD = 5%, n = 9). Interestingly, when snc2-M42A under regulation of SNC2 promotor and terminator elements was integrated at the LEU2 locus of a snc1Δ snc2Δ strain, the cells had a wild-type growth rate.
Figure 2.

Slow growth of SNC1 snc2Δ and snc1-M43A snc2Δ colonies. Wild-type (SP1), SNC1 snc2Δ (NY2264), snc1-M43A snc2Δ (NY2265), SNC2 snc1Δ (NY2204), and snc2-M42A snc1Δ (NY2205) strains were streaked out to form single colonies on YPD plates. The plates were incubated for 3 d at 25°C or for 2 d at 34°C.
Membrane Trafficking Defects in snc1-M43A SNC2Δ Cells
Cells were observed by transmission electron microscopy to examine the ultrastructure of intracellular membranes in snc1-M43A snc2Δ cells (Figure 3). Compared with wild-type cells, snc1-M43A snc2Δ cells have a massive accumulation of 100-nm vesicles that are often observed to be concentrated in bud tips. An accumulation of 250-1000-nm vesicles was also observed. SNC1 snc2Δ cells accumulate a lesser number of vesicles than snc1-M43A cells.
Figure 3.

Vesicle accumulation in snc1-M43A mutant yeast. (A) Electron micrographs of thin sections. Wild-type (SP1), SNC1 snc2Δ (NY2264), and snc1-M43A snc2Δ (NY2265) yeast were grown to early log phase at 25°C and then shifted to 37°C for 20 min before fixation. Bar = 1 μm. (B) Quantitation of secretory vesicle (100-nm) and fragmented (250–1000-nm) vacuole accumulation. Secretory vesicle and fragmented vacuole profiles were counted in 60 wild-type, 119 SNC1, and 75 snc1-M43A cells. The number of vesicles in each class was divided by the total surface area.
Cells were stained with the lipophilic endocytic tracer FM4-64 to determine whether the 250-1000-nm vesicles were of endocytic origin (Figure 4). After a 15-min incubation of living cells at 37°C followed by a 45-min chase, FM4-64 is internalized to vacuoles. In most wild-type cells, the vacuoles appear as a single large ring. In dividing cells, smaller vesicles were also observed corresponding to the vacuole fragments that segregate into the daughter cell. In the snc1-M43A snc2Δ mutant, a large number of smaller doughnut-shaped vesicles were observed in most cells, but large vacuoles were also occasionally observed. SNC1 snc2Δ cells have an intermediate phenotype that often includes a single large vacuole surrounded by several smaller vesicles. No obvious difference in the rate or extent of FM4-64 uptake was observed. The vacuole fragmentation phenotype suggests that Sncp endocytosis also contributes to membrane fusion in the endosomal pathway.
Figure 4.

Vacuole fragmentation in snc1-M43A yeast. Wild-type (SP1), SNC1 snc2Δ (NY2264), and snc1-M43A snc2Δ (NY2265) yeast were grown to early log phase at 25°C. The cells were incubated at 37°C for 15 min with 50 mM FM4-64, washed, and chased at 37°C for an additional 45 min. FM4-64 fluorescence was visualized by using a digital imaging microscope.
The growth defect and accumulation of 100-nm vesicles suggests that snc1-M43A snc2Δ cells have a defect in secretion. To measure secretion more directly, cells were pulse labeled with [35S]methionine for 15 min at 37°C. Proteins secreted into the media were collected by TCA precipitation, separated on a polyacrylamide gel, and detected by using a phosphor imaging system (Figure 5). Secretion of p190 by the snc1-M43A snc2Δ cells was reduced by 71% compared with SNC1 snc2Δ cells, and by 76% compared with wild-type cells. After correcting for the 52% reduction in the amount of total protein synthesis during the 15-min [35S]methionine pulse, secretion was still reduced by 50% in the snc1-M43A snc2Δ cells compared with both wild-type and SNC1 snc2Δ cells (Figure 5B). No secretion defect was observed in snc1Δ snc2-M42A cells.
Figure 5.


Secretion defect in snc1-M43A yeast. (A) Wild-type (SP1), SNC1 snc2Δ (NY2264), and snc1-M43A snc2Δ (NY2265) yeast were pulse labeled for 15 min at 37°C with [35S]methionine. Cells were removed from the media by centrifugation and secreted proteins were concentrated by TCA precipitation. 35S-labeled proteins in the media (top) and cell pellet (bottom) were separated on polyacrylamide gels. (B) 35S incorporated in the 190-kDa band identified with an arrow in A was quantified using a phosphor imager. The amount of 35S-p190 secreted was compared with the amount of total 35S-labeled proteins in the lysate. (C) Invertase secretion. Wild-type (SP1), SNC1 snc2Δ (NY2264), snc1-M43A snc2Δ (NY2265), snc1 Δ SNC2 (NY2204), and snc1Δ snc2-M42A (NY2205) yeast were shifted to 0.1% glucose media for 20 min at 30°C to derepress invertase synthesis. External invertase was measured from intact cells and internal invertase was measured after preparing spheroplasts.
To confirm the secretion defect of snc1-M43A cells, an invertase secretion assay was performed (Nair et al., 1990). Twenty minutes after shifting to low glucose media to derepress invertase synthesis, the total amount of newly synthesized invertase was similar in wild-type and mutant cells. At this early time point, 55% of the newly synthesized invertase in the wild-type cells had been secreted. Compared with the wild type, there was a 23% reduction in invertase secretion in the SNC1 snc2Δ mutant and a 59% reduction in the snc1-M43A snc2Δ mutant (Figure 5C). The secretion defect in snc1-M43A cells suggests that recycling of Sncp by endocytosis is important for the generation of functional secretory vesicles.
