

Abstract
Bcl-2 is a critical suppressor of apoptosis that is overproduced in many types of cancer. Phosphorylation of the Bcl-2 protein is induced on serine residues in tumor cells arrested by microtubule-targeting drugs (paclitaxel, vincristine, nocodazole) and has been associated with inactivation of antiapoptotic function through an unknown mechanism. Comparison of a variety of pharmacological inhibitors of serine/threonine-specific protein kinases demonstrated that the cyclin-dependent kinase inhibitor, flavopiridol, selectively blocks Bcl-2 phosphorylation induced by antimicrotubule drugs. Bcl-2 could also be coimmunoprecipitated with the kinase Cdc2 in M-phase-arrested cells, suggesting that Cdc2 may be responsible for phosphorylation of Bcl-2 in cells treated with microtubule-targeting drugs. Examination of several serine! alanine substitution mutants of Bcl-2 suggested that serine 70 and serine 87 represent major sites of Bcl-2 phosphorylation induced in response to microtubuletargeting drugs. Both these serines are within sequence contexts suitable for proline-directed kinases such as Cdc2. Phosphorylated Bcl-2 protein was discovered to associate in M-phase-arrested cells with Pin1, a mitotic peptidyl prolyl isomerase (PPIase) known to interact with substrates of Cdc2 during mitosis. In contrast, phosphorylation of Bcl-2 induced by microtubuletargeting drugs did not alter its ability to associate with Bcl-2 (homodimerization), Bax, BAG1, or other Bcl-2-binding proteins. Since the region in Bcl-2 containing serine 70 and serine 87 represents a proline-rich loop that has been associated with autorepression of its antiapoptotic activity, the discovery of Pin1 interactions with phosphorylated Bcl-2 raises the possibility that Pin1 alters the conformation of Bcl-2 and thereby modulates its function in cells arrested with antimicrotubule drugs.
Keywords: Bcl-2, mitosis, phosphorylation, Cdc2, Pin1
Introduction
Bcl-2 is a central regulator of apoptosis that is overexpressed in many types of cancer (reviewed in Ref. [1]). High levels of Bcl-2 protein are associated with resistance of tumor cells to apoptosis induction by multiple anticancer drugs and X-irradiation [2]. Thus, great interest has emerged in understanding the molecular mechanisms by which Bcl-2 suppresses apoptosis and devising strategies for combating Bcl-2 in cancer.
The ∼26-kDa Bcl-2 protein contains a membrane-anchoring domain near its carboxyl terminus that causes its insertion into intracellular membranes of mitochondria and other organelles [3–5]. Though a three-dimensional structure of Bcl-2 is not yet available, comparisons with its close homologue Bcl-XL imply that the nonmembranous portion of Bcl-2 is likely comprised of a seven α-helical bundle, where the first and second α-helices are separated by a long proline-rich (presumably flexible) loop of ∼60 amino acids [6]. The “loop region” (LR) of Bcl-2 and Bcl-XL has been reported to negatively regulate their functions, inasmuch as deletion of the LR increases the apparent antiapoptotic function of these proteins [7,8]. LR-deficient mutants of Bcl-2 have been shown to protect cells under conditions were the wild-type (full-length) protein is inadequate, including after exposure of cells to the microtubule -targeting drug paclitaxel [7,9–12].
How the LR domain suppresses the apoptosis-blocking function of Bcl-2 is unclear, but this region (residues ∼30–90) has been reported to undergo phosphorylation on serine or threonine residues. For example, drugs affecting microtubule polymerization such as paclitaxel and taxotere have been shown to induce phosphorylation of Bcl-2 on serine 70, serine 87, and possibly (to a small extent) on threonine 69 [11–14]. Similarly, deletion of the LR (residues 32–80) from Bcl-2 prevents paclitaxel-induced incorporation of 32P-orthophosphate and associated shifts in Bcl-2 protein mobility in SDS-PAGE experiments [7,10,12]. In cells overexpressing active Rac1 and p54-SAPKβ, Bcl-2 undergoes phosphorylation on threonine 56, serine 70, threonine 74, and serine 87 [15]. Also, PKCα reportedly induces phosphorylation of Bcl-2 on serine 70 in lymphoid and hematopoietic cells, and appears to be responsible for Bcl-2 phosphorylation induced by interleukin-3 and bryostatin in lymphoid and hematopoietic cells [16,17].
Though phosphorylation site mapping has not been uniformly performed, inducible phosphorylation of the Bcl-2 protein has been described following exposure of many types of malignant cell lines in vitro to microtubule-targeting drugs, including those that depolymerize (vincristine; vinblastine; nocodazole; colchicine; colcemid; 3-iodoacetamido-benzyolurea; dolastatin-15) and those that aggregate microtubules (paclitaxel; taxotere; 2-methoxy-estradiol) [10,13,14,18–26]. This correlation has implied that phosphorylation of Bcl-2 inactivates this protein, and permits apoptosis. Indeed, mutant Bcl-2 proteins in which serine 70 or serine 87 are replaced with alanines display enhanced suppression of apoptosis in response to paclitaxel [11,12]. Interestingly, several reports have provided evidence that phosphorylation of Bcl-2 is normally induced during transit through M-phase, suggesting that the effects of microtubule-targeting drugs seen in cycling tumor cells are merely a reflection of their ability to induce mitotic arrest [11,23,24]. The concept thus has emerged that phosphorylation-induced inactivation of Bcl-2 during mitosis may define a checkpoint that permits apoptosis if aberrant chromosome segregation or defective cytokinesis occurs.
A variety of protein kinases have been claimed to mediate the phosphorylation of Bcl-2 during mitotic arrest, including Raf1, PKA, Cdc2, and JNK [11,12,21,27–29]. In this report, we further explore the mechanisms surrounding the phosphorylation of Bcl-2 in cells arrested in mitosis by microtubule-targeting drugs, providing additional evidence implicating Cdc2 and demonstrating for the first time an inducible interaction with Pin1, a PPIase that binds Cdc2 substrates in a phosphorylation-dependent manner [30,31].
Materials and Methods
Antibodies
Antipeptide rabbit antisera and monoclonal antibodies (4D7 or 6C8) specific for Bcl-2 have been described previously [32,33], and were obtained from PharMingen (San Diego, CA). Antipeptide antiserum recognizing Bax has been described [34] (PharMingen). Antibodies specific for the unique C-terminal region of Cdc2 were obtained from Upstate Biotechnology. Polyclonal anti-Pin1 antibodies have been described [30].
Cell Lines, Cultures, Transfections, and Treatments
Stably transfected Bcl-2-expressing 697, Jurkat, 32D, and HEK293 cells have been described previously [35–38]. HEK293T cells were obtained from ATCC (American Type Culture Collection, Rockville, MD). RS11846 cells were a kind gift of C. Croce (Philadelphia, PA) [39]. Cells were cultured at 37°C in 5%CO2:95% air in either RPMI1640 or Dulbecco's modified Eagles medium (DMEM) with 10% heat-inactivated fetal bovine serum (FBS), 1 mM l-glutamine, and antibiotics, then treated while in log-phase growth with various concentrations of various anticancer drugs, including paclitaxel (Bristol-Myers-Squibb), vincristine, vinblastine, nocodazole, daunomycin, adriamycin, and chlorambucil (Sigma, St. Louis, MO). In some cases, protein kinase inhibitors were added to the cultures 0.5 hour before addition of anticancer drugs, including flavopiridol (Aventis), PD098059 (New England Biolabs, Beverly, MA), SB203580 (Calbiochem-Novobiochem, San Diego, CA), or GF109203X (bisindolylmaleimide I) (CalBiochem). In some experiments, cells were metabolically labeled with 32PO4 as described previously [40].
