

Abstract
Recent discoveries shed light on the importance of prostaglandin (PG) production in the development of skin cancer. Work by Fischer et al. demonstrates that skin tumor promotion caused by ultraviolet B radiation can be decreased by up to 89% by blocking cyclooxygenase-2 (COX-2) with the drug Celecoxib. A similar study showed that Celecoxib can decrease new tumor formation by 44% in mice that already have tumors. These studies demonstrate the importance of COX-2 and PGs in the development of squamous cell carcinoma. We have explored growth signaling in a model of skin tumor progression. Because changes in PG production have been implicated in skin carcinogenesis, we examined this pathway. We found that malignant cell lines secrete more prostaglandin E2 (PGE2) than the parental cells. We observed increased expression of COX-1 and -2. We also found that these cells express the PGE2 receptors EP1 and EP4. When the cells are grown in the presence of indomethacin, the growth rate of the malignant cells is decreased. This effect can be reversed by addition of PGE2 or an EP1 agonist to the medium. Thus, we have shown that skin tumor cells depend in part on PGE2 signaling through the EP1 prostanoid receptor for their in vitro growth.
Keywords: COX-1, COX-2, PGE2, EP1, tumor progression, keratinocyte growth
Introduction
Prostaglandins (PGs) mediate numerous physiological responses including inflammatory and immune responses, bone development, wound healing, hemostasis, glomerular filtration, production of the extracellular matrix, and cell proliferation [1–5]. Prostaglandin E2 (PGE2) alone has been shown to be active in many of these processes including induction of matrix metalloproteinases, bone resorption, induction of oncogenes, and induction of cyclooxygenase-2 (COX-2) [6–16]. PG production involves activation of phospholipase A2 (PLA2) releasing arachidonic acid (AA) from the cell membrane. AA then serves as a substrate for either COX-1 or -2, or PGH synthase. These enzymes display the same enzymatic activity, but are the products of two different genes [17,18]. COX-2 has been shown to be inducible by various factors including ultraviolet B and 12-O-tetradecanoylphorbol-13-acetate (TPA) [19–22]. There are also reports that COX-1, although generally constitutively expressed, can be induced by agents such as 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone derived from nicotine and PGE2 [23,24]. The activity of the COX enzymes produces PGH2. This is then further metabolized to other PGs or thromboxanes. The production of PGE2 from PGH2 is dependent on PGE synthase, also referred to as PGH-PGE isomerase [25–27]. Thus, PG production requires several enzymatic steps, some of which can be regulated.
PG production in the skin is inducible by numerous stimuli including UV radiation [28,29] and treatment with the phorbol ester TPA [30–32]. Both of these can act as tumor-promoting agents [19,33]. There is evidence to suggest that at least some of the tumor-promoting effects of these agents are mediated by stimulation of PGE2 production [19,28,30–32,34–36]. Work by Fischer et al. [37] has shown that blocking COX-2 activity with a selective inhibitor (Celecoxib) significantly decreased the tumor yield produced by ultraviolet B irradiation of hairless mice. This decrease was dose dependent with respect to Celecoxib. They also showed that the nonselective COX inhibitor, indomethacin, could produce a similar effect [37]. Müller-Decker et al. [38] also provided evidence that induction of COX-2 is an essential step for mouse skin two-stage carcinogenesis. They showed increased PGE2 production due to COX-2 induction in skin treated with TPA. In this study, they found that repeated applications of TPA resulted in stationary hyperplasia and a loss of the PG response. However, in papillomas and carcinomas, constitutive overexpression of COX-2 and increased PG production persisted. This result was confirmed in keratinocyte cell lines corresponding to different stages of tumor development. It is of interest that inhibition of COX-2 can inhibit growth of a human cutaneous squamous cell carcinoma cell line [39].
Other work indicates that tumor promoters like TPA stimulate PGE2 production in primary cultures of mouse epidermal cells [40]. Also of interest is the fact that smokeless tobacco extracts can increase PGE2 production in human gingival keratinocytes [41]. PGE2 production has also been linked to several events important to cancer formation and spread. Addition of PGE2 to growth medium can increase the growth rate of human keratinocytes [42]. Expression of the oncogenes JunB and p65 can be induced by PGE2 [43]. PGE2 production can also increase migration and adherence in a head-and-neck squamous cell carcinoma cell line [44].
PGE2 has been shown to signal through several pharmacologically distinct receptors [45]. These are G-protein-coupled receptors classified according to their binding affinity for various agonists or antagonists, and are called EP1, EP2, EP3, and EP4 prostanoid receptors. The EP2 and EP4 prostanoid receptors both signal through increased adenylyl cyclase activity, whereas the EP3 prostanoid receptor decreases the activity of this enzyme [45]. The EP1 prostanoid receptor signals through increased activity of PLC-β [45]. This results in release of diacylglycerol (DAG) and inositol triphosphate (IP3) [46]. These receptors are most likely responsible for the downstream effects of PGE2 and thus could be important in several disease states including skin cancer.
Dysregulation of PGE2 production is a common finding in skin cancer [28,30,31,47]. In the present study, we assessed potential dysregulation of PGE2 production in a murine model of skin tumor progression developed in our laboratory [48]. We observed increased expression of the COX enzymes in the malignant SCC progressed cell lines compared to the precursor benign papilloma-producing cell line. Total PGE2 measurements showed that these enzymes are active. We further observed that the cells express the EP1 and EP4 prostanoid receptors. We then established that signaling through the EP1 prostanoid receptor is partially responsible for the in vitro growth of these cells.
Materials and Methods
Cell Culture
The murine keratinocyte cell line 308 was progressed with LD90 doses of γ rays (750 cGy) from a 60Co source. The resulting cells were injected subcutaneously into athymic nude mice, and a cell line started from the resulting tumors. This is cell line 6R90 [48]. Cell line 6RI (6RInvasive) was established at a later date from an unusually invasive 6R90 tumor. The cells were maintained in minimal essential medium supplemented with 7.5% fetal calf serum, 2.5% calf serum, and 100 U/ml penicillin /streptomycin (Life Technologies, Rockville, MD) at 37°C in a humidified atmosphere containing 7% CO2.
Isolation of Cytoplasmic Protein and Western Blot Analysis
Cytoplasmic protein was isolated from subconfluent cultures using published protocols [48]. Briefly, the cells were rinsed with PBS and buffer A (10 mM HEPES pH 7.9, 1.5 mM MgCl2, 10 mM KCl, 0.5 mM DTT), and then lysed by adding 0.1% NP-40 in buffer A to the cells on the plate and scraping the plate. The lysate was then transferred to a 1.5-ml plastic tube. Nuclei were pelleted by centrifugation and the proteins extracted in buffer C (20 mM HEPES pH 7.9, 25% glycerol, 420 mM NaCl, 1.5 mM MgCl2, 0.2 mM EDTA, 0.5 mM DTT, 0.5 mM PMSF). The supernatant was diluted 1:6 with buffer D (20 mM HEPES pH 7.9, 20% glycerol, 1.5 mM MgCl2, 100 mM KCl, 0.2 mM EDTA, 0.5 mM DTT, 0.5 mM PMSF) and the extracts were stored at -80°C until used. The protein concentrations of the extracts were determined with Bio-Rad reagent (Bio-Rad Laboratories, Hercules, CA). Forty micrograms of cytoplasmic extract was resolved on a 10% SDS-polyacrylamide gel and analyzed by Western blot using antibodies against COX-1 (1:250 dilution, cat# sc-1752; Santa Cruz Biotechnology, Santa Cruz, CA) or COX-2 (1:3000 dilution, cat# PG26; Oxford Biochemicals, Oxford, MI) or PGE synthase (1:250 dilution, cat# 160140; Caymen Chemical, Ann Arbor, MI).