Reduced expression of Sncp is an additional factor that may contribute to the membrane trafficking phenotypes of SNC mutant cells. Sncp expression was observed using an anti-Snc1p antibody that cross-reacts with Snc2p (Figure 6). Native Snc1p and Snc2p comigrate on polyacrylamide gels, so when both Snc proteins are expressed, only the combined expression level can be determined. By densitomety, Snc1p expression in SNC1 snc2Δ cells is 60% less that the combined expression of Snc1p and Snc2p in the wild-type control. Reduced Sncp expression may explain the partial secretion defect found in the SNC1 snc2Δ cells. Surprisingly, the expression level of Snc1-M43Ap was significantly less than the expression level of wild-type Snc1p despite the fact that the two proteins are expressed from the same genetic locus. Similarly, there is more Snc2p than Snc2-M42Ap expressed when otherwise identical SNC2 genes are integrated with SNC2 upstream and downstream regulatory sequences at the LEU2 locus. Because cells expressing 70% of the wild-type level of the endocytosis-deficient Snc2-M42Ap mutant grow and secrete as fast as wild-type cells, Sncp endocytosis is only rate limiting at low Sncp expression levels.
Figure 6.
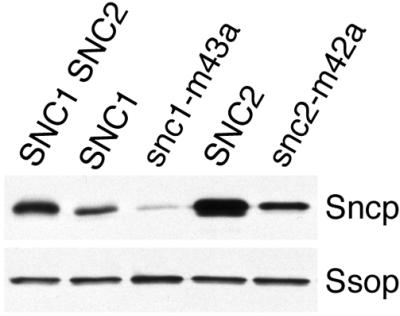
Sncp expression. A Western blot prepared from samples of equal protein concentration from wild-type (SP1), SNC1 snc2Δ (NY2264), snc1-M43A snc2Δ (NY2265), snc1 Δ SNC2 (NY2204), and snc1Δ snc2-M42A (NY2205) yeast was probed for Sncp and Ssop. The anti-Sncp antiserum may bind preferentially to Snc1p because the immunogen included an N-terminal domain of Snc1p that is poorly conserved in Snc2p.
Growth and Secretion after Repression of Snc2-M42Ap Synthesis
For a more stringent test of the requirement for Sncp endocytosis, growth and secretion were compared in strains expressing either Snc2p or Snc2-M42Ap under control of the GAL1 promotor in a snc1Δ snc2Δ host strain. Transcription from the GAL1 promotor is induced by galactose and repressed by glucose. In cells grown in glucose media, only recycled Snc2p can be incorporated into secretory vesicles once the remaining pool of newly synthesized Snc2p has been cleared from the early secretory pathway. The two strains selected for this experiment expressed equal amounts of Snc2p or Snc2-M43Ap when grown on galactose media. After transferring the cells to glucose media, Snc2p synthesis was reduced to background levels (by >95%) after 2 h. The GAL1p-SNC2 cells grew at a constant rate for 10 h after being transferred to glucose media (Figure 7A). At later time points, the growth rate declined as Snc2p became limiting. The GAL1p-snc2-M42A cells grew at the same rate as the GAL1p-SNC2 cells for the first 5 h in glucose media, and then grew more slowly at later times. To determine whether differences in the rate of Snc2p degradation were responsible for the earlier onset of slower growth in the GAL1p-snc2-M42A strain, Sncp expression was compared at each time point (Figure 7B). During the first 9 h, Snc2p and Snc2-M42Ap expression was similar at each time point. The decrease in Snc2p expression was roughly proportional to the increase in the number of cells. Thus, both Snc2p and Snc2-M42Ap have a long half-life compared with the cell cycle time, and the decrease in Snc2p concentration is simply a consequence of diluting Snc2p into a larger cell volume with each cell division. At later time points, there was more Snc2-M42Ap per cell because there were fewer cells. Secretion of 35S-p190 synthesized during a 20-min pulse 8 h after the transfer to glucose media was reduced by 27% in the GAL1p-snc2-M42A cells compared with the GAL1p-SNC2 control (Figure 7C). Because growth continues for several hours after repressing Snc2-M42Ap synthesis, we conclude either that sufficient recycling of Snc2-M42Ap occurs despite the absence of an endocytosis signal, or that Snc2-M42Ap can function on the plasma membrane. However, because Snc2-M42Ap becomes limiting for growth and secretion at a higher concentration (earlier time point) than Snc2p, endocytosis potentiates an essential Snc2p function.
Figure 7.


Phenotypes arising after repression of Snc2p and Snc2-M42Ap synthesis. (A) Growth curves. Snc2p (NY2270) and Snc2-M42Ap (NY2271) were expressed in a snc1Δ snc2Δ host strain under control of the GAL1 promotor. Cells were grown to early log phase in galactose media and then shifted to glucose media at t = 0. Cell density was measured by reading the adsorbance at 600 nm and adjusted for dilution of the cultures to prepare a growth curve. (B) Snc2p expression. Snc2p expression level was measured by densitometry from a Western blot. The combined expression level of Snc1p and Snc2p in wild-type cells (SP1) was defined as 1. (C) 35S-secretion. After 8 h of growth in glucose media cells were pulse labeled with [35S]methionine for 20 min. Secreted proteins collected from the media by TCA precipitation and total cellular proteins prepared by glass bead lysis were resolved on a polyacrylamide gel, and detected by autoradiography.