For transient transfection assays, 293T cells in log-phase were transfected with plasmids encoding wild-type or mutant Bcl-2 proteins [13,14,41], using Superfect transfection reagent (Qiagen).
Immunoblot and Immunoprecipitation Assays
Cells were lysed in ice-cold RIPA buffer (10 mM Tris pH 7.4, 142.5 mM NaCl, 1% Na deoxycholate, 0.1% SDS, 1% Triton X-100, 1 mM EDTA) or NP-40 lysis buffer (10 mM Tris pH 7.4, 142.5 mM KCl, 0.5% NP-40, 5 mM EGTA), containing protease inhibitors and phosphatase inhibitors (10 mM sodium β-glycerophosphate, 1 mM Na3VO4, 5 mM NaF, 2 mM Na4P2O7, 50 mM and 4-nitrophenyl phosphate). After normalization for either input cell number or total protein content, the resulting lysates were subjected to immunoprecipitation with rabbit antipeptide anti-Bcl-2 or anti-Bax antibodies using Protein A Sepharose (from Sigma) or with anti-Bcl-2 monoclonals using Protein G Sepharose (Zymed, San Francisco, CA). Alternatively, lysates were analyzed directly by immunoblotting. Proteins were separated in 12% SDS polyacrylamide gels, followed by transfer to either nitrocellulose (Biorad, Hercules, CA) or Immobilon-PPVDF membranes (Millipore, Bedford, MA). Immunodetection was accomplished by an enhanced chemiluminescence (ECL) method (Amersham/Pharmacia, Piscataway, NJ).
In Vitro Protein Interaction Assay
Binding of proteins in vitro to GST-Bcl-2, GST-Bax, or GST-BAG1 proteins immobilized on glutathione-Sepharose (Pharmacia) was assayed as described, using cell lysates normalized for total protein content and 1 µg GST-fusion protein per 20 µl beads in 0.1 ml of either RIPA or NP40 lysis buffer. Beads were washed extensively in lysis buffer [40,41], and analyzed by SDS-PAGE/immunoblotting as above.
Subcellular Fractionation Studies
Stably transfected 293-Bcl-2 cells were washed twice in ice-cold PBS and resuspended in 2 ml of hypotonic buffer (5 mM Tris pH 7.4, 5 mM KCl, 1.5 mM MgCl2, 0.1 mM EGTA, 1 mM DTT) containing 0.2 mM PMSF, 5 µg/ml leupeptin, 5 µg/ml aprotinin, 0.7 µg/ml pepstatin A, and incubated on ice for 30 minutes. After homogenization using 30 to 40 strokes of a Dounce homogenizer, samples were transferred to Eppendorf centrifuge tubes and centrifuged at 500xg for 5 minutes at 4°C to collect nuclei. The resulting supernatant was centrifuged at 10,000xg for 0.5 hour at 4°C to obtain the heavy membrane (HM) fraction (pellet), and this supernatant was then centrifuged for 1.5 hour at 150,000xg to obtain the light membrane (LM) (pellet) and cytosolic (supernatant) fractions [38,42]. The HM, LM and nuclear material were resuspended in 0.1 ml of Triton X-100 lysis buffer (10 mM Tris pH 7.4, 150 mM NaCl, 1% Triton X-100, 1 mM EDTA) containing protease and phosphatase inhibitors. Samples from equivalent numbers of cells were subjected to SDS-PAGE/immunoblot analysis.
Cell-Cycle and Apoptosis Assays
Cell-cycle analysis based on DNA content of cells as determined by flow-cytometry analysis of propidium-iodide-stained fixed cells was performed as described [37]. Apoptosis was measured either by TUNEL assay or by UV-microscopic examination of fixed cells stained with DAPI, as described [43].
Results
Antimicrotubule Drugs Selectively Induce Phosphorylation of Bcl-2
Previously it has been reported that Bcl-2 undergoes phosphorylation in cells during mitosis and in cells arrested in M-phase by antimicrotubule drugs. We confirmed an increase in the molecular weight of the Bcl-2 protein, as determined by immunoblot analysis of lysates prepared from a variety of neoplastic cell lines subjected to treatment with the microtubule-depolymerizing agents vincristine, vinblastine and nocodazole and the microtubule-aggregating drug, paclitaxel (Figure 1 and data not shown). The shift in Bcl-2 gel-mobility typically resulted in the appearance of two or three higher molecular weight bands in SDS-PAGE analysis (Figure 1), suggesting multiple sites of post-translational modification in the Bcl-2 protein. Metabolic labeling with 32PO4 demonstrated the incorporation of phosphate into all these higher molecular mass forms of Bcl-2 but not the ∼26-kDa unmodified Bcl-2 protein (not shown). Treatment of extracts with phosphatases (not shown) also reduced the amounts of these higher mobility Bcl-2 bands, further suggesting that they arise due to phosphorylation, as previously reported [13,14,18–26]. These Bcl-2 modifications induced by antimicrotubule drugs were observed in several cell lines, including RS11846 lymphoma cells, which contain high endogenous levels of Bcl-2 due to the presence of a t (14;18) translocation [39], as well as Jurkat T-cell leukemia, 697 pre-B-cell leukemia, 32D myeloid progenitor, and HEK293 epithelial cells transfected with plasmids encoding Bcl-2 (Figure 1 and not shown). Induction of Bcl-2 modification by antimicrotubule drugs such as paclitaxel was dose-dependent, and generally correlated with the percentage of cells arrested in G2/M-phase, as determined by DNA content analysis (Figure 1 and not shown). Modifications of Bcl-2 protein were not induced by a variety of other anticancer drugs, including chlorambucil (alkylating agent), daunomycin and adriamycin (anthracyclines), methotrexate (dihydrofolate reductase inhibitor), and fludarabine (purine nucleoside analogue) (Figure 1 and not shown). Modifications were also not induced in noncycling cells, such as chronic lymphocytic leukemia (CLL) cells freshly isolated from peripheral blood (unpublished observations).
Figure 1.