Growth Curves
A total of 1x104 cells were plated in six-well plates and grown for 2 days as described above. Triplicate wells were treated with ethanol or 10 µM indomethacin or 10 µM indomethacin and PGE2 or agonist. The amount of PGE2 or selective agonist added was based on the amount of PGE2 present in the medium of normally grown cells as shown in Figure 3, and indexed to reflect cell number in the growth curve experiment. Every 12 hours, medium was replaced, and PGE2 or agonist was added based on the amount that would be present at the end of the following 12 hours. The maximum dose was 1.67 nM. These cells were trypsinized every 12 hours in triplicate and the cells were counted on a Coulter Counter. Mean cell numbers were plotted against time to generate growth curves for all three cell lines. The selective EP1 and EP3 agonist 17-phenyl trinor PGE2 was purchased from Caymen Chemical (Ann Arbor, MI).
Figure 3.
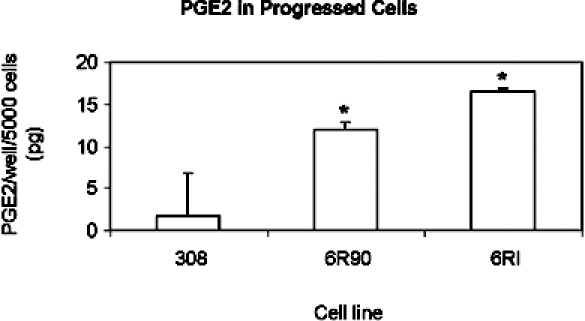
PGE2 production. EIA was performed to measure PGE2 production. The cells were grown under normal culture conditions in 96-well plates and lysed in the plate. A sample was then taken for analysis by EIA. The bars indicate 95% confidence intervals. The asterisk indicates a significant difference compared to 308 cells with a P value <.01 by analysis of variance.
PGE2 Measurement
Total cellular PGE2 was measured by enzyme immunosorbent assay (EIA). The EIA apparatus was purchased from Amersham (Piscataway, NJ). Briefly, cells were lysed in medium and a sample was taken. The sample was incubated with antibody and a PGE2 conjugate. The plate was then washed and the substrate was added. The plate was incubated for 1 hour. The reaction was then halted by addition of 1 M sulfuric acid, and the results were read at 450 nm.
In Vivo Tumor Growth
5x106 Cells from the cell lines were injected subcutaneously at four sites into athymic nude mice (BALB/c, nu/nu, Harlan Sprague-Dawley, Indianapolis, IN). Tumors were harvested and sections were stained with hematoxylin and eosin (H&E).
EP Receptor Staining
Antibodies to human EP1 and EP4 receptors were generated in chickens using recombinant fusion proteins containing glutathione-S-transferase and a portion of the carboxy terminus of the EP4 receptor or the third intracellular loop of the EP1 receptor [49]. Cells were grown on coverslips to 60% confluence. Medium was aspirated, and coverslips were washed in PBS. Cells were fixed with 4% paraformaldehyde. Coverslips were washed in PBS, and incubated in 30 mM SSC. Coverslips were incubated in 300 mM glycine, then in permeabilization buffer (0.1% Triton X-100 in 30 mM SSC). Coverslips were placed in primary antibody diluted 1:250 in antibody dilution buffer, and incubated overnight at 4°C. Coverslips were washed, and then placed in fluorescein 5-isothiocyanate (FITC)-conjugated rabbit anti-chicken secondary antibody (cat# F-8888; Sigma Chemical Co., St. Louis, MO) diluted 1:1000. Samples were then washed. Two microliters of 4′,6′-diamidino-2-phenylindole (DAPI) was added to the final wash. The coverslips were mounted on glass slides for viewing, and the stained cells were then imaged.
RNA Preparation
Cells were cultured from a 10-cm dish and centrifuged at 3000 rpm for 5 minutes at 4°C, then suspended in 400 µl of ice-cold lysis buffer and placed on ice. Lysis buffer contained 140 mM NaCl, 1.5 mM MgCl2, 10 mM Tris-HCl (pH 8.6), 0.5% NP-40, and ribonuclease (RNase) inhibitor (10 mM vanadyl ribonucleotide complexes). The cell mixture was slowly loaded onto 400 µl of sucrose lysis buffer and centrifuged at 3000 rpm for 10 minutes at 4°C. Sucrose lysis buffer was lysis buffer with the addition of 24% sucrose to the solution. The supernatant was mixed with 400 µl of 0.2 M Tris-HCl (pH 7.5), 25 mM EDTA, 0.3 M NaCl, 2% SDS, and 200 µg/ml of proteinase K, incubated at 37°C for 40 minutes, extracted with phenol, and precipitated with sodium acetate and ethanol.
Reverse Transcription-Polymerase Chain Reaction (RT-PCR)
Cytoplasmic RNA (20 µg) was treated with DNase for 15 minutes at room temperature, then 25 mM EDTA was added, and the reaction was inactivated at 65°C for 10 minutes. Four microliters of oligo(dT) was added, and the reaction was heated for 10 minutes at 70°C. First strand synthesis was carried out by adding 10 µl of the RNA (10 µg) mix to 9 µl of the RT mix. The RT mix contained 5 µl of first strand buffer (250 mM Tris-HCl pH 8.3, 375 mM KCl, 15 mM MgCl2), 1 µl dNTP (10 mM), 2 µl DTT (0.1 M), and 2 µl DEPC-H20 per 10 µl RNA. For each sample, two treatment groups were performed, one with reverse transcriptase (RT+) and one without (RT-). The samples were then incubated for 50 minutes at 42°C followed by 15 minutes at 70µC and ice for 5 minutes. RNase H (2 µl) was added to each tube, and was then incubated for 20 minutes at 37°C.