Normal SNARE Complex Assembly and Disassembly
Because the endocytosis mutations are located in the same domain of Sncp that interacts with t-SNAREs, assembly and disassembly of SNARE complexes were compared in cells expressing the endocytosis mutant Snc2-M42Ap. The exocytic SNARE complex between Ssop and the wild-type Snc2 or Snc2 proteins was detected by coimmunoprecipitation of Ssop with Sncp. We have previously established that binding of Ssop to Sncp depends on flux through the secretory pathway, and that the coimmunoprecipitation assay detects only SNARE complexes that exist before cells are lysed (Grote and Novick, 1999). An equal amount of Ssop was bound to the wild-type and mutant Snc proteins despite the fact that less Snc1-M43Ap was detected in the lysate (Figure 8). Exocytic SNARE complexes are disassembled by Sec18p if ATP and an ATP-regenerating system are added to the lysate (Carr et al., 1999; Grote and Novick, 1999). Both wild-type and mutant SNARE complexes were disassembled upon the addition of ATP (Figure 8). Thus, the phenotypes of the snc1-M43A mutant are not due to an obvious defect in SNARE complex assembly or disassembly.
Figure 8.

Assembly and disassembly of SNARE complexes containing Snc2-M42Ap. Cells expressing Snc2p (NY2204) or Snc2-M42Ap (NY2205), but not Snc1p, were lysed in the absence or presence of ATP. A Western blot of anti-Ssop immunoprecipitates was probed for coprecipitating Snc proteins (top). On a second immunoblot, an aliquot of each lysate was probed for Sncp and Ssop (bottom).
DPL1 Is a Multicopy snc Suppressor
A yeast genomic library of multicopy plasmids was screened for snc1-M43A suppressors in an attempt to identify a receptor for the endocytosis signal. In principal, if a mutation reduces the affinity of a signal for its receptor, increasing the concentration of the receptor might compensate for the reduced affinity. Spontaneous recessive suppressors were observed at high frequency in the haploid snc1-M43A snc2Δ strain. To avoid characterizing plasmids in these spontaneous revertants, the screen was carried out in a snc1-M43A/snc1Δ snc2Δ/snc2Δ diploid strain. In a preliminary experiment, the SNC1 and SNC2 genes were found to suppress the snc1-M43A growth defect, whereas the SEC1, SEC2, SEC3, SEC4, SEC6, SEC9, SEC10, and SSO2 genes did not. In a screen of 330,000 independent transformants, 69 snc1-M43A/snc1Δ snc2Δ/snc2Δ suppressing plasmids were isolated. Diagnostic PCR revealed that 53 suppressing plasmids contained the SNC1 gene, and 13 contained the SNC2 gene. The remaining three plasmids were partial suppressors of the snc1-M43A growth defect and each contained the same 10-kb insert with three long open reading frames, SSD1, DPL1, and YDR925C (Figure 9). By deleting fragments of this plasmid flanked by convenient restriction sites (see MATERIALS AND METHODS), the suppressing activity was mapped to DPL1, the structural gene for dihydrosphingosine phosphate lyase (see DISCUSSION). High expression of DPL1 is essential for its suppressing activity because a low copy number CEN plasmid containing the DPL1 gene failed to suppress snc1-M43A. The Dpl1 protein has a single predicted membrane spanning domain and a consensus sequence for pyridoxyl phosphate binding. Mutation of the critical lysine (K370) in the pyridoxyl phosphate-binding site to arginine or asparagine ablates the snc1-M43A-suppressing activity. Thus, a pyridoxyl phosphate-dependent enzymatic activity suppresses snc1-M43A.
Figure 9.

Suppression of the snc1-M43A growth defect by DPL1 overproduction. Various plasmids were transformed into a snc1-M43A strain (NY2258). Transformants were streaked to single colonies on a YPD plate and incubated for 3 d at 30°C. The plasmids tested included an empty high-copy number 2 μ vector (pRS425), pRS425 derivatives containing the wild-type DPL1 (pNB1048) and dpl1-K370N (pNB1049) and dpl1-K370R (pNB1050) mutant genes, a CEN plasmid containing the DPL1 gene that is present as a single copy in each cell (pNB1047), and the original plasmid containing the SSD1, DPL1, and YDR925C genes that was selected from a 2 μ genomic library (pNB1040).
A receptor for the Sncp endocytosis signal might be expected to reside on the plasma membrane. To determine whether Dpl1p is concentrated on the plasma membrane, the localization of Dpl1p was determined by using an N-terminal Myc-tagged version with full suppressing activity. Cells were homogenized in detergent-free lysis buffer and fractionated into 1,000 × g, 10,000 × g, and 100,000 × g pellet fractions and a 100,000 × g supernatant fraction. Myc-Dpl1p was concentrated in the 10,000 × g pellet (Figure 10). Interestingly, approximately 10-fold more Myc-Dpl1p was expressed in snc mutant cells than in wild-type cells transformed with the same multicopy plasmid. This could reflect selection for increased DPL1 copy number. To further refine the localization of Dpl1p, Myc-Dpl1p was stained in transformed cells by indirect immunofluorescence microscopy using an anti-Myc monoclonal antibody. Myc-Dpl1p was concentrated in the nuclear envelope and in punctate structures in the cytoplasm that partially overlapped with the endoplasmic reticulum marker Kar2p (Figure 11). Thus, at high expression levels, Myc-Dpl1p is concentrated in intracellular organelles, including the endoplasmic reticulum.
Figure 10.

Fractionation of Myc-Dpl1p. Wild-type (NY2261) and sncΔ (JG8) cells transformed with a Myc-Dpl1p overproducing plasmid were grown in selective media and then lysed in detergent-free buffer. The lysate was fractionated into P1, P10, and P100 pellet fractions and a S100 supernatant fraction. Myc-Dpl1p was detected on a Western blot prepared from equal aliquots of each fraction with an anti-Myc monoclonal antibody.