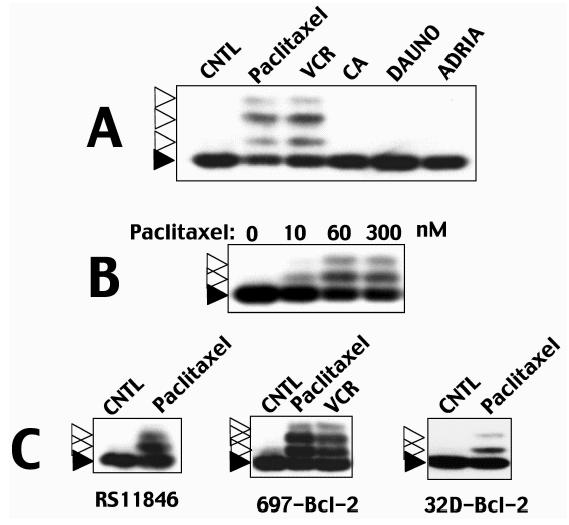
Antimicrotubule drugs induce Bcl-2 protein modifications. Examples of Bcl-2 protein modifications induced by antimicrotubule drugs are presented. Cells were treated with various anticancer drugs for ∼1 day before preparing cell lysates and analyzing Bcl-2 protein by SDS-PAGE/immunoblotting using anti-Bcl-2 antibody with ECL-based detection. All lysates were normalized for total protein content before analysis. Dark and open arrowheads indicate unmodified and modified Bcl-2, respectively. In (A), Jurkat-Bcl-2 cells [36] were cultured for ∼1 day with 300 nM paclitaxel, 1 µM vincristine (VCR), 10 ∼M chlorambucil (CA), 3.5 µM daunomycin (DAUNO), or 1 µM adriamycin (ADRIA). In (B), Jurkat-Bcl-2 cells were treated with ∼10, ∼60, or ∼300 µM paclitaxel. In (C), RS11846 lymphoma cells that contain high endogenous levels of Bcl-2 protein [39], Bcl-2 retrovirus-transduced 697 pre-B-cell leukemia [35], and human Bcl-2-transfected 32D murine myeloid progenitor cells (maintained in interleukin-3-containing medium) [37] were treated with 300 nM paclitaxel or 1 µM vincristine.
Selective Inhibition of Bcl-2 Phosphorylation by a Cyclin-Dependent Kinase Inhibitor
The kinase(s) responsible for phosphorylation of Bcl-2 in cells arrested with antimicrotubule drugs remain unclear, though several candidates have been suggested including Raf1, PKA, Cdk-family members, and JNK. We used pharmacological inhibitors of several serine/threoninekinases to preliminarily explore their relevance to the antimicrotubule drug-induced modification of Bcl-2, including (a) flavopiridol, an inhibitor of cyclin-dependent kinases, (b) PD098059, a MEK inhibitor, (c) SB203580, an inhibitor of p38 kinase, and (d) GF109203X (bisindolylmaleimide I), a protein kinase C inhibitor. All kinase inhibitors were employed at 1 µM, a concentration at which they exhibit relatively selective activity against target kinases in intact cells. Among these compounds, only flavopiridol prevented the modification of Bcl-2 induced by antimicrotubule drugs (Figure 2). All of these kinase inhibitors, however, induced cell cycle arrest (not shown), suggesting that the effect of flavopiridol are not necessarily a secondary result of growth suppression.
Figure 2.
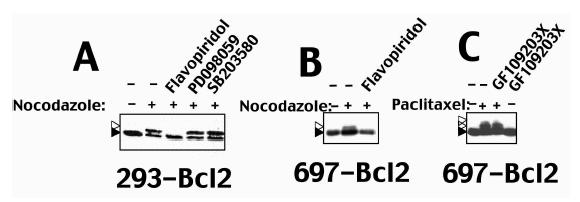
Cdk inhibitor flavopiridol selectively inhibits Bcl-2 modification in cells arrested by antimicrotubule drugs. Bcl-2-overexpressing cells were treated with antimicrotubule drugs alone or in combination with various kinase inhibitors (1 µM). After ∼1 day, cell lysates were prepared, normalized for either cell equivalents of total protein content, and subjected to SDS-PAGE/immunoblot analysis using anti-Bcl-2 antibodies with ECL-based detection. Dark and open arrowheads indicate unmodified and modified Bcl-2, respectively. In (A), detergent-solubilized lysates (2x106 cells/sample) were prepared from 293-Bcl-2 cells that were cultured without treatment (-), ∼2 µM nocodazole (+) or the combination of nocodazole and Cdk-inhibitor flavopiridol, MEK inhibitor PD098059, or p38 kinase inhibitor SB203580. In (B), 697-Bcl-2 cells were treated with nocodazole alone or in combination with flavopiridol as indicated. In (C), 697-Bcl-2 cells were cultured with 300 nM paclitaxel, 1 µM PKC-inhibitor GF109203X, or combinations of these reagents, followed by preparation of lysates (normalized for total protein content) and SDS-PAGE/immunoblot analysis of Bcl-2.
Bcl-2 Associates with the Kinase Cdc2 in M-Phase-Arrested Cells
Because the kinase Cdc2 is the major cyclin-dependent kinase active during mitosis (reviewed in Ref. [44]), we explored whether a physical interaction of Bcl-2 with Cdc2 could be detected by coimmunoprecipitation assays (Figure 3). In untreated cycling cells, no detectable association of Bcl-2 and Cdc2 was observed. However, when cells were arrested with antimicrotubule drug nocodazole, Bcl-2 could be recovered in association with anti-Cdc2 immunoprecipitates. Control immunoprecipitates lacked associated Bcl-2, confirming the specificity of these results. DNA content analysis indicated that the majority of cells in these cultures were arrested in G2/M-phase (not shown), suggesting that interaction of Bcl-2 with Cdc2 is cell cycle dependent. Interestingly, when the Cdc2 inhibitor flavopiridol was used, Bcl-2 association with Cdc2 was prevented in nocodazole-treated cells. Thus, association with Bcl-2 may require that Cdc2 is active. Though merely correlative, these findings demonstrating Bcl-2/Cdc2 complex formation provide evidence that Cdc2 may be directly responsible for phosphorylation of Bcl-2 in M-phase-arrested cells.
Figure 3.
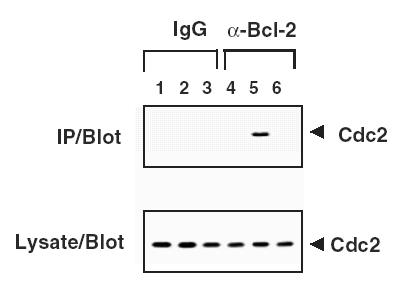
Bcl-2 associates with Cdc2 in cells arrested with antimicrotubule drugs. Detergent-solubilized lysates (2x106 cells/sample) were prepared from 293-Bcl-2 cells cultured without treatment (lanes 1 and 4), ∼2 µM nocodazole (lanes 2 and 5) or nocodazole and 1 µM flavopiridol (lanes 3 and 6). Samples were either analyzed directly by immunoblotting with anti-Cdc2 antibody (lower panel) or subjected to immunoprecipitation with anti-mIgG (lanes 1–3) or with anti-Bcl-2 antibodies (lanes 4–6) followed by SDS-PAGE/immunoblot analysis with anti-Cdc2 antibody (upper panel).
Structure-Function Analysis of Bcl-2 Modifications Induced by Antimicrotubule Drugs
Previous studies have documented that antimicrotubule drugs induce phosphorylation of Bcl-2 predominantly on serine residues [13,18]. To explore which residues in the Bcl-2 protein might undergo modification following treatment of cells with antimicrotubule drugs, all serine residues in the Bcl-2 protein were systemically mutated to nonphosphorylatable alanine. Plasmids encoding these Bcl-2 mutants were expressed by transient transfection in HEK293T cells, and then the cells were arrested with nocodazole and analyzed.