PCR was then performed using primers designed for the murine EP1, EP4, and GAPDH nucleotide sequences. Primer sequences are as follows: EP1 sense (574–602 coding sequence, CTGGGTCCCCCGGGCGGCTGGCGC-CAGGC), EP1 anti-sense (980–1005, CAGGAACAGTGGCCGCTGCAGGGAGG), EP4 sense (49–72, CTGAA-CAGCCCAGTGACCATCCCG), EP4 antisense (391–411, GCCCGCCAATCGCTTGTCCAC), mouse GAPDH sense (43–67, CAAAATGGTGAAGGTCGGTGTGAAC), mouse GAPDH antisense (294-265, ATGTTAGTGGGGTCTCGCTCCTGGAAGATG). PCRs were amplified using 1 µl of cDNA from RT reactions. Each PCR contained 10 mM of the respective primers (EP1, EP4, or GAPDH), 0.2 mM dNTP, 9 mM or 1.5 mM (EP/GAPDH, respectively) MgCl2, 1xPCR buffer (20 mM Tris-HCl pH 8.4, 50 mM KCl), 0.5 µl Taq polymerase (5 U/µl; Gibco, Rockville, MD), and nuclease free water per 50 µl reaction. The PCR amplification cycle consisted of 95°C for 5 minutes initial denaturing step, followed by 95°C for 1 minute, 63°C for 1 minute, 72°C for 1 minute for 30 cycles, and ending with 72°C for 15 minutes. Twenty microliters of sample was analyzed by electrophoresis on a 1.2% agarose gel. The EP1, EP4, and GAPDH primers should produce PCR product sizes of 431, 362, and 222 base pairs, respectively.
Quantification of EP Receptors
Cells were harvested and fixed in 4% paraformaldehyde. Cells were washed in PBS and 30 mM SSC. Cells were incubated in 300 mM glycine, then in permeabilization buffer. A 1:300 dilution of primary antibody was added, and the cells were rotated overnight at 4°C. Cells were washed in SSC. FITC-conjugated rabbit anti-chicken secondary antibody (cat# F-8888; Sigma) diluted 1:250 was added, and cells were incubated for 1 hour. Cells were washed, and resuspended in 30 mM SSC. Then, relative fluorescence units per cell were read on a flow cytometer.
Results
Cell Line 6RI is Invasive
The cell line 6R90 was derived from the benign papilloma-producing mouse keratinocyte cell line, 308 [48]. An invasive variant of the 6R90 cell line was subsequently isolated from an invasive tumor formed by injecting 6R90 cells subcutaneously into athymic nude mice. To test the stability of the invasive phenotype, the cell line 6RI was injected subcutaneously into athymic nude mice. All 12 6RI injection sites developed tumors, and these tumors appeared in 1 to 2 weeks. All 16 6R90 injection sites developed tumors in 2 to 3 weeks. Tumors were harvested at 10 weeks and examined by H&E stain to determine if the tumors had invaded the underlying muscle. In Figure 1, a photomicrograph is shown of typical margins for 6R90 and 6RI tumors. The 6R90 tumor had a pushing margin, whereas all the 6RI tumors had clearly invaded the underlying muscle.
Figure 1.
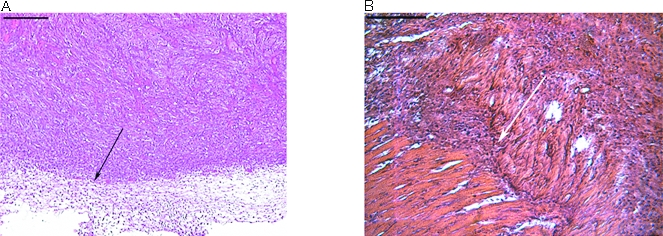
The cell line 6RI is invasive. A total of 5x106 cells were injected subcutaneously into athymic nude mice. Tumors were harvested and H&E-stained. A representative margin is shown. The bars in the upper left corners represent 500 µm. (A) Tumor formed by the cell line 6R90. The arrow shows a regular margin that does not invade the underlying tissue. (B) Tumor formed by the cell line 6RI. The arrow shows the tumor invading the underlying muscle.
6R90 and 6RI Cells Have Increased COX Expression
We examined the expression of COX proteins while looking for possible sources of reactive oxygen species in 6R90 cells. COX-1 and -2 were examined by Western blot analysis. The COX enzymes are responsible for converting AA into PGH2. Western blots for either COX-1 or -2 showed elevated levels in both malignant variants over the parental 308 cells as seen in Figure 2. The 6RI cells expressed higher levels of both COX-1 and -2 than the 6R90 cells. The 6R90 cells showed an increase in enzyme expression over the 308 cells. Thus, there was a trend of increasing amounts of enzyme expression as the cells progress from benign to malignant to highly invasive.
Figure 2.
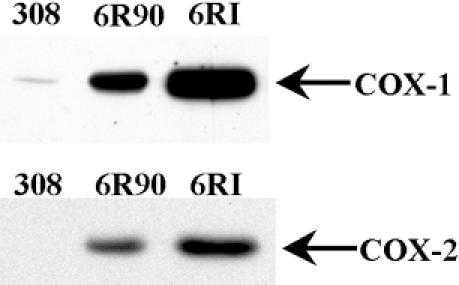
The malignant cell lines have increased COX expression. This is a Western blot analysis of COX-1 and -2 in 308, 6R90, and 6RI cells. Forty micrograms of protein lysate was fractionated by SDS-PAGE. After transferring the protein to PVDF membrane, the blots were probed with antibody specific to either COX-1 or -2 as indicated.
Expression of PGE synthase, also called PGH-PGE isomerase, was also examined by Western blot analyses. This enzyme is responsible for converting PGH2 into PGE2. This analysis showed no increase in protein expression in either malignant cell line compared to the benign 308 cells (data not shown).
To test whether these enzymes were active, PGE2 was measured in cultured cells. Cells were lysed and combined cellular and secreted PGE2 was measured by EIA. The results shown in Figure 3 demonstrate that the 6RI cells have the greatest PGE2 production at 16.45 pg/well per 5000 cells, followed by the 6R90 cells at 12.03 pg/well per 5000 cells. The cell line 308 produced little, if any, PGE2 with an average of 1.82 pg/well per 5000 cells. As the cells progressed in terms of their malignant phenotype, the amount of PGE2 produced increased.
Murine Keratinocyte Cell Lines Express PGE2 Receptors
The cell lines were examined by observing immunofluorescence to determine if they expressed receptors for PGE2. The staining for these receptors (Figure 4) indicated that all three cell lines expressed the EP1 and EP4 prostanoid receptors. Staining for the EP2 and EP3 prostanoid receptors was negative (data not shown). To confirm these results, RT-PCR was performed on RNA isolated from the cell lines. This confirmed that the EP1 and EP4 prostanoid receptors were transcribed in all three cell lines (data not shown). RT-PCR was not performed for EP2 or EP3. Because these cells also produce the ligand for these receptors, there could be autocrine or paracrine PG regulation of cellular functions, including growth.
Figure 4.