Figure 11.

Partial colocalization of Myc-Dpl1p and Kar2p. Wild-type cells overproducing Myc-Dpl1p were fixed with formaldehyde and stained with a mouse anti-Myc antibody and a rabbit anti-Kar2 antibody.
DPL1 is not an allele-specific snc suppressor. In addition to suppressing the snc1-M43A mutation, it also suppresses the growth defect of snc1Δ snc2Δ cells. Dpl1p overproduction also partially suppressed the secretion defect of sncΔ cells, as expected because secretion is essential for growth (Table 3). Thus, overexpression of Dpl1p allows cells to bypass the normal requirement for Sncp in exocytosis. The lack of allele specificity confirms that DPL1 does not suppress the snc1-M43A growth defect by ameliorating an endocytosis defect.
Table 3.
DPL1 overproduction restores secretion from sncΔ cells
| p190 secretion (% wild-type) | Total protein synthesis (% wild-type) | Normalized secretiona (% wild-type) | |
|---|---|---|---|
| Wild-type | 100 | 100 | 100 |
| sncΔ | 23 | 41 | 58 |
| sncΔ 2μ DPL1 | 81 | 107 | 67 |
Wild-type (SP1), sncΔ (JG8, snc1∷URA3 snc2∷ADE8 GAL1p-HA-SNC1) (Protopopov et al., 1993), and sncΔ cells overproducing DPL1 (pNB1047) were grown to early log phase at 30 °C in SC galactose media lacking tryptophan to maintain the GAL1p-HA-SNC1 plasmid or leucine to maintain the DPL1 plasmid, as appropriate. The cells were then shifted to methionine-free SC glucose media for 11 h to repress HA-Snc1p synthesis in the sncΔ cells. At this time point, the sncΔ cells have a reduced growth rate, but the sncΔ cells suppressed by DPL1 grow at the same rate as wild-type cells. Secretion of radiolabeled p190 and the rate of total protein synthesis were measured as described in Figure 5. At this time point, eight major radiolabeled proteins ranging from 30 to 190 kDa were secreted in similar relative amounts by all three strains.
Secretion/synthesis × 100%.
Disruption of the DPL1 gene in a wild-type strain had no effect on growth or invertase secretion. To test for genetic interactions between DPL1 and genes coding for components of the secretory pathway, a dpl1 deletion strain was crossed with strains containing a variety of sec mutations that inhibit fusion of secretory vesicles with the plasma membrane. Deletion of DPL1 neither increased nor decreased the growth rate of the sec1-1, sec4-8, sec5-24, or sec9-4 temperature-sensitive mutants or a snc2Δ mutant. Furthermore, overproducing DPL1 did not suppress the sec1-1, sec4-8, sec5-24, or sec9-4 mutants. Because DPL1 overexpression has no effect on secretion of sec mutant strains that express normal levels of Sncp, DPL1 cannot activate a bypass secretory pathway independent of these other Sec proteins. Thus, secretion in the DPL1-suppressed snc mutants is likely to use the plasma membrane t-SNAREs Sec9p and Ssop and the standard SEC-dependent secretory pathway.
DISCUSSION
Endocytosis of exocytic v-SNAREs in yeast and mammalian cells requires a conserved targeting signal that is unique to this class of v-SNAREs. This signal is centered on a methionine residue on the hydrophobic face of the conserved amphipathic α-helix that binds to t-SNAREs. Mutation of this critical methionine to alanine reduced endocytosis of both Snc1p and Snc2p in yeast by >95% without directly affecting exocytic SNARE complex assembly or disassembly. Lewis et al. (2000) have used an alternative method to observe the effect of mutations in the Snc1p cytoplasmic domain. Their results show that GFP-Snc1-V40A-M43Ap does not redistribute from the cell surface to intracellular vesicles in sec6-4 cells shifted to 37°C (Lewis et al., 2000). These results complement our direct evidence of reduced endocytosis. Conservation of this sorting signal between Sncp and VAMP suggests that yeast and mammalian cells share fundamentally similar mechanisms of sorting signal recognition and endocytosis (Geli and Riezman, 1998).
Our expectation was that preventing Sncp endocytosis would block transport of Sncp from the plasma membrane back to the Golgi after exocytosis. This, in turn, would inhibit the formation of fusogenic secretory vesicles, either because vesicles that bud from Sncp-depleted Golgi are unable to fuse, or because Sncp itself is required for secretory vesicle budding. Sncp clearly recycles because yeast continue growing for at least 10 h after Sncp synthesis is repressed, and cycling of green fluorescent protein-Snc1p between the plasma membrane and intracellular compartments has been observed by fluorescence microscopy (Lewis et al., 2000). The growth and secretion phenotypes associated with endocytosis deficient snc mutants support the proposal that endocytosis is involved in Sncp recycling. However, despite a 90% reduction in the endocytosis rate and a defect in transport to the Golgi (Lewis et al., 2000), ablation of the Snc endocytosis signal only partially inhibited Sncp recycling defined functionally as the ability of Sncp to support more than one round of secretion. These data suggest that the number of Snc proteins targeted to secretory vesicles is normally in excess of that required for secretion such that a sufficient amount of Sncp recycles to the Golgi even without signal-mediated endocytosis. An alternative possibility is that “reverse topology” SNARE complexes between Sncp on the plasma membrane and newly synthesized “t-SNAREs” on secretory vesicles can mediate secretion when Sncp is retained on the plasma membrane. In this situation, fusion of post-Golgi secretory vesicles with the plasma membrane may be mechanistically more similar to homotypic fusion than to fusion of endoplasmic reticulum-derived vesicles with the Golgi where v- and t-SNAREs function asymmetrically in an in vitro assay (Cao and Barlowe, 2000).