In HEK293T cells expressing wild-type Bcl-2, SDS-PAGE/immunoblot analysis revealed a single ∼26-kDa form of Bcl-2. When these cells were treated with nocodazole for 2 days, in contrast, two additional, slower-migrating bands were detected (Figure 4A). Alanine substitution of serine 70 abolished the nocodazole-induced appearance of both slower-migrating forms of Bcl-2. In comparison, alanine replacement of serine 87 prevented the induction of the slowest migrating band but not the appearance of the intermediate band. None of the other alanine replacement mutations, including Serines 24, 50, 51, 62, 105, 116, 117, 161, and 167, were associated with failure of nocodazole to induce Bcl-2 modifications (Figure 4). These data thus provide indirect evidence that serines 70 and 87 in the LR domain of Bcl-2 are required for Bcl-2 modifications induced by antimicrotubule drugs. Interestingly, both serine 70 and serine 87 reside proximal to proline residues, thus representing an amino acid sequence context characteristic of Cdc2 substrates.
Figure 4.
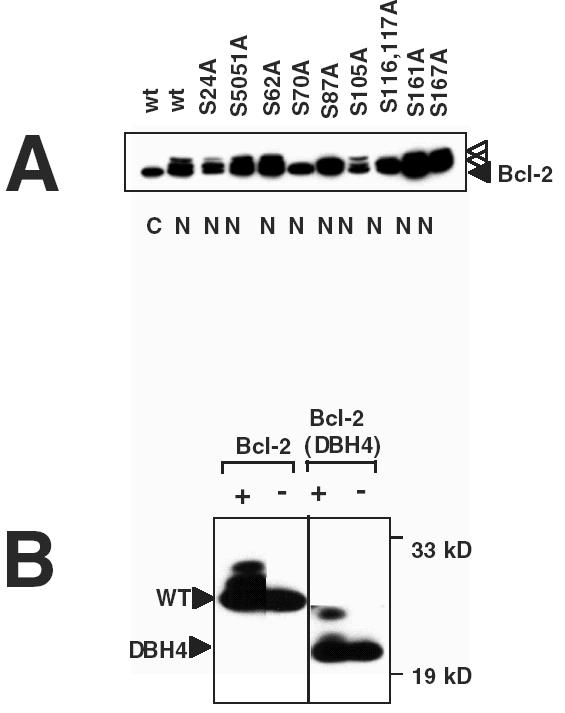
Structure-function analysis of Bcl-2 modifications induced by antimicrotubule drugs. (A) 293T cells in 100-mm dishes were transiently transfected with 10 µg of total DNA, including 2.5 µg of plasmid encoding wild-type (wt) or mutant Bcl-2 proteins, 0.5 µg pEGPF, and 7 µg pcDNA3 plasmid, then either cultured without further treatment (C) or with ∼2 µM nocodazole (N). Detergent-solubilized lysates were prepared from the transfectants and subjected to immunoprecipitation with anti-Bcl-2 anti-peptide antibody (2x106 cells/sample) followed by SDS-PAGE/immunoblot analysis with anti-Bcl-2 monoclonal (4D7) antibody. Examination of transfected cells by UV-microscopy confirmed >90% transfection efficiency (GFP positive cells) for all plasmid combinations (not shown). (B) 32D cells stably transfected with Bcl-2 or Bcl-2 (ΔBH4) (42) were cultured for ∼1 day with (+) or without (-) ∼300 nM paclitaxel before preparing lysates, which were normalized for total protein content and subjected to SDS-PAGE/immunoblot analysis using anti-Bcl-2 antibody with ECL-based detection. Arrow heads indicate unmodified and modified Bcl-2 or Bcl-2 (ΔBH4), respectively. Molecular weight markers are indicated in kilodaltons.
Raf1 is the only other kinase previously demonstrated to directly interact with Bcl-2 in cells [42] and it has been suggested that Raf1 mediates Bcl-2 phosphorylation in response to paclitaxel [21,22]. As an additional approach for exploring the structure-function relationships of the Bcl-2 protein with respect to antimicrotubule drug-induced modifications, we took advantage of a previously described Bcl-2 mutant that lacks the N-terminal region containing the BH4 domain required for association with Raf-1 [41,42]. Despite absence of its Raf1-binding domain, Bcl-2 (ΔBH4) still became modified in cells treated with antimicrotubule drugs, giving rise to two drug-inducible bands (as demonstrated by in SDS-PAGE/immunoblot analysis) analogous to wild-type Bcl-2 (Figure 4B). Thus, the BH4 domain of Bcl-2 is not required for the protein modifications induced by antimicrotubules drugs, though this domain is required for interactions with Raf1 [42].
Subcellular Location of Modified Bcl-2
During mitosis, the nuclear envelope breaks down, with nuclear and cytosolic proteins sharing a single compartment. Though best known for its mitochondrial location, quantitative analysis of the subcellular localization of the Bcl-2 protein has previously demonstrated that up to half the Bcl-2 protein can be found in the nuclear envelope and contiguous endoplasmic reticulum (ER) membranes [5]. We were therefore curious to know the location of the Bcl-2 protein when it undergoes posttranslational modifications in cells that have been arrested in mitosis by antimicrotubule drugs.
Subcellular fractionation analysis of cycling untreated cells demonstrated the presence of unmodified Bcl-2 protein in the nuclear and mitochondria-containing heavy membrane (HM) fractions, but not in the cytosolic, and very little in light-membrane fractions (Figure 5). In nocodazole-arrested cells, in contrast, both modified and unmodified Bcl-2 were located predominantly in the HM fraction. These findings suggest that both modified and unmodified Bcl-2 localize predominantly to mitochondria-containing HM fractions during mitotic arrest.
Figure 5.

Subcellular localization of Bcl-2 in control and M-phase-arrested cells. 293-Bcl-2 cells were either cultured without treatment (lanes 1–4) or arrested by treatment with ∼2 µM nocodazole (lanes 5–8) for ∼16 hours. Subcellular fractions were prepared, including HM (heavy membrane), LM (light membrane), N (nuclear), and C (cytosolic) fractions. Fractions were normalized for cell equivalents (107 cells each) and subjected to SDS-PAGE/immunoblot analysis using anti-Bcl-2 antibody with ECL-based detection. Reprobing the same blot with an antibody specific for mitochondrial F1β-ATPase confirmed successful fractionation (not shown).
A small proportion of Bcl-2 molecules in nocodazole-treated cells resided in the LM fraction (Figure 5). Interestingly, the Bcl-2 in this LM fraction was completely modified, as evidenced by its slower migration in SDS-PAGE.
Antimicrotubule Drug-Induced Modifications of Bcl-2 Induce Association with Pin1 without Effecting Bcl-2's Interactions with Other Proteins
Several substrates of Cdc2 are known to bind Pin1 in a phosphorylation-dependent manner during mitosis (reviewed in Ref. [45]).We therefore explored the possibility of a Pin1 interaction with Bcl-2 in cells arrested with antimicrotubule drugs, using coimmunoprecipitation assays. In untreated cycling cells, immunoprecipitation of Bcl-2 did not reveal associated Pin1 protein (Figure 6). In contrast, after inducing M-phase arrest by nocodazole-treatment, Pin1 protein was associated with Bcl-2 immunoprecipitates. Immunoblot analysis of cell lysates demonstrated equivalent amounts of Pin1 protein in both untreated and nocodazole-exposed cells, excluding variations in the amounts of this protein as a possible explanation for the results.