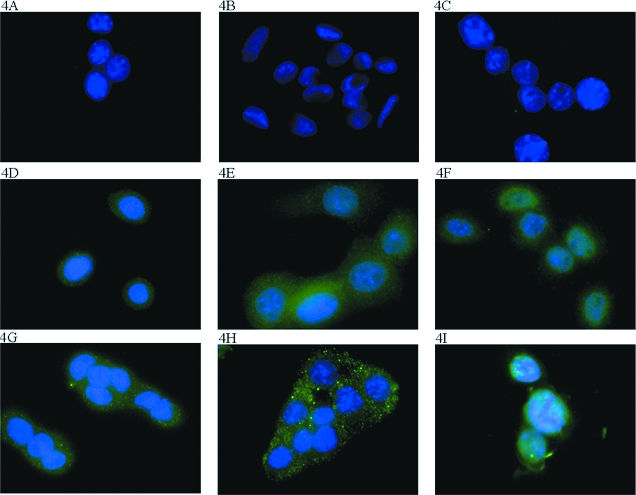
Immunohistochemical staining for EP receptors. Cells were grown on cover slips under normal culture conditions and stained for the EP receptors. The counterstain was DAPI staining of the nucleus. (A) 308 Cells incubated with secondary antibody. (B) 6R90 Cells incubated with secondary antibody. (C) 6RI Cells incubated with secondary antibody. (D) 308 Cells stained for EP1. (E) 6R90 Cells stained for EP1. (F) 6RI Cells stained for EP1. (G) 308 Cells stained for EP4. (H) 6R90 Cells stained for EP4. (I) 6RI Cells stained for EP4.
To compare expression levels of these receptors, flow cytometry was used. In these experiments, a FITC-conjugated secondary antibody labeled the receptors, and the average intensity of fluorescence per cell was recorded. The data shown in Figure 5 indicated that the 308 and 6R90 cell lines had equivalent expression of receptors. The 6RI cells demonstrated a two-fold increase in EP1 and EP4 prostanoid receptor expression.
Figure 5.
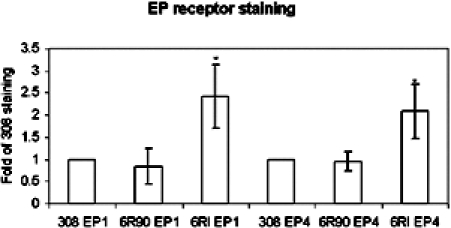
Quantification of EP receptors. A total of 106 cells were stained for the EP1 and EP4 receptors. Data are expressed as fold of 308 staining. The bars indicate the standard deviation from five experiments.
Malignant Variants of 308 Cells Depend on PGE2 for Growth
Because the cells produce PGE2 and express receptors for PGE2, we tested the cells for changes in growth rate after changes in PG production. The cells were treated with 10 µM indomethacin, a nonselective COX inhibitor, and various amounts of PGE2. The amount of PGE2 added to the medium was based on the projected cell count and the amount of PGE2 that would be produced by that number of cells as predicted by the PGE2 production measured in Figure 3. This amount varied between 0.025 and 1.67 nM. The results of these treatments (Figure 6) showed that the growth of the 6R90 and 6RI cells was reduced by 62% and 46%, respectively, in the presence of indomethacin. This growth inhibition was reversed by the addition of PGE2 to the medium. This shows that, although production of other eicosanoids could be inhibited by indomethacin, the growth of these cells depends on PGE2 production.
Figure 6.
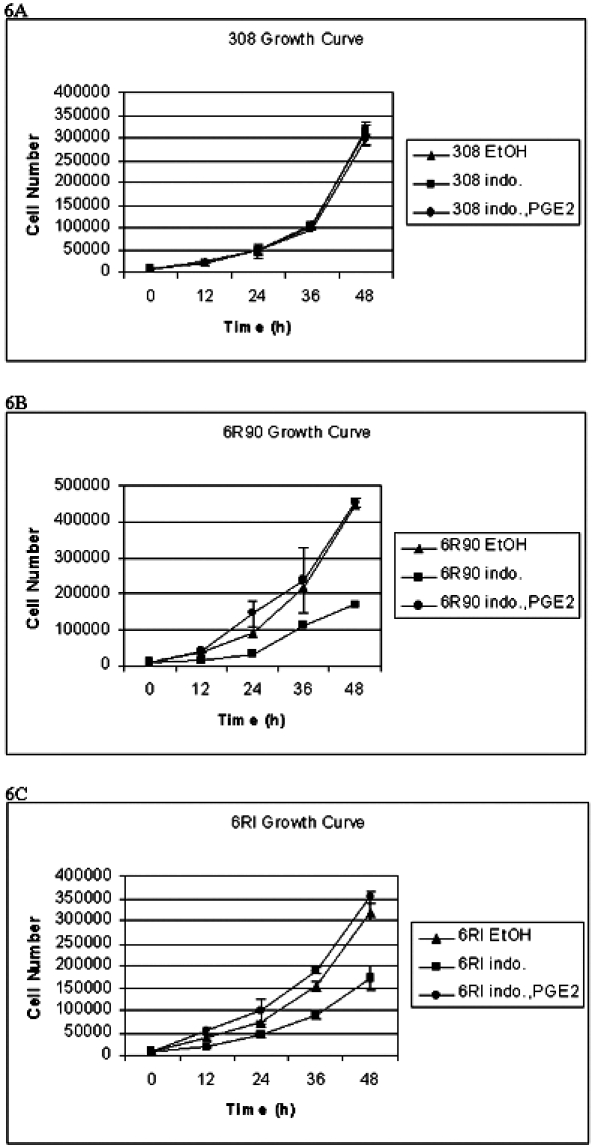
A total of 104 cells were plated and counted every 12 hours for 48 hours. Cells were treated with vehicle control (ethanol, EtOH) or 10 µM indomethacin (indo.) or 10 µM indomethacin and PGE2 (indo., PGE2). The bars indicate the standard deviation from three experiments. (A) Growth curve of 308 cells. (B) Growth curve of 6R90 cells. (C) Growth curve of 6RI cells.
A similar experiment was then performed to determine if the PGE2-induced growth of these cells depends on activation of a specific receptor. Rescue from indomethacin-induced growth inhibition was performed with 17-phenyl trinor PGE2, an EP1 and EP3 selective agonist with concentrations as derived for PGE2. Figure 7 shows that 17-phenyl trinor PGE2 can rescue the cells from indomethacin-induced growth inhibition. Because the cells do not express the EP3 prostanoid receptor, the growth signaling is through the EP1 prostanoid receptor.
Figure 7.