In addition to their secretion defect, snc1-M43A mutant yeast also have fragmented vacuoles and an accumulation of multivesicular endosomes. A delay in processing the vacuolar protein carboxypeptidase Y was also observed (Grote, unpublished observation). These phenotypes suggest that Sncp is required on a pathway leading to vacuole fusion. Sncp may be a component of SNARE complexes that catalyze fusion on the endosomal pathway because it binds to the endosomal t-SNAREs Tlg1p, Tlg2p, Pep12p, and Vam3p (Abeliovich et al., 1998; Holthuis et al., 1998; Grote and Novick, 1999). Interestingly, fragmented vacuoles were also observed in vam3Δ and tlg2Δ yeast, but were not found in yeast with a deletion of the gene for the vacuolar v-SNARE Nyv1p (Nichols et al., 1997; Wada et al., 1997; Holthuis et al., 1998; Seron et al., 1998). However, because the vacuole fragmentation observed in snc1-M43A cells is a chronic phenotype, it may be an indirect effect of the mutation. Vacuole fragmentation has also been observed in mutants that affect budding from endosomes and the Golgi (Banta et al., 1988; Seaman et al., 1998; Walch-Solimena and Novick, 1999). However, fragmented vacuoles have not been noted in any of the post-Golgi sec mutants. A defect at an earlier step in the endocytosis pathway was observed in sec1-1 yeast after 1 h at 37°C (Vida and Emr, 1995). One interpretation of this result is that a failure to deliver Sncp to the plasma membrane results in the formation of defective endosomes lacking v-SNAREs.
DPL1 Is a Multicopy snc Bypass Suppressor
The DPL1 gene was isolated in a screen for multicopy suppressors of the snc1-M43A mutant. The original aim of this screen was to identify a sorting receptor that interacts with the Sncp endocytosis signal. Overexpression of a sorting receptor might compensate for the endocytosis defect of Snc1-M43Ap by mass action, but improving the efficiency of Sncp recycling would be irrelevant if no Sncp is expressed. Because DPL1 overexpression suppressed a snc deletion mutant as well as snc1-M43A, it cannot be a sorting receptor. Furthermore, myc-Dpl1p is concentrated in the endoplasmic reticulum rather than at sites of endocytosis on the plasma membrane. A more direct approach involving a screen for proteins that bind to the wild-type, but not mutant, Sncp cytoplasmic domain may be successful at identifying a sorting receptor (Heilker et al., 1999).
Dihydrosphingosine phosphate lyase, the product of the DPL1 gene, is a catabolic enzyme that degrades dihydrosphingosine-1-phosphate and phytosphingosine-1-phosphate to ethanolamine and fatty aldehydes (Saba et al., 1997). In mammalian cells, sphingosine-1-phosphates serve both as intracellular second messengers and as extracellular ligands for G protein-coupled receptors (Van Brocklyn et al., 1998). In yeast, sphingosine-1-phosphates are signaling molecules that modulate the heat stress response (Skrzypek et al., 1999). Sphingosine-1-phosphate synthesis is also required to incorporate exogenously added sphingosine into ceramides (Mao et al., 1997; Qie et al., 1997). DPL1 overproduction could suppress the snc defect either by down-regulating a dihydrosphingosine-1-phosphate signal that recruits or activates an enzyme or by changing the composition and physical properties of cellular membranes. The recent identification of yeast enzymes for synthesis (Nagiec et al., 1998), dephosphorylation (Skrzypek et al., 1999), and degradation (Saba et al., 1997) of sphingosine-1-phosphates should help elucidate the suppression mechanism of DPL1 in sncΔ cells and aid in the identification of sphingosine-1-phosphate effectors. Incidentally, because ceramide is synthesized in the endoplasmic reticulum, many sphingosine metabolism enzymes are likely to colocalize with Dpl1p (Dickson and Lester, 1999).
The sncΔ growth defect can also be suppressed by loss of function mutations in the ELO2 or ELO3 genes (David et al., 1998). Like DPL1 overexpression, the elo2 and elo3 mutations do not suppress mutations in the plasma membrane t-SNAREs or other secretory mutants. Thus, the two classes of sncΔ suppressors are likely to share a common mechanism. Another connection is that both suppressors are involved in ceramide metabolism. Ceramides are synthesized by the condensation of very-long chain fatty acids with dihydrosphingosine (Dickson and Lester, 1999). The Elo2 and Elo3 proteins catalyze elongation of long-chain fatty acids to very long chain fatty acids (Oh et al., 1997). Thus, the two sncΔ suppressor classes may reduce the availability of different ceramide precursors. The actual mechanism of suppression is likely to be more complicated however, because reducing dihydrosphingosine synthesis by inhibiting the rate-limiting biosynthetic enzyme serine palmityoltransferase with myriosin or temperature-sensitive mutations (Zhao et al., 1994; Beeler et al., 1998) did not suppress snc mutations (Grote, unpublished observation).