Figure 6.
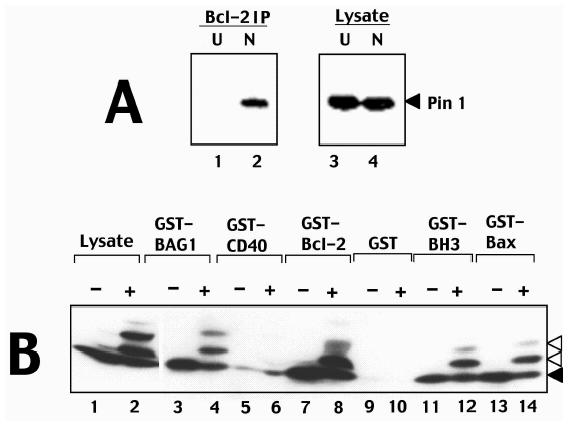
Phosphorylated Bcl-2 interacts with Pin1. (A) Detergent-solubilized lysates (2x106 cells/sample) were prepared either from 293-Bcl-2 cells that were cultured without treatment (lanes 1 and 3) or with nocodazole (lanes 2 and 4). Samples were either analysed directly by immunoblotting with anti-Pin1 antibodies (lanes 3 and 4) or were subjected to immunoprecipitation with anti-Bcl-2 antibodies followed by immunoblotting with anti-Pin1 antibodies. Immunodetection was accomplished by an ECL method. (B) GST-fusion proteins (1 µg) were immobilized on glutathione-Sepharose and incubated with lysates prepared from 697-Bcl-2 cells (100 µg total protein) cultured for ∼1 day with (+) or without (-) 300 nM paclitaxel. Beads were washed extensively and associated proteins were analyzed by SDS-PAGE/immunoblotting using anti-Bcl-2 antibody. An equivalent aliquot of lysate was loaded directly in gels for comparison.
To explore whether modifications induced in Bcl-2 by mitotic arrest influence its ability to interact with other proteins, in vitro protein-binding assays were conducted in which GST-fusion proteins representing various known Bcl-2-interacting proteins were tested for binding to unmodified and modified Bcl-2 in cell lysates derived from cells arrested with antimicrotubule drugs. As shown in Figure 6B, both unmodified and modified Bcl-2 bound to GST-Bcl-2, GST-Bax, and GST-BAG1, with little or no binding to control proteins GST and GST-CD40 cytosolic domain. A GST-fragment of Bax containing the dimerizing BH3 domain also bound to both modified and unmodified Bcl-2 (Figure 6B). Similar experiments demonstrated that modified Bcl-2 also is not impaired in its ability to associate with Raf1 or Nip3 (not shown). We conclude therefore that modifications of Bcl-2 induced by antimicrotubule drugs do not cause a generalized alternation in the ability of Bcl-2 to interact with other proteins, but do specifically promote associations of this protein with Pin1.
Discussion
In this report, we provide preliminary evidence that modifications induced in Bcl-2 during cell cycle arrest induced by antimicrotubule drugs promote association with Pin1. This PPIase binds through its WW domain to proline-containing phosphoserine and/or phosphothreonine motifs in target proteins, inducing conformational changes by cis-trans isomerization of peptide bonds proximal to proline residues, thereby altering protein bioactivity or stability [30,45]. The LR domain in the Bcl-2 protein that has been demonstrated to undergo phosphorylation during treatment of cells with antimicrotubule drugs consists of a proline-rich flexible loop of ∼60-amino-acids length. The predominant phosphorylations at serine 70 and serine 87 occur immediately proximal to proline residues, typical of Pin1-binding sites [30]. We speculate therefore that Pin1-mediated isomerization at prolyl residues in the LR of phosphorylated Bcl-2 alters the conformation of the LR domain, resulting somehow in a change in the bioactivity of the Bcl-2 protein.
Modifications of Bcl-2 induced by antimicrotubule drugs have been associated with suppression of the antiapoptotic activity of Bcl-2 [11,12,14,18], but the mechanisms remain unclear. Using in vitro protein-binding assays, we failed to observe a difference in the ability of unmodified and modified Bcl-2 to bind Bcl-2, Bax, BAG1, Raf1, or Nip3. Thus, modifications of the LR of Bcl-2 do not appear to alter association of this protein with multiple known binding partners. This observation is consistent with data that have suggested that binding of Bcl-2, Bax, BAG1, and Raf1 to Bcl-2 is mediated by or dependent on other domains in the Bcl-2 protein, particularly either the N-terminal BH4 domain or a hydrophobic crevice formed on the surface of the protein by interfaces involving the BH1, BH2, and BH3 domains [42,46].
In addition to binding other proteins, Bcl-2 also possesses a biochemical activity as an ion channel [47]. Thus, Pin1-induced isomerization of peptidyl bonds in the LR of Bcl-2 conceivably could alter this activity of Bcl-2, perhaps making it more difficult for Bcl-2 to undergo the profound conformational changes required for membrane insertion of α-helices required for ion-channel activity.
The Bcl-2 protein recovered from cells arrested using antimicrotubule drugs displays several modified forms that migrate differently in SDS-PAGE. However, only two major sites of phosphorylation of Bcl-2 have been reproducibly detected, serine 70 and serine 87, which fails to account for the multiple bands sometimes observed [11,13]. We speculate therefore that some of the anomalous migrating forms of Bcl-2 may arise as a result of Pin1-induced isomerization of peptidyl bonds in the LR of Bcl-2, producing Bcl-2 proteins with alternative SDS-resistant conformations. It is also a possibility, however, that a subpopulation of Bcl-2 molecules in microtubule drug-arrested cells undergo modifications on other residues, besides serine 70 and 87, thus explaining the origin of these additional bands in SDS-PAGE analysis. In this regard, threonine 56 and 69 have been identified as possible additional (but probably minor) phosphorylation sites in Bcl-2 [11,28].
The LR of Bcl-2 has been shown to act as a negative-regulatory domain that autorepresses the antiapoptotic function of Bcl-2 [7–10]. It is unclear, however, whether LR-dependent autoinhibitory activity is related to Pin1 interactions. For example, Bcl-2 (ΔLR) mutants display enhanced activity after treatment with anti-immunoglobulin in cells [7], which arrest in G0G1 phase, where the modifications associated with M-phase arrest and Pin1 binding are not observed. Bcl-2 (ser70ala) and Bcl-2 (ser87ala) mutants have also been shown to provide enhanced protection compared to wild-type Bcl-2 against apoptosis induced by anti-Fas antibody, a cell-cycle-independent cell death stimulus [11]. In addition, serine 70 in the LR of Bcl-2 has been reported to undergo PKC-dependent phosphorylation in response to growth factors and PKC agonists, though this modification was associated with enhancement rather than inhibition of Bcl-2's antiapoptotic function [16,48]. Thus, further analysis is required for understanding the mechanism(s) by which the LR regulates Bcl-2 function.