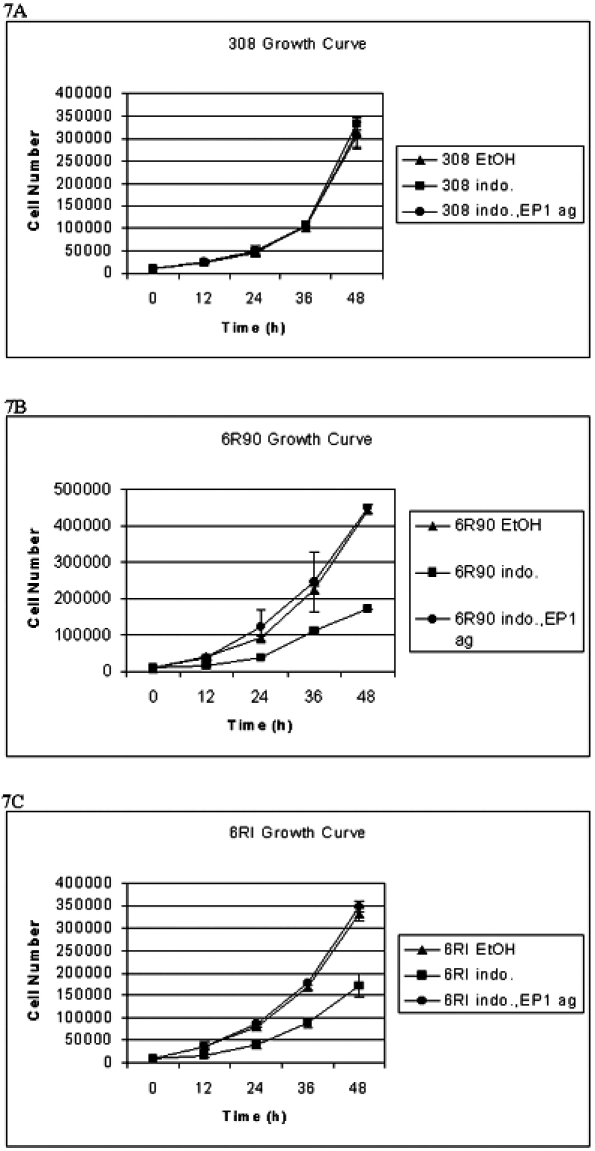
A total of 104 cells were plated and counted every 12 hours for 48 hours. Cells were treated with vehicle control (ethanol, EtOH) or 10 µM indomethacin (indo.) or 10 µM indomethacin and 17-phenyl trinor PGE2 (an EP1 and EP3 agonist) (indo., EP1 ag). The bars indicate the standard deviation from three experiments. (A) Growth curve of 308 cells. (B) Growth curve of 6R90 cells. (C) Growth curve of 6RI cells.
Discussion
Our results indicate that the model of skin tumor progression examined has all of the components necessary to comprise an autocrine growth-signaling loop. The cells exhibit increased expression of the enzymes necessary to produce PGE2 including COX-1 and -2. The activity of these enzymes was deduced by measurements of PGE2 production. The cells also express the EP1 and EP4 prostanoid receptors for this eicosanoid. Thus, there could be autocrine or paracrine PG signaling in these cell lines. Additionally, these cells exhibit an increased growth rate when ligand binding activates the EP1 prostanoid receptor.
Elevation of COX expression and activity has been observed in squamous cell carcinomas [19,20]. This is also a common finding in colon cancer [50]. The fact that nonsteroidal anti-inflammatory drugs can decrease colon tumor incidence [4] indicates that some products of these enzymes influence tumor growth. Recent studies by Fischer et al. [37] also indicate that inhibition of COX-2 can decrease the rate of skin tumor promotion. This influence could be through downstream products of further metabolism of PGH2, or it could be due to the reactive oxygen species produced by the activity of the COX enzymes [51]. In the work reported here, we present evidence that the growth of the cell lines studied depends on PGE2 production; and that the PGE2 can engage the EP1 prostanoid receptor to exert its effects on growth.
PGE2 production can be regulated at several levels. It is interesting to note that there are reports suggesting that the various enzymes involved in production of PGE2 are coregulated. PLA2 activation by phosphorylation has been shown to occur concurrently with COX-2 production [52]. It has also been shown that dexamethasone can simultaneously downregulate COX-2 and PGE synthase [53]. Thus, upregulation of one part of the PGE2 biosynthesis pathway may occur in the presence of some sort of upregulation of one or more of the other members of the pathway. When an increase in COX-2 and PGE synthase activity occurs, it has been shown to lead to an increased growth rate [53].
PGE2 in the tumor could come from several sources, including the tumor cells themselves, cells surrounding the tumor or infiltrating immune cells. Because PGE2 is unstable, it is unlikely that PGE2 produced at distant sites would affect the tumor cells. Squamous cell carcinomas often form at sites of chronic inflammation [51], thus the production of PGE2 by immune cells could stimulate growth of cells that still have normal regulation of COX enzymes. In this context, PGE2 could act as a tumor promoter by engaging the EP1 receptor on cells that have been initiated. It is of interest that addition of PGE2 to growth medium can increase the growth rate of human keratinocytes [42].
Although the EP4 prostanoid receptor does not appear to play a role in growth regulation of the cells, it could be involved in other aspects of the malignant phenotype. The EP4 prostanoid receptor has been shown to localize to the nuclear membrane [54] where its activation could influence transcription factors. In rat large granular lymphocytes, a model of natural killer cells, activation of the EP4 prostanoid receptor has been shown to be responsible for increased production of matrix metalloproteinases [55]. PGE2 can also influence head and neck squamous cell carcinoma migration and adherence to fibronectin. The authors of this study also show that this adherence is dependent on protein kinase A (PKA) activity [44], although they did not demonstrate specific involvement of either the EP2 or the EP4 receptor. Although the EP4 prostanoid receptor is not responsible for the growth of the cells, it could influence transcription factor activity and possibly invasion.
The EP1 prostanoid receptor is coupled to the Gqα-type G-protein. This protein acts by activation of PLC-β. The products of activation of PLC-β are DAG and IP3 [46]. IP3 causes the release of calcium from intracellular stores [46]. This release of calcium can influence signal transduction at several levels. It has recently been demonstrated that calcium can activate the Ras pathway through proline-rich tyrosine kinase 2 [46]. Together with DAG, calcium can also activate PKC [46]. Both of these pathways can then activate the mitogen-activated protein kinase pathways (MAPK) [46]. Many MAPKs have transcription factors as substrates [46], and thus activation of MAPKs can change the transcription of genes, including those involved in cell division [46]. Thus, EP1 prostanoid receptors may modulate cell growth by activation of the Ras and PKC pathways.
Some of the cellular effects of activating the EP1 prostanoid receptor may be due to the induced expression of proto-oncogenes. In a recent report, it was shown that activation of the EP1 prostanoid receptor could induce expression of the proto-oncogenes, c-jun and c-fos, thus activating the AP-1 transcription factor [56]. It is reasonable to think that this induction could lead to activation of genes controlled by a TPA responsive element. Of interest, Young et al. [57] recently demonstrated that AP-1 activation is required for TPA-induced skin tumor promotion. Therefore, antagonists of the EP1 prostanoid receptor could, in principle, block activation of the AP-1 transcription factor complex.
The EP1 prostanoid receptor has recently been implicated in colon carcinogenesis. Using a pharmacologic antagonist, Watanabe et al. [58] have shown that this antagonist can inhibit the formation of aberrant crypt foci after azoxymethane treatment of mice. The antagonist was given in various amounts in the diet. This resulted in a dose-dependent inhibition of aberrant crypt foci formation up to 36% of control. Additional evidence of a role for EP1 prostanoid receptors in colon cancer comes from experiments in which Min mice (mice with a nonsense mutation in the adenomatous polyposis coli gene) were fed a diet that included an EP1 antagonist. In these studies, the appearance of aberrant crypt foci was decreased by 43% [59]. The same group used EP1 knockout mice to examine azoxymethane-induced aberrant crypt foci. They found that the EP1 knockout mice demonstrated 40% fewer aberrant crypt foci than the wild-type controls, in agreement with the antagonist studies [59]. Because aberrant crypt foci are putative premalignant lesions, this gives rise to the interesting possibility that signaling through the EP1 prostanoid receptor is involved in cancer initiation or promotion in the colon.