Secretion in the DPL1 suppressed snc mutants probably occurs via the conventional secretory pathway because the sncΔ suppressors do not suppress mutations in other late-acting SEC genes. It will be of interest to identify the SNARE protein on secretory vesicles that interacts with Ssop and Sec9p in these strains. Candidates include Sec22p, Ykt6p, and Nyv1p, the three remaining v-SNAREs with an arginine at a central position in the SNARE-binding domain (Weimbs et al., 1997), and Vti1p, a promiscuous “v-SNARE” with a glutamine at the center of its SNARE-binding domain (von Mollard et al., 1997). We were unable to detect an enhanced interaction between Ssop and these SNAREs in a suppressed sncΔ strain (Grote, unpublished observation). An alternative possibility is that a t-SNARE–t-SNARE interaction mediates exocytosis in sncΔ strains (Patel et al., 1998; Rabouille et al., 1998). We have observed coimmunoprecipitation of untagged Ssop with HA-Ssop (Grote et al., 2000b), but this interaction is not enhanced in a suppressed sncΔ strain (Grote, unpublished observation). Thus, it is unlikely that the SNARE complexes in sncΔ cells contain Ssop masquerading as a v-SNARE. Identifying the functional secretory vesicle v-SNARE in the sncΔ cells and determining how DPL1 overproduction allows this v-SNARE to function remain challenges for the future.
ACKNOWLEDGMENTS
We thank Regis B. Kelly (University of California, San Francisco, CA) for supporting the initial development of this project; Jeffry Gerst (Wietzman Institute, Tel Aviv, Israel), Ira Herskowitz (University of California, San Francisco, CA), Mark Rose (Princeton University, Princeton NJ), Teresa Dunn and Troy Beeler (Uniformed Services University of the Health Sciences, Bethesda, MD), and Kim Nasmth for strains, plasmids, and antibodies; and Michael Sacher for critical reading of the manuscript. This work was supported by a National Institutes of Health grant to Peter Novick and a National Research Service Award postdoctoral fellowship to Eric Grote.
REFERENCES
- Abeliovich H, Grote E, Novick P, Ferro-Novick S. Tlg2p, a yeast syntaxin homolog that resides on the Golgi and endocytic structures. J Biol Chem. 1998;273:11719–11727. doi: 10.1074/jbc.273.19.11719. [DOI] [PubMed] [Google Scholar]
- Banta LM, Robinson JS, Klionsky DJ, Emr SD. Organelle assembly in yeast: characterization of yeast mutants defective in vacuolar biogenesis and protein sorting. J Cell Biol. 1988;107:1369–1383. doi: 10.1083/jcb.107.4.1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beeler T, Bacikova D, Gable K, Hopkins L, Johnson C, Slife H, Dunn T. The Saccharomyces cerevisiae TSC10/YBR265w gene encoding 3-ketosphinganine reductase is identified in a screen for temperature-sensitive suppressors of the Ca2+-sensitive csg2Delta mutant. J Biol Chem. 1998;273:30688–30694. doi: 10.1074/jbc.273.46.30688. [DOI] [PubMed] [Google Scholar]
- Cao X, Barlowe C. Asymmetric requirements for a Rab GTPase and SNARE proteins in fusion of COPII vesicles with acceptor membranes. J Cell Biol. 2000;149:55–66. doi: 10.1083/jcb.149.1.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carr CM, Grote E, Munson M, Hughson FM, Novick PJ. Sec1p binds to SNARE complexes and concentrates at sites of secretion. J Cell Biol. 1999;146:333–344. doi: 10.1083/jcb.146.2.333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cremona O, De Camilli P. Synaptic vesicle endocytosis. Curr Opin Neurobiol. 1997;7:323–330. doi: 10.1016/s0959-4388(97)80059-1. [DOI] [PubMed] [Google Scholar]
- David D, Sundarababu S, Gerst JE. Involvement of long chain fatty acid elongation in the trafficking of secretory vesicles in yeast. J Cell Biol. 1998;143:1167–1182. doi: 10.1083/jcb.143.5.1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Wit H, Lichtenstein Y, Geuze HJ, Kelly RB, van der Sluijs P, Klumperman J. Synaptic vesicles form by budding from tubular extensions of sorting endosomes in PC12 cells. Mol Biol Cell. 1999;10:4163–4176. doi: 10.1091/mbc.10.12.4163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickson RC, Lester RL. Yeast sphingolipids. Biochim Biophys Acta. 1999;1426:347–357. doi: 10.1016/s0304-4165(98)00135-4. [DOI] [PubMed] [Google Scholar]
- Edelmann L, Hanson PI, Chapman ER, Jahn R. Synaptobrevin binding to synaptophysin: a potential mechanism for controlling the exocytotic fusion machine. EMBO J. 1995;14:224–231. doi: 10.1002/j.1460-2075.1995.tb06995.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geli MI, Riezman H. Endocytic internalization in yeast and animal cells: similar and different. J Cell Sci. 1998;111:1031–1037. doi: 10.1242/jcs.111.8.1031. [DOI] [PubMed] [Google Scholar]
- Gerst JE, Rodgers L, Riggs M, Wigler M. SNC1, a yeast homolog of the synaptic vesicle-associated membrane protein/synaptobrevin gene family: genetic interactions with the RAS and CAP genes [published erratum appears in Proc. Natl. Acad. Sci. USA (1992) 89, 7287] Proc Natl Acad Sci USA. 1992;89:4338–4342. doi: 10.1073/pnas.89.10.4338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gotte M, von Mollard GF. A new beat for the SNARE drum. Trends Cell Biol. 1998;8:215–218. doi: 10.1016/s0962-8924(98)01272-0. [DOI] [PubMed] [Google Scholar]