We provide correlative evidence here linking the cyclin-dependent kinase Cdc2 to phosphorylation of Bcl-2 in cells arrested with antimicrotubule drugs. This evidence includes: (a) suppression of Bcl-2 protein modifications by flavopiridol, a selective inhibitor of Cdc2 family kinases; (b) association of Cdc2 with Bcl-2 in antimicrotubule drug-arrested cells; (c) mutagenesis-based mapping of phosphorylation sites in Bcl-2 to serine 70 and 87, which reside in sequence contexts typical of Cdc2 substrates; and (d) association of Bcl-2 with Pin1, which is known to bind substrates of Cdc2. Clearly, the assertion that Cdc2 is responsible for phosphorylation of Bcl-2 in antimicrotubule drug-arrested cells is subject to several caveats, including limitations in the selectivity of flavopiridol, which interacts with ATP-binding pocket of kinases and thus exhibits “nonspecific” inhibitory activity against other kinases besides Cdc2 and its relatives [49]. It is also possible that multiple kinases could participate in phosphorylating Bcl-2 in M-phase-arrested cells, and indeed, others have provided data implicating Raf1, PKA, JNK [11,12,21,27,29,50]. Nevertheless, the data provided here strongly suggest that Cdc2 is among the potential kinases responsible for modifications of Bcl-2 in tumor cells arrested with antimicrotubule drugs. Interestingly, others have also recently implicated Cdc2 in phosphorylation of Bcl-2 during mitotic arrest induced by okadaic acid or nocodazole in combination with okadaic acid, and have shown that Cdc2 can phosphorylate the Bcl-2 protein in vitro [23,28]. However, in one of these studies [28], Bcl-2 phosphorylation occurred on threonine 56. Furthermore, Bcl-2 phosphorylation can be induced by Cdks activated as a result of herpes virus v-cyclin [51].
Normally Cdc2 is located in the nucleus, and would not have access to Bcl-2. However, during mitosis and anaphase, the nuclear envelope is absent, placing mitochondria-associated Bcl-2 and active Cdc2 in the same compartment. Interestingly, it has been suggested that the Bcl-2 protein may associate with the condensed chromosomes during mitosis [52], which (if true) would place Bcl-2 into close proximity with the sites targeted by Cdc2. However, we observed Bcl-2 (including modified Bcl-2) in the mitochondria-enriched HM fraction in nocodazole-treated cells, suggesting that Bcl-2 protein molecules associated with this organellar fraction do become modified and implying that they gain access to Cdc2 during this phase of the cell cycle. Interestingly, essentially all the Bcl-2 protein molecules resident in the LM fraction of nocodazole-arrested cells appeared to be modified, as opposed to the HM fraction where unmodified Bcl-2 was more abundant than modified protein. This observation might be relevant to the disposition of the nuclear envelope during mitosis, where this membrane breaks into small vesicles that associate with mitotic chromosomes [53]. These chromosome-associated vesicles might be expected to have more chances for direct contact with active Cdc2.
What is the potential clinical significance of these observations concerning mechanisms of Bcl-2 modification in cancer cells treated with antimicrotubule drugs? The cyclin-dependent kinase inhibiting drug flavopiridol is presently undergoing testing in clinical trials for patients with cancer. Suppression of cyclin-dependent kinases has been demonstrated to produce cell cycle arrest. However, it has been reported that Cdc2 or related kinases (Cdk family proteins) are required for apoptosis induction in some settings [54,55]. Furthermore, flavopiridol prevents the modifications of Bcl-2 that are normally induced during arrest of cycling tumor cells by antimicrotubule drugs. Inasmuch as these modifications of Bcl-2 associated with mitotic arrest are believed to inactivate the Bcl-2 protein, flavopiridol might guard against apoptosis by allowing Bcl-2 to retain its antiapoptotic activity. The predicted result thus would be cell cycle arrest without concomitant apoptosis. Empiric observations of the effects of flavopiridol on neoplastic cells, however, have demonstrated apoptosis induction in a variety of circumstances. For example, in noncycling B-cell chronic lymphocytic leukemia (CLL) cells, we and others have demonstrated that flavopiridol can induce apoptosis [56,57], implying that this compound is capable of inhibiting cell-cycle-independent kinases required for cell survival. Thus, the benefit of flavopiridol in the clinical setting must be empirically determined. However, mechanism-based interactions of this kinase inhibitory drug with antimicrotubule agents such as paclitaxel should be carefully considered in designing trials that combine flavopiridol with this class of agents
Acknowledgements
We thank K. P. Lu for supplying anti-Pin antibody, R. Cornell and A. Fernandez for manuscript preparation, the Leukemia and Lymphoma Society of America, and the NIH (GM 60554, CA 77328), for generous support.
Footnotes
Present address: IDEC Pharmaceuticals, Inc, 3010 Science Park Road, La Jolla, CA 92191-9080.
Present address: Editions Elsevier, 23, rue Linois, 75724 Paris cedex 15, France.
References
- 1.Reed J. Bcl-2 family proteins. Oncogene. 1998;17:3225–3236. doi: 10.1038/sj.onc.1202591. [DOI] [PubMed] [Google Scholar]
- 2.Reed JC. Bcl-2 family proteins: regulators of apoptosis and chemoresistance in hematologic malignancies. Semin Hematol. 1997;34(4):9–19. [PubMed] [Google Scholar]
- 3.Nguyen M, Branton PE, Walton PA, Oltvai ZN, Korsmeyer SJ, Shore GC. Role of membrane anchor domain of Bcl-2 in suppression of apoptosis caused by E1B-defective adenovirus. J Biol Chem. 1994;269:16521–16524. [PubMed] [Google Scholar]
- 4.Tanaka S, Saito K, Reed J. Structure-function analysis of the bcl2 oncoprotein. Addition of a heterologous transmembrane domain to a portions of the bcl2 beta protein restore function as a regulator of cell survival. J Biol Chem. 1993;268:10920–10926. [PubMed] [Google Scholar]