There is strong evidence that production of PGs plays a role in tumor development. This work, and the work of others, indicates that PGE2 could play an important role in the growth of squamous cell carcinomas. This work further demonstrates that the EP1 prostanoid receptor is important for tumor cell growth in the model examined. Thus, the EP1 prostanoid receptor could become an important target for therapeutic intervention.
Abbreviations
- COX
cyclooxygenase
- PG
prostaglandin
- TPA
12-O-tetradecanoylphorbol-13-acetate
Footnotes
This work was supported in part by National Cancer Institute grants RO1 CA 40584 (to G.T.B.) and P30 CA 23074, and National Institute of Environmental Health Sciences grants T32 ES07091 and P30 ES 06694 and an Arizona Disease Control Research Commission Grant (to J.W.R.).
References
- 1.Morham SG, Langenbach R, Loftin CD, Tiano HF, Vouloumanos N, Jennette JC, Mahler JF, Kluckman KD, Ledford A, Lee CA. Prostaglandin synthase 2 gene disruption causes severe renal pathology in the mouse. Cell. 1995;83:473–482. doi: 10.1016/0092-8674(95)90125-6. [DOI] [PubMed] [Google Scholar]
- 2.Langenbach R, Morham SG, Tiano HF, Loftin CD, Ghanayem BI, Chulada PC, Mahler JF, Lee CA, Goulding EH, Kluckman KD. Prostaglandin synthase 1 gene disruption in mice reduces arachidonic acid-induced inflammation and indomethacin-induced gastric ulceration. Cell. 1995;83:483–492. doi: 10.1016/0092-8674(95)90126-4. [DOI] [PubMed] [Google Scholar]
- 3.Tsujii M, DuBois RN. Alterations in cellular adhesion and apoptosis in epithelial cells overexpressing prostaglandin endoperoxide synthase 2. Cell. 1995;83:493–501. doi: 10.1016/0092-8674(95)90127-2. [DOI] [PubMed] [Google Scholar]
- 4.Marnett LJ. Aspirin and the potential role of prostaglandins in colon cancer. Cancer Res. 1992;52:5575–5589. [PubMed] [Google Scholar]
- 5.Smith WL, Dewitt DL. Prostaglandin endoperoxide H synthases-1 and -2. Adv Immunol. 1996;62:167–215. doi: 10.1016/s0065-2776(08)60430-7. [DOI] [PubMed] [Google Scholar]
- 6.Zahner G, Harendza S, Muller E, Wolf G, Thaiss F, Stahl RA. Prostaglandin E2 stimulates expression of matrix metalloproteinase 2 in cultured rat mesangial cells. Kidney Int. 1997;51:1116–1123. doi: 10.1038/ki.1997.154. [DOI] [PubMed] [Google Scholar]
- 7.Zeng L, An S, Goetzl EJ. Regulation of expression of matrix metalloproteinase-9 in early human T cells of the HSB.2 cultured line by the EP3 subtype of prostaglandin E2 receptor. J Biol Chem. 1996;271:27744–27750. doi: 10.1074/jbc.271.44.27744. [DOI] [PubMed] [Google Scholar]
- 8.Mehindate K, al-Daccak R, Aoudjit F, Damdoumi F, Fortier M, Borgeat P, Mourad W. Interleukin-4, transforming growth factor beta 1, and dexamethasone inhibit superantigen-induced prostaglandin E2- dependent collagenase gene expression through their action on cyclooxygenase -2 and cytosolic phospholipase A2. Lab Invest. 1996;75:529–538. [PubMed] [Google Scholar]
- 9.Lindsey JD, Kashiwagi K, Boyle D, Kashiwagi F, Firestein GS, Weinreb RN. Prostaglandins increase proMMP-1 and proMMP-3 secretion by human ciliary smooth muscle cells. Curr Eye Res. 1996;15:869–875. doi: 10.3109/02713689609017628. [DOI] [PubMed] [Google Scholar]
- 10.Singhal PC, Sagar S, Garg P, Bansal V. Vasoactive agents modulate matrix metalloproteinase -2 activity by mesangial cells. Am J Med Sci. 1995;310:235–241. [PubMed] [Google Scholar]
- 11.Ushikubi F, Sugimoto Y, Ichikawa A, Narumiya S. Roles of prostanoids revealed from studies using mice lacking specific prostanoid receptors [In Process Citation] Jpn J Pharmacol. 2000;83:279–285. doi: 10.1254/jjp.83.279. [DOI] [PubMed] [Google Scholar]
- 12.Li X, Okada Y, Pilbeam CC, Lorenzo JA, Kennedy CR, Breyer RM, Raisz LG. Knockout of the murine prostaglandin EP2 receptor impairs osteoclastogenesis in vitro. Endocrinology. 2000;141:2054–2061. doi: 10.1210/endo.141.6.7518. [DOI] [PubMed] [Google Scholar]
- 13.Miyaura C, Inada M, Suzawa T, Sugimoto Y, Ushikubi F, Ichikawa A, Narumiya S, Suda T. Impaired bone resorption to prostaglandin E2 in prostaglandin E receptor EP4-knockout mice. J Biol Chem. 2000;275:19819–19823. doi: 10.1074/jbc.M002079200. [DOI] [PubMed] [Google Scholar]
- 14.Sakuma Y, Tanaka K, Suda M, Yasoda A, Natsui K, Tanaka I, Ushikubi F, Narumiya S, Segi E, Sugimoto Y, Ichikawa A, Nakao K. Crucial involvement of the EP4 subtype of prostaglandin E receptor in osteoclast formation by proinflammatory cytokines and lipopolysaccharide. J Bone Miner Res. 2000;15:218–227. doi: 10.1359/jbmr.2000.15.2.218. [DOI] [PubMed] [Google Scholar]
- 15.Sakuma Y, Tanaka K, Suda M, Komatsu Y, Yasoda A, Miura M, Ozasa A, Narumiya S, Sugimoto Y, Ichikawa A, Ushikubi F, Nakao K. Impaired bone resorption by lipopolysaccharide in vivo in mice deficient in the prostaglandin E receptor EP4 subtype. Infect Immun. 2000;68:6819–6825. doi: 10.1128/iai.68.12.6819-6825.2000. [In Process Citation] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Suzawa T, Miyaura C, Inada M, Maruyama T, Sugimoto Y, Ushikubi F, Ichikawa A, Narumiya S, Suda T. The role of prostaglandin E receptor subtypes (EP1, EP2, EP3, and EP4) in bone resorption: an analysis using specific agonists for the respective EPs. Endocrinology. 2000;141:1554–1559. doi: 10.1210/endo.141.4.7405. [DOI] [PubMed] [Google Scholar]
- 17.Yokoyama C, Tanabe T. Cloning of human gene encoding prostaglandin endoperoxide synthase and primary structure of the enzyme. Biochem Biophys Res Commun. 1989;165:888–894. doi: 10.1016/s0006-291x(89)80049-x. [DOI] [PubMed] [Google Scholar]