- Grote E, Baba M, Ohsumi Y, Novick PJ. Geranylgeranylated SNAREs are dominant inhibitors of membrane fusion. J Cell Biol. 2000b;151:453–465. doi: 10.1083/jcb.151.2.453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grote E, Hao JC, Bennett MK, Kelly RB. A targeting signal in VAMP regulating transport to synaptic vesicles. Cell. 1995;81:581–589. doi: 10.1016/0092-8674(95)90079-9. [DOI] [PubMed] [Google Scholar]
- Grote E, Kelly RB. Endocytosis of VAMP is facilitated by a synaptic vesicle targeting signal. J Cell Biol. 1996;132:537–547. doi: 10.1083/jcb.132.4.537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grote E, Novick PJ. Promiscuity in rab-SNARE interactions. Mol Biol Cell. 1999;10:4149–4161. doi: 10.1091/mbc.10.12.4149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grote E, Carr CM, Novick PJ. Ordering the final events in yeast exocytosis. J Cell Biol. 2000a;151:439–451. doi: 10.1083/jcb.151.2.439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guthrie C, Fink GR. Guide to Yeast Genetics and Molecular Biology. San Diego: Academic Press; 1991. [Google Scholar]
- Heilker R, Spiess M, Crottet P. Recognition of sorting signals by clathrin adaptors. BioEssays. 1999;21:558–567. doi: 10.1002/(SICI)1521-1878(199907)21:7<558::AID-BIES4>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- Holthuis JC, Nichols BJ, Dhruvakumar S, Pelham HR. Two syntaxin homologues in the TGN/endosomal system of yeast. EMBO J. 1998;17:113–126. doi: 10.1093/emboj/17.1.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis MJ, Nichols BJ, Prescianotto-Baschong C, Riezman H, Pelham HR. Specific retrieval of the exocytic SNARE snc1p from early yeast endosomes. Mol Biol Cell. 2000;11:23–38. doi: 10.1091/mbc.11.1.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis MJ, Rayner JC, Pelham HR. A novel SNARE complex implicated in vesicle fusion with the endoplasmic reticulum. EMBO J. 1997;16:3017–3024. doi: 10.1093/emboj/16.11.3017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lustgarten V, Gerst JE. Yeast VSM1 encodes a v-SNARE binding protein that may act as a negative regulator of constitutive exocytosis. Mol Cell Biol. 1999;19:4480–4494. doi: 10.1128/mcb.19.6.4480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao C, Wadleigh M, Jenkins GM, Hannun YA, Obeid LM. Identification and characterization of Saccharomyces cerevisiae dihydrosphingosine-1-phosphate phosphatase. J Biol Chem. 1997;272:28690–28694. doi: 10.1074/jbc.272.45.28690. [DOI] [PubMed] [Google Scholar]
- Nagiec MM, Skrzypek M, Nagiec EE, Lester RL, Dickson RC. The LCB4 (YOR171c) and LCB5 (YLR260w) genes of Saccharomyces encode sphingoid long chain base kinases. J Biol Chem. 1998;273:19437–19442. doi: 10.1074/jbc.273.31.19437. [DOI] [PubMed] [Google Scholar]
- Nair J, Muller H, Peterson M, Novick P. Sec2 protein contains a coiled-coil domain essential for vesicular transport and a dispensable carboxy terminal domain. J Cell Biol. 1990;110:1897–1909. doi: 10.1083/jcb.110.6.1897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nichols BJ, Ungermann C, Pelham HR, Wickner WT, Haas A. Homotypic vacuolar fusion mediated by t- and v-SNAREs. Nature. 1997;387:199–202. doi: 10.1038/387199a0. [DOI] [PubMed] [Google Scholar]
- Oh CS, Toke DA, Mandala S, Martin CE. ELO2 and ELO3, homologues of the Saccharomyces cerevisiae ELO1 gene, function in fatty acid elongation and are required for sphingolipid formation. J Biol Chem. 1997;272:17376–17384. doi: 10.1074/jbc.272.28.17376. [DOI] [PubMed] [Google Scholar]
- Patel SK, Indig FE, Olivieri N, Levine ND, Latterich M. Organelle membrane fusion: a novel function for the syntaxin homolog Ufe1p in ER membrane fusion. Cell. 1998;92:611–620. doi: 10.1016/s0092-8674(00)81129-0. [DOI] [PubMed] [Google Scholar]
- Pfeffer SR. Transport vesicle docking: SNAREs and associates. Annu Rev Cell Biol. 1996;12:441–461. doi: 10.1146/annurev.cellbio.12.1.441. [DOI] [PubMed] [Google Scholar]
- Protopopov V, Govindan B, Novick P, Gerst JE. Homologs of the synaptobrevin/VAMP family of synaptic vesicle proteins function on the late secretory pathway in S. cerevisiae. Cell. 1993;74:855–861. doi: 10.1016/0092-8674(93)90465-3. [DOI] [PubMed] [Google Scholar]
- Qie L, Nagiec MM, Baltisberger JA, Lester RL, Dickson RC. Identification of a Saccharomyces gene, LCB3, necessary for incorporation of exogenous long chain bases into sphingolipids. J Biol Chem. 1997;272:16110–16117. doi: 10.1074/jbc.272.26.16110. [DOI] [PubMed] [Google Scholar]
- Rabouille C, Kondo H, Newman R, Hui N, Freemont P, Warren G. Syntaxin 5 is a common component of the NSF- and p97-mediated reassembly pathways of Golgi cisternae from mitotic Golgi fragments in vitro. Cell. 1998;92:603–610. doi: 10.1016/s0092-8674(00)81128-9. [DOI] [PubMed] [Google Scholar]
- Rothman JE, Warren G. Implications of the SNARE hypothesis for intracellular membrane topology and dynamics. Curr. Biol. 1994;4:220–233. doi: 10.1016/s0960-9822(00)00051-8. [DOI] [PubMed] [Google Scholar]