- 5.Krajewski S, Tanaka S, Takayama S, Schibler MJ, Fenton W, Reed JC. Investigation of the subcellular distribution of the bcl-2 oncoprotein: residence in the nuclear envelope, endoplasmic reticulum, and outer mitochondrial membranes. Cancer Res. 1993;53:4701–4714. [PubMed] [Google Scholar]
- 6.Muchmore SW, Sattler M, Liang H, et al. X-ray and NMR structure of human Bcl-XL, an inhibitor of programmed cell death. Nature. 1996;381:335–341. doi: 10.1038/381335a0. [DOI] [PubMed] [Google Scholar]
- 7.Chang BS, Minn AJ, Muchmore SW, Fesik SW, Thompson CB. Identification of a novel regulatory domain in Bcl-xL and Bcl-2. EMBO J. 1997;16(5):968–977. doi: 10.1093/emboj/16.5.968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Uhlmann E, D'Sa-Eipper C, Subramanian T, Wagner A, Hay N, Chinnadurai G. Deletion of a nonconserved region of Bcl-2 confers a novel gain of function: suppression of apoptosis with concomitant cell proliferation. Cancer Res. 1996;56(11):2506–2509. [PubMed] [Google Scholar]
- 9.Wang S, Wang Z, Boise L, Dent P, Grant S. Loss of the bcl-2 phosphorylation loop domain increases resistance of human leukemia cells (U937) to paclitaxel-mediated mitochondrial dysfunction and apoptosis. Biochem Biophys Res Commun. 1999;259:67–72. doi: 10.1006/bbrc.1999.0669. [DOI] [PubMed] [Google Scholar]
- 10.Fang G, Chang BS, Kim CN, Perkins C, Thompson CB, Bhalla KN. Loop domain is necessary for taxol-induced mobility shift and phosphorylation of Bcl-2 as well as for inhibiting taxol-induced cytosolic accumulation of cytochrome c and apoptosis. Cancer Res. 1998;58:3202–3208. [PubMed] [Google Scholar]
- 11.Yamamoto K, Ichijo H, Korsmeyer SJ. Bcl-2 is phosphorylated and inactivated by an ask1/jun n-terminal protein kinase pathway normally activated at g (2)/m. Mol Cell Biol. 1999;19:8469–8478. doi: 10.1128/mcb.19.12.8469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Srivastava RK, Mi QS, Hardwick JM, Longo DL. Deletion of the loop region of Bcl-2 completely blocks paclitaxel-induced apoptosis. Proc Natl Acad Sci USA. 1999;96:3775–3780. doi: 10.1073/pnas.96.7.3775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Haldar S, Basu A, Croce CM. Serine-70 is one of the critical sites for drug-induced Bcl2 phosphorylation in cancer cells. Cancer Res. 1998;58(8):1609–1615. [PubMed] [Google Scholar]
- 14.Basu A, Haldar J. Microtubule-damaging drugs triggered bcl2 phosphorylation-requirement of phosphorylation on both serine-70 and serine-87 residues of bcl2 protein. Int J Oncol. 1998;13:659–664. doi: 10.3892/ijo.13.4.659. [DOI] [PubMed] [Google Scholar]
- 15.Maundrell K, Antonsson B, Magnenat E, et al. Bcl-2 undergoes phosphorylation by c-Jun N-terminal kinase/stress-activated protein kinases in the presence of the constitutively active GTP-binding protein Rac1. J Biol Chem. 1997;272:25238–25242. doi: 10.1074/jbc.272.40.25238. [DOI] [PubMed] [Google Scholar]
- 16.Ruvolo P, Deng X, Carr B, May W. A functional role for mitochondrial protein kinase Cα in Bcl-2 phosphorylation and suppression of apoptosis. J Biol Chem. 1998;273(39):25436–25442. doi: 10.1074/jbc.273.39.25436. [DOI] [PubMed] [Google Scholar]
- 17.May WS, Tyler PG, Ito T, Armstrong DK, Qatsha KA, Davidson NE. Interleukin-3 and bryostatin-1 mediate hyperphosphorylation of Bcl-2 α in association with suppression of apoptosis. J Biol Chem. 1994;269:26865–26870. [PubMed] [Google Scholar]
- 18.Haldar S, Jena N, Croce CM. Inactivation of Bcl-2 by phosphorylation. Proc Natl Acad Sci USA. 1995;92:4507–4511. doi: 10.1073/pnas.92.10.4507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Haldar S, Chintapallli J, Croce CM. Taxol induces Bcl-2 phosphorylation and death of prostate cancer cells. Cancer Res. 1996;56:1253–1255. [PubMed] [Google Scholar]
- 20.Haldar S, Basu A, Croce CM. Bcl2 is the guardian of microtubule integrity. Cancer Res. 1997;57:229–233. [PubMed] [Google Scholar]
- 21.Blagosklonny M, Schulte T, Nguyen P, Trepel J, Neckers L. Taxol-induced apoptosis and phosphorylation of Bcl-2 protein involves c-Raf-1 and represents a novel c-Raf-1 transduction pathway. Cancer Res. 1996;56:1851–1854. [PubMed] [Google Scholar]
- 22.Blagosklonny MV, Giannakakou P, el-Deiry WS, et al. Raf-1/bcl-2 phosphorylation: a step from microtubule damage to cell death. Cancer Res. 1997;57 (1):130–135. [PubMed] [Google Scholar]
- 23.Ling Y-H, Tornos C, Perez-Soler R. Phosphorylation of Bcl-2 is a marker of M phase events and not a determinant of apoptosis. J Biol Chem. 1998;273:18984–18991. doi: 10.1074/jbc.273.30.18984. [DOI] [PubMed] [Google Scholar]
- 24.Scatena C, Stewart Z, Mays D, et al. Mitotic phosphorylation of Bcl-2 during normal cell cycle progression and taxol-induced growth arrest. J Biol Chem. 1998;273:30777–30784. doi: 10.1074/jbc.273.46.30777. [DOI] [PubMed] [Google Scholar]
- 25.Jiang JD, Davis AS, Middleton K, et al. 3-(Iodoacetamido)-benzoylurea: a novel cancericidal tubulin ligand that inhibits microtubule polymerization, phosphorylates bcl-2, and induces apoptosis in tumor cells. Cancer Res. 1998;58:5389–5395. [PubMed] [Google Scholar]
- 26.Attalla H, Westberg JA, Andersson LC, Adlercreutz H, Makela TP. 2-Methoxyestradiol-induced phosphorylation of Bcl-2: uncoupling from JNK/SAPK activation. Biochem Biophys Res Commun. 1998;247:616–619. doi: 10.1006/bbrc.1998.8870. [DOI] [PubMed] [Google Scholar]
- 27.Srivastava RK, Srivastava AR, Korsmeyer SJ, Nesterova M, Cho-Chung YS, Longo DL. Involvement of microtubules in the regulation of Bcl-2 phosphorylation and apoptosis through cyclic AMP-dependent protein kinase. Mol Cell Biol. 1998;18:3509–3517. doi: 10.1128/mcb.18.6.3509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Furukawa Y, Iwase S, Kikuchi J, et al. Phosphorylation of Bcl-2 protein by CDC2 kinase during G2/M phases and its role in cell cycle regulation. J Biol Chem. 2000;275:21661–21667. doi: 10.1074/jbc.M906893199. [DOI] [PubMed] [Google Scholar]
- 29.Basu A, You SA, Haldar S. Regulation of Bcl2 phosphorylation by stress response kinase pathway. Int J Oncol. 2000;16:497–500. doi: 10.3892/ijo.16.3.497. [DOI] [PubMed] [Google Scholar]