- 18.Fletcher BS, Kujubu DA, Perrin DM, Herschman HR. Structure of the mitogen-inducible TIS10 gene and demonstration that the TIS10-encoded protein is a functional prostaglandin G/H synthase. J Biol Chem. 1992;267:4338–4344. [PubMed] [Google Scholar]
- 19.Buckman SY, Gresham A, Hale P, Hruza G, Anast J, Masferrer J, Pentland AP. COX-2 expression is induced by UVB exposure in human skin: implications for the development of skin cancer. Carcinogenesis. 1998;19:723–729. doi: 10.1093/carcin/19.5.723. [DOI] [PubMed] [Google Scholar]
- 20.Fosslien E. Molecular pathology of cyclooxygenase-2 in neoplasia. Ann Clin Lab Sci. 2000;30:3–21. [PubMed] [Google Scholar]
- 21.Wilgus TA, Ross MS, Parrett ML, Oberyszyn TM. Topical application of a selective cyclooxygenase inhibitor suppresses UVB mediated cutaneous inflammation. Prostaglandins Other Lipid Mediators. 2000;62:367–384. doi: 10.1016/s0090-6980(00)00089-7. [in process citation] [DOI] [PubMed] [Google Scholar]
- 22.Williams CS, Tsujii M, Reese J, Dey SK, DuBois RN. Host cyclooxygenase-2 modulates carcinoma growth. J Clin Invest. 2000;105:1589–1594. doi: 10.1172/JCI9621. [see comments] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rioux N, Castonguay A. The induction of cyclooxygenase-1 by a tobacco carcinogen in U937 human macrophages is correlated to the activation of NF-kappaB. Carcinogenesis. 2000;21:1745–1751. doi: 10.1093/carcin/21.9.1745. [DOI] [PubMed] [Google Scholar]
- 24.Maldve RE, Kim Y, Muga SJ, Fischer SM. Prostaglandin E(2) regulation of cyclooxygenase expression in keratinocytes is mediated via cyclic nucleotide-linked prostaglandin receptors. J Lipid Res. 2000;41:873–881. [PubMed] [Google Scholar]
- 25.Nugteren DH, Christ-Hazelhof E. Chemical and enzymic conversions of the prostaglandin endoperoxide PGH2. Adv Prostaglandin Thromboxane Res. 1980;6:129–137. [PubMed] [Google Scholar]
- 26.Yamamoto S, Ohki S, Ogino N, Shimizu T, Yoshimoto T, Watanabe K, Hayaishi O. Enzymes involved in the formation and further transformations of prostaglandin endoperoxides. Adv Prostaglandin Thromboxane Res. 1980;6:27–34. [PubMed] [Google Scholar]
- 27.Moonen P, Buytenhek M, Nugteren DH. Purification of PGH-PGE isomerase from sheep vesicular glands. Methods Enzymol. 1982;86:84–91. doi: 10.1016/0076-6879(82)86173-9. [DOI] [PubMed] [Google Scholar]
- 28.Ruzicka T, Walter JF, Printz MP. Changes in arachidonic acid metabolism in UV-irradiated hairless mouse skin. J Invest Dermatol. 1983;81:300–303. doi: 10.1111/1523-1747.ep12519287. [DOI] [PubMed] [Google Scholar]
- 29.Karmali RA, Safai B. Ultraviolet-evoked prostaglandin biosynthesis in varying stages of keratinocyte differentiation in guinea pig skin. Prostaglandins, Leukotrienes Med. 1984;15:277–286. doi: 10.1016/0262-1746(84)90127-6. [DOI] [PubMed] [Google Scholar]
- 30.Brune K, Kalin H, Schmidt R, Hecker E. Inflammatory, tumor initiating and promoting activities of polycyclic aromatic hydrocarbons and diterpene esters in mouse skin as compared with their prostaglandin releasing potency in vitro. Cancer Lett. 1978;4:333–342. doi: 10.1016/s0304-3835(78)95612-4. [DOI] [PubMed] [Google Scholar]
- 31.Fischer SM, Furstenberger G, Marks F, Slaga TJ. Events associated with mouse skin tumor promotion with respect to arachidonic acid metabolism: a comparison between SENCAR and NMRI mice. Cancer Res. 1987;47:3174–3179. [PubMed] [Google Scholar]
- 32.Hamasaki Y, Eling TE. EGF and TPA stimulate de novo synthesis of PGHS-1 and PGHS-2 through different signal transduction pathways. Prostaglandins, Leukotrienes Essent Fatty Acids. 1995;53:225–229. doi: 10.1016/0952-3278(95)90121-3. [DOI] [PubMed] [Google Scholar]
- 33.Marks F, Furstenberger G, Kownatzki E. Prostaglandin E-mediated mitogenic stimulation of mouse epidermis in vivo by divalent cation ionophore A 23187 and by tumor promoter 12-O-tetradecanoylphorbol-13-acetate. Cancer Res. 1981;41:696–702. [PubMed] [Google Scholar]
- 34.Miller CC, Hale P. Pentland AP, Ultraviolet B injury increases prostaglandin synthesis through a tyrosine kinase-dependent pathway. Evidence for UVB-induced epidermal growth factor receptor activation. J Biol Chem. 1994;269:3529–3533. [PubMed] [Google Scholar]
- 35.Chen X, Gresham A, Morrison A, Pentland AP. Oxidative stress mediates synthesis of cytosolic phospholipase A2 after UVB injury. Biochim Biophys Acta. 1996;1299:23–33. doi: 10.1016/0005-2760(95)00166-2. [DOI] [PubMed] [Google Scholar]
- 36.Zakar T, Teixeira FJ, Hirst JJ, Guo F, MacLeod EA, Olson DM. Regulation of prostaglandin endoperoxide H synthase by glucocorticoids and activators of protein kinase C in the human amnion. J Reprod Fertil. 1994;100:43–50. doi: 10.1530/jrf.0.1000043. [DOI] [PubMed] [Google Scholar]
- 37.Fischer SM, Lo HH, Gordon GB, Seibert K, Kelloff G, Lubet RA, Conti CJ. Chemopreventive activity of Celecoxib, a specific cyclooxygenase-2 inhibitor, and indomethacin against ultraviolet light-induced skin carcinogenesis. Mol Carcinog. 1999;25:231–240. [PubMed] [Google Scholar]
- 38.Müller-Decker K, Scholz K, Marks F, Furstenberger G. Differential expression of prostaglandin H synthase isozymes during multistage carcinogenesis in mouse epidermis. Mol Carcinog. 1995;12:31–41. doi: 10.1002/mc.2940120106. [DOI] [PubMed] [Google Scholar]
- 39.Higashi Y, Kanekura T, Kanzaki T. Enhanced expression of cyclooxygenase (COX)-2 in human skin epidermal cancer cells: evidence for growth suppression by inhibiting COX- 2 expression. Int J Cancer. 2000;86:667–671. doi: 10.1002/(sici)1097-0215(20000601)86:5<667::aid-ijc10>3.0.co;2-y. [DOI] [PubMed] [Google Scholar]