- Saba JD, Nara F, Bielawska A, Garrett S, Hannun YA. The BST1 gene of Saccharomyces cerevisiae is the sphingosine-1-phosphate lyase. J Biol Chem. 1997;272:26087–26090. doi: 10.1074/jbc.272.42.26087. [DOI] [PubMed] [Google Scholar]
- Salem N, Faundez V, Horng JT, Kelly RB. A v-SNARE participates in synaptic vesicle formation mediated by the AP3 adaptor complex. Nat Neurosci. 1998;1:551–556. doi: 10.1038/2787. [DOI] [PubMed] [Google Scholar]
- Salminen A, Novick PJ. A ras-like protein is required for a post-Golgi event in yeast secretion. Cell. 1987;49:527–538. doi: 10.1016/0092-8674(87)90455-7. [DOI] [PubMed] [Google Scholar]
- Seaman MN, McCaffery JM, Emr SD. A membrane coat complex essential for endosome-to-Golgi retrograde transport in yeast. J Cell Biol. 1998;142:665–681. doi: 10.1083/jcb.142.3.665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seron K, Tieaho V, Prescianotto-Baschong C, Aust T, Blondel MO, Guillaud P, Devilliers G, Rossanese OW, Glick BS, Riezman H, Keranen S, Haguenauer-Tsapis R. A yeast t-SNARE involved in endocytosis. Mol Biol Cell. 1998;9:2873–2889. doi: 10.1091/mbc.9.10.2873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shih SC, Sloper-Mold KE, Hicke L. Monoubiquitin carries a novel internalization signal that is appended to activated receptors. EMBO J. 2000;19:187–198. doi: 10.1093/emboj/19.2.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skehel JJ, Wiley DC. Coiled coils in both intracellular vesicle and viral membrane fusion. Cell. 1998;95:871–874. doi: 10.1016/s0092-8674(00)81710-9. [DOI] [PubMed] [Google Scholar]
- Skrzypek MS, Nagiec MM, Lester RL, Dickson RC. Analysis of phosphorylated sphingolipid long-chain bases reveals potential roles in heat stress and growth control in Saccharomyces. J Bacteriol. 1999;181:1134–1140. doi: 10.1128/jb.181.4.1134-1140.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutton RB, Fasshauer D, Jahn R, Brunger AT. Crystal structure of a SNARE complex involved in synaptic exocytosis at 2.4 A resolution [see comments] Nature. 1998;395:347–353. doi: 10.1038/26412. [DOI] [PubMed] [Google Scholar]
- Trowbridge IS, Collawn JF, Hopkins CR. Signal-dependent membrane protein trafficking in the endocytic pathway. Annu Rev Cell Biol. 1993;9:129–161. doi: 10.1146/annurev.cb.09.110193.001021. [DOI] [PubMed] [Google Scholar]
- Van Brocklyn JR, Lee MJ, Menzeleev R, Olivera A, Edsall L, Cuvillier O, Thomas DM, Coopman PJ, Thangada S, Liu CH, Hla T, Spiegel S. Dual actions of sphingosine-1-phosphate: extracellular through the Gi-coupled receptor Edg-1 and intracellular to regulate proliferation and survival. J. Cell Biol. 1998;142:229–240. doi: 10.1083/jcb.142.1.229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vida TA, Emr SD. A new vital stain for visualizing vacuolar membrane dynamics and endocytosis in yeast. J Cell Biol. 1995;128:779–792. doi: 10.1083/jcb.128.5.779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Mollard GF, Nothwehr SF, Stevens TH. The yeast v-SNARE Vti1p mediates two vesicle transport pathways through interactions with the t-SNAREs Sed5p and Pep12p. J Cell Biol. 1997;137:1511–1524. doi: 10.1083/jcb.137.7.1511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wada Y, Nakamura N, Ohsumi Y, Hirata A. Vam3p, a new member of syntaxin related protein, is required for vacuolar assembly in the yeast Saccharomyces cerevisiae. J Cell Sci. 1997;110:1299–1306. doi: 10.1242/jcs.110.11.1299. [DOI] [PubMed] [Google Scholar]
- Walch-Solimena C, Novick P. The yeast phosphatidylinositol-4-OH kinase Pik1 regulates secretion at the Golgi. Nat Cell Biol. 1999;1:523–525. doi: 10.1038/70319. [DOI] [PubMed] [Google Scholar]
- Warren RA, Green FA, Stenberg PE, Enns CA. Distinct saturable pathways for the endocytosis of different tyrosine motifs. J Biol Chem. 1998;273:17056–17063. doi: 10.1074/jbc.273.27.17056. [DOI] [PubMed] [Google Scholar]
- Weber T, Zemelman BV, McNew JA, Westermann B, Gmachl M, Parlati F, Sollner TH, Rothman JE. SNAREpins: minimal machinery for membrane fusion. Cell. 1998;92:759–772. doi: 10.1016/s0092-8674(00)81404-x. [DOI] [PubMed] [Google Scholar]
- Weimbs T, Low SH, Chapin SJ, Mostov KE, Bucher P, Hofmann K. A conserved domain is present in different families of vesicular fusion proteins: a new superfamily. Proc Natl Acad Sci USA. 1997;94:3046–3051. doi: 10.1073/pnas.94.7.3046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang B, Gonzalez L, Jr, Prekeris R, Steegmaier M, Advani RJ, Scheller RH. SNARE interactions are not selective. Implications for membrane fusion specificity. J Biol Chem. 1999;274:5649–5653. doi: 10.1074/jbc.274.9.5649. [DOI] [PubMed] [Google Scholar]
- Zhao C, Beeler T, Dunn T. Suppressors of the Ca(2+)-sensitive yeast mutant (csg2) identify genes involved in sphingolipid biosynthesis. Cloning and characterization of SCS1, a gene required for serine palmitoyltransferase activity. J Biol Chem. 1994;269:21480–21488. [PubMed] [Google Scholar]