- 30.Yaffe MB, Schutkowski M, Shen M, et al. Sequence-specific and phosphorylation-dependent proline isomerizaion: a potential mitotic regulatory mechanism. Science. 1997;278:1957–1960. doi: 10.1126/science.278.5345.1957. [DOI] [PubMed] [Google Scholar]
- 31.Winkler KE, Swenson KI, Kornbluth S, Means AR. Requirement of the prolyl isomerase pin1 for the replication checkpoint. Science. 2000;287:1644–1647. doi: 10.1126/science.287.5458.1644. [DOI] [PubMed] [Google Scholar]
- 32.Reed JC, Tanaka S, Cuddy M, et al. A strategy for generating monoclonal antibodies against recombinant baculovirus-produced proteins: application to the Bcl-2 oncoprotein. Anal Biochem. 1992;205(1):70–76. doi: 10.1016/0003-2697(92)90580-z. [DOI] [PubMed] [Google Scholar]
- 33.Hockenbery DM, Zutter M, Hickey W, Nahm M, Korsmeyer SJ. Bcl-2 protein is topographically restricted in tissues characterized by apoptotic cell death. Proc Natl Acad Sci USA. 1991;88:6961–6965. doi: 10.1073/pnas.88.16.6961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Krajewski S, Blomvqvist C, Franssila K, et al. Reduced expression of pro-apoptotic gene Bax is associated with poor response rates to combination chemotherapy and shorter survival in women with metastatic breast adenocarcinoma. Cancer Res. 1995;55:4471–4478. [PubMed] [Google Scholar]
- 35.Miyashita T, Reed JC. Bcl-2 oncoprotein blocks chemotherapy-induced apoptosis in a human leukemia cell line. Blood. 1993;81(1):151–157. [PubMed] [Google Scholar]
- 36.Torigoe T, Millan JA, Takayama S, Taichman R, Miyashita T, Reed JC. Bcl-2 inhibits T-cell-mediated cytolysis of leukemia cell line. Cancer Res. 1994;54:4851–4854. [PubMed] [Google Scholar]
- 37.Wang H-G, Miyashita T, Takayama S, et al. Apoptosis regulation by interaction of Bcl-2 protein and Raf-1 kinase. Oncogene. 1994;9:2751–2756. [PubMed] [Google Scholar]
- 38.Xu Q, Reed JC. Bax inhibitor-1, a mammalian apoptosis suppressor identified by functional screening in yeast. Mol Cell. 1998;1(3):337–346. doi: 10.1016/s1097-2765(00)80034-9. [DOI] [PubMed] [Google Scholar]
- 39.Gauwerky CE, Hoxie J, Nowell PC, Croce CM. Pre-B-cell leukemia with a t (14;18) translocation is preceded by follicular lymphoma. Oncogene. 1988;2:431–435. [PubMed] [Google Scholar]
- 40.Wang H-G, Pathan N, Ethell I, et al. Calcineurin promotes apoptosis by dephosphorylating BAD. Science. 1998;284(5412):339–343. doi: 10.1126/science.284.5412.339. [DOI] [PubMed] [Google Scholar]
- 41.Hanada M, Aime-Sempe C, Sato T, Reed JC. Structure-function analysis of Bcl-2 protein. Identification of conserved domains important for homodimerization with Bcl-2 and heterodimerization with Bax. J Biol Chem. 1995;270(20):11962–11969. doi: 10.1074/jbc.270.20.11962. [DOI] [PubMed] [Google Scholar]
- 42.Wang HG, Rapp UR, Reed JC. Bcl-2 targets the protein kinase Raf-1 to mitochondria. Cell. 1996;87(4):629–638. doi: 10.1016/s0092-8674(00)81383-5. [DOI] [PubMed] [Google Scholar]
- 43.Wang H-G, Millan JA, Cox AD, et al. R-ras promotes apoptosis caused by growth factor deprivation via a Bcl-2 suppressible mechanism. J Cell Biol. 1995;129:1103–1114. doi: 10.1083/jcb.129.4.1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Koepp DM, J.W. H, S.J. E. How the cyclin became a cyclin: regulated proteolysis in the cell cycle. Cell. 1999;97:431–434. doi: 10.1016/s0092-8674(00)80753-9. [DOI] [PubMed] [Google Scholar]
- 45.Hunter T. Prolyl isomerases and nuclear function. Cell. 1998;92:141–143. doi: 10.1016/s0092-8674(00)80906-x. [DOI] [PubMed] [Google Scholar]
- 46.Kelekar A, Thompson CB. Bcl-2-family proteins — the role of the BH3 domain in apoptosis. Trends Cell Biol. 1998;8(8):324–330. doi: 10.1016/s0962-8924(98)01321-x. [DOI] [PubMed] [Google Scholar]
- 47.Schendel SL, Xie Z, Montal MO, Matsuyama S, Montal M, Reed JC. Channel formation by antiapoptotic protein Bcl-2. Proc Natl Acad Sci USA. 1997;94:5113–5118. doi: 10.1073/pnas.94.10.5113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Itoh T, Deng X, Carr B, May WS. Bcl-2 phosphorylation required for anti-apoptosis function. J Biol Chem. 1997;272(18):11671–11673. doi: 10.1074/jbc.272.18.11671. [DOI] [PubMed] [Google Scholar]
- 49.Roberts CJ, Nelson B, Marton MJ, et al. Signaling and circuitry of multiple MAPK pathways revrealed by a matrix of global gene expression profiles. Science. 2000;287:873–880. doi: 10.1126/science.287.5454.873. [DOI] [PubMed] [Google Scholar]
- 50.Blagosklonny MV, Chuman Y, Bergan RC, Fojo T. Mitogen-activated protein kinase pathway is dispensable for microtubule-active drug-induced Raf-1/Bcl-2 phosphorylation and apoptosis in leukemia cells. Leukemia. 1999;13:1028–1036. doi: 10.1038/sj.leu.2401449. [DOI] [PubMed] [Google Scholar]
- 51.Ojala PM, Yamamoto K, Korsmeyer SJ, Makela TP. The apoptotic v-cyclin-CDK6 complex phosphorylates and inactivates Bcl-2. Nat Cell Biol. 2000;2:819–825. doi: 10.1038/35041064. [DOI] [PubMed] [Google Scholar]
- 52.Tang C, Willingham MC, Reed JC, et al. High levels of p26Bcl-2 oncoprotein retard taxol-induced apoptosis in human pre-B leukemia cells. Leukemia. 1994;8:1960–1969. [PubMed] [Google Scholar]
- 53.Melchior F, Gerace L. Mechanisms of nuclear protein import. Curr Opin Cell Biol. 1995;7:310–318. doi: 10.1016/0955-0674(95)80084-0. [DOI] [PubMed] [Google Scholar]
- 54.Shi L, Chen G, He D, Bosc D, Litchfield D, Greenberg A. Granzyme B induces apoptosis and cyclin a-associated cyclin-dependent kinase activity in all stages of the cell cycle. J Immunol. 1996;157:2381–2385. [PubMed] [Google Scholar]
- 55.Harvey KJ, Lukovic D, Ucker DS. Caspase-dependent Cdk activity is a requisite effector of apoptotic death events. J Cell Biol. 2000;148:59–72. doi: 10.1083/jcb.148.1.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Konig A, Schwartz GK, Mohammad RM, Katib AA, Gabrilove JL. The novel cyclin-dependent kinase inhibitor flavopiridol downregulates Bcl-2 and induces growth arrest and apoptosis in chronic B-cell leukemia lines. Blood. 1997;90(11):4307–4312. [PubMed] [Google Scholar]
- 57.Kitada S, Andreeff M, Reed JC. Protein kinase inhibitors flavopiridol and 7-hydroxy-staurosporine down-regulate antiapoptosis proteins in B-cell chronic lymphocytic leukemia. Blood. 2000;96:393–397. [PubMed] [Google Scholar]