- 40.Aizu E, Yamamoto S, Nakadate T, Kato R. Differential effects of various skin tumor-promoting agents on prostaglandin E2 release from primary cultures of mouse epidermal cells. Eur J Pharmacol. 1990;182:19–28. doi: 10.1016/0014-2999(90)90489-s. [DOI] [PubMed] [Google Scholar]
- 41.Johnson GK, Poore TK, Payne JB, Organ CC. Effect of smokeless tobacco extract on human gingival keratinocyte levels of prostaglandin E2 and interleukin-1. J Periodontol. 1996;67:116–124. doi: 10.1902/jop.1996.67.2.116. [DOI] [PubMed] [Google Scholar]
- 42.Pentland AP, Needleman P. Modulation of keratinocyte proliferation in vitro by endogenous prostaglandin synthesis. J Clin Invest. 1986;77:246–251. doi: 10.1172/JCI112283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nusing RM, Klein T, Pfeilschifter J, Ullrich V. Effect of cyclic AMP and prostaglandin E2 on the induction of nitric oxide- and prostanoid-forming pathways in cultured rat mesangial cells. Biochem J. 1996;313:617–623. doi: 10.1042/bj3130617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lozano Y, Taitz A, Petruzzelli GJ, Djordjevic A, Young MR. Prostaglandin E2-protein kinase A signaling and protein phosphatases-1 and -2A regulate human head and neck squamous cell carcinoma motility, adherence, and cytoskeletal organization. Prostaglandins. 1996;51:35–48. doi: 10.1016/0090-6980(95)00155-7. [DOI] [PubMed] [Google Scholar]
- 45.Ichikawa A, Sugimoto Y, Negishi M. Molecular aspects of the structures and functions of the prostaglandin E receptors. J Lipid Mediators Cell Signalling. 1996;14:83–87. doi: 10.1016/0929-7855(96)00512-3. [DOI] [PubMed] [Google Scholar]
- 46.Gawler DJ. Points of convergence between Ca2+ and Ras signalling pathways. Biochim Biophys Acta. 1998;1448:171–182. doi: 10.1016/s0167-4889(98)00141-4. [DOI] [PubMed] [Google Scholar]
- 47.Vanderveen EE, Grekin RC, Swanson NA, Kragballe K. Arachidonic acid metabolites in cutaneous carcinomas. Evidence suggesting that elevated levels of prostaglandins in basal cell carcinomas are associated with an aggressive growth pattern. Arch Dermatol. 1986;122:407–412. doi: 10.1001/archderm.122.4.407. [DOI] [PubMed] [Google Scholar]
- 48.Gupta A, Rosenberger SF, Bowden GT. Increased ROS levels contribute to elevated transcription factor and MAP kinase activities in malignantly progressed mouse keratinocyte cell lines. Carcinogenesis. 1999;20:2063–2073. doi: 10.1093/carcin/20.11.2063. [DOI] [PubMed] [Google Scholar]
- 49.Anthony TL, Pierce KL, Regan JW. Characterization of EP prostanoid receptor subtypes in primary cultures of bovine ciliary epithelial cells by immunofluorescent microscopy and functional studies. Curr Eye Res. 2000;20:394–404. [PubMed] [Google Scholar]
- 50.Sano H, Kawahito Y, Wilder RL, Hashiramoto A, Mukai S, Asai K, Kimura S, Kato H, Kondo M, Hla T. Expression of cyclooxygenase-1 and -2 in human colorectal cancer. Cancer Res. 1995;55:3785–3789. [PubMed] [Google Scholar]
- 51.Perchellet JP, Perchellet EM, Gali HU, Gao XM. Oxidant Stress and Multistage Skin Carcinogenesis. In: Mukhtar H, editor. Skin Cancer: Mechanisms and Human Relevance. Boca Raton, FL: CRC Press; 1995. pp. 149–155. [Google Scholar]
- 52.Croxtal JD, Newman SP, Choudhury Q, Flower RJ. The concerted regulation of cPLA2, COX2, and lipocortin 1 expression by IL-1 beta in A549 cells. Biochem Biophys Res Commun. 1996;220:491–495. doi: 10.1006/bbrc.1996.0432. [DOI] [PubMed] [Google Scholar]
- 53.Murakami M, Naraba H, Tanioka T, Semmyo N, Nakatani Y, Kojima F, Ikeda T, Fueki M, Ueno A, Oh-Ishi S, Kudo I. Regulation of prostaglandin E2 biosynthesis by inducible membrane-associated prostaglandin E2 synthase that acts in concert with cyclooxygenase-2 [in process citation] J Biol Chem. 2000;275:32783–32792. doi: 10.1074/jbc.M003505200. [DOI] [PubMed] [Google Scholar]
- 54.Bhattacharya M, Peri K, Ribeiro-da-Silva A, Almazan G, Shichi H, Hou X, Varma DR, Chemtob S. Localization of functional prostaglandin E2 receptors EP3 and EP4 in the nuclear envelope. J Biol Chem. 1999;274:15719–15724. doi: 10.1074/jbc.274.22.15719. [DOI] [PubMed] [Google Scholar]
- 55.Zeng L, An S, Goetzl EJ. Selective regulation of RNK-16 cell matrix metalloproteinases by the EP4 subtype of prostaglandin E2 receptor. Biochemistry. 1996;35:7159–7164. doi: 10.1021/bi960036x. [DOI] [PubMed] [Google Scholar]
- 56.Suda M, Tanaka K, Sakuma Y, Yasoda A, Ozasa A, Fukata J, Tanaka I, Narumiya S, Nakao K. Prostaglandin E(2) (PGE(2)) induces the c-fos and c-jun expressions via the EP(1) subtype of PGE receptor in mouse osteoblastic MC3T3-E1 cells. Calcif Tissue Int. 2000;66:217–223. doi: 10.1007/s002230010043. [DOI] [PubMed] [Google Scholar]
- 57.Young MR, Li JJ, Rincon M, Flavell RA, Sathyanarayana BK, Hunziker R, Colburn N. Transgenic mice demonstrate AP-1 (activator protein-1) transactivation is required for tumor promotion. Proc Natl Acad Sci USA. 1999;96:9827–9832. doi: 10.1073/pnas.96.17.9827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Watanabe K, Kawamori T, Nakatsugi S, Ohta T, Ohuchida S, Yamamoto H, Maruyama T, Kondo K, Narumiya S, Sugimura T, Wakabayashi K. Inhibitory effect of a prostaglandin E receptor subtype EP(1) selective antagonist, ONO-8713, on development of azoxymethane-induced aberrant crypt foci in mice. Cancer Lett. 2000;156:57–61. doi: 10.1016/s0304-3835(00)00440-7. [DOI] [PubMed] [Google Scholar]
- 59.Watanabe K, Kawamori T, Nakatsugi S, Ohta T, Ohuchida S, Yamamoto H, Maruyama T, Kondo K, Ushikubi F, Narumiya S, Sugimura T, Wakabayashi K. Role of the prostaglandin E receptor subtype EP1 in colon carcinogenesis. Cancer Res. 1999;59:5093–5096. [PubMed] [Google Scholar]