

Abstract
We have investigated a single telomere expansion in a case of acute lymphoblastic B-cell leukemia (B-ALL), where half of the cells in the bone marrow sample appeared with a Philadelphia chromosome. Comparing telomere sizes in Philadelphia-positive versus -negative cells, we found generally shorter telomeres in the Philadelphia-positive cells, but with an expansion of the telomere on the long arm of one chromosome 11 homologue. This expansion was also found in a minority of Philadelphia-negative cells. The telomeres in these cells were of the same overall size as the telomeres in the Philadelphia-negative cells without the 11q expansion. Together, these findings suggest that the order of events was: 11q telomere expansion, Philadelphia translocation, overall telomere shortening. The expanded 11q telomere contained the standard telomeric (AGGGTT)n repeat, but also variant repeat sequences. The single telomere expansion suggests a non-telomerase mechanism behind the expansion which may also explain the presence of variant repeats in the expanded telomere. The present case illustrates that telomere changes may occur at only some chromosome ends in a subset of cells. To reveal such changes, telomere morphology should be studied with in situ methodology.
Keywords: telomere repeats, telomere expansion, Philadelphia chromosome, PRINS, B-ALL
Introduction
All human chromosome ends are capped with tandem repetitions of the six-base repeat unit, AGGGTT [1]. These repeats are added to the chromosome ends in germ line cells by the specialized enzyme telomerase [2]. In most somatic cells, telomerase is not active, and telomeres shorten with every round of cell division, eventually causing mitotic arrest [3,4]. To avoid this cell cycle arrest, cancer cells are forced to find ways of compensating for the telomere shortening [5]. This may obviously be accomplished by reactivation of the telomerase [5,6]. It has, however, been reported that tumor samples may be negative for telomerase, and still have the same average telomere size as samples positive for telomerase [7]. Consequently, these tumor cells must have maintained their telomeres by some alternative mechanisms [7]. Except that Terminal Restriction Fragment (TRF) analysis demonstrated an increased heterogeneity in telomeric repeat sizes in tumor samples negative for telomerase [7], nothing seems known about telomere appearance after telomerase-mediated versus alternative telomere elongation. Unfortunately, since TRF assays generate data on average telomere sizes over a large number of pooled cells, it is not possible to determine whether the observed increase reflects an increased difference in telomere sizes within the individual cells, or between cells.
Unlike the de novo synthesis accomplished by telomerase, telomere elongation by alternative mechanisms must necessarily move or copy pre-existing repeats. The sources or templates available for the alternative mechanisms are cryptic telomeric sequences at internal positions and telomeric repeats at chromosomal termini. Telomeric repeats at internal sites are generally quite degenerate, and terminal telomeric repeat domains contain a region of variant repeats, centromeric to the perfect repeats [8]. It could, therefore, be speculated that unless the alternative mechanisms are capable of discriminating perfect from variant repeats, copying only the former, they would give an elongation product of a mixed sequence composition. If this is indeed the case, it should be possible to distinguish between telomerase-mediated and alternative telomere elongation from the sequence composition of the product. Unfortunately, no data on such a direct finger-print of the mode of telomere lengthening has been published.
The mode of telomeric reconstruction could have implications for how the cancer progresses and how it responds to therapy, both because of the supposedly more uneven telomere elongation by alternative mechanism [7], and because variant telomeric repeats bind key proteins differently from the perfect AGGGTT repeat [9–11].
Recently, methodology has been developed enabling the sizes of telomeric repeat domains on single chromosome ends to be measured [12,13]. Our assay is based on telomere staining by dideoxy-PRINS [14], a novel version of the Primed In Situ (PRINS) labeling technique [15]. An attractive feature of the dideoxy-PRINS technique is that it can selectively visualize variant repeat sequences in situ [14,16]. Fingerprinting of individual telomere expansion products should thus be theoretically possible, employing dideoxy-PRINS with various repeat probes to “pseudo-sequence” individual telomeres in situ. In this work, we investigate in greater detail a case of single telomere expansion first noticed in connection with the first presentation of the dideoxy-PRINS for measurement of telomere sizes [13]. In an effort to establish the identity and the sequence contents of the expanded telomere, as well as a likely order of events, we combined fluorescence in situ hybridization (FISH), telomere sizing after dideoxy-PRINS, and “pseudo-sequencing” in situ with PRINS probes for both standard and variant repeats.
Materials and Methods
The Case
A 31-year-old male presented with B-ALL in May 1994. The initial white blood cell count was high (112.000/µL). Immune phenotyping revealed positivity for HLA-DR, CD 10, CD 19, and CD 20 (B-ALL). Cytogenetic analysis found only one cell in metaphase. This cell was positive for the 9;22 Philadelphia translocation. In concordance, multiplex polymerase chain reaction analysis demonstrated a BCR-abl rearrangement of the b3a2 subtype. Induction therapy with prednisone, vincristine, daunorubicin, cyclophosphamide, l-asparaginase, and intrathecal methotrexate resulted in complete remission morphologically and immunophenotypically on day 29. Intensive consolidation was given in three cycles immediately after remission induction. After regeneration from the third course (September 1994), the patient received oral maintenance therapy with 6-mercaptopurine and methotrexate. In November 1994, a routine pretransplant bone marrow examination showed a cytogenetic relapse with 100% Ph-positive metaphases (8/8). One week later, haematologic relapse (leukopenia and blast foci) was evident. A second remission was obtained with high-dose amsacrine and cytarabin, although 50% (5/10) of the bone marrow cells in metaphase remained Philadelphia-positive. The only bone marrow cells available to the present study were from this admission. After two courses of CHOP, a successful bone marrow transplantation was carried out. The patient remains well 4 years after diagnosis and 3.3 years after the bone marrow transplantation. No cytogenetic follow-up has been performed.
Dideoxy-PRINS and FISH
An overnight culture of bone marrow cells was synchronized with metotrexate, and harvested and fixed in methanol-acetic acid according to standard methodology. Spreads of metaphase chromosomes were used fresh. The suspensions of fixed cells used in this study were 3 1/2 years old. Slides of the same age, spread at the time of admission, did not give telomeric signals upon dideoxy-PRINS.
The probes used in the dideoxy-PRINS reactions were: “Telo2” (CCCTAA)7 [17], corresponding to the main telomeric repeat in human chromosomes, “Telo3” (GGG(TTGGGG)2TTG) [16], corresponding to the telomeric repeat from Tetrahymena, “Telo5” (AGGGTTT5) [16], corresponding to the telomeric sequence in the malaria parasite Plasmodium falciparum, “Telo7” (GGTGAG)4-GGTG [14], corresponding to a repeat variant that commonly occurs as the AGGGTT repeat diverges at interstitial sites in the human genome and “Telo8” (TCAC(CCTCAC)4) [16], which is complementary to Telo7.
Dideoxy-PRINSes with Telo probes were performed as described in detail elsewhere [13,14,16]. In brief, the dideoxy-PRINS does not differ from the standard PRINS [18], except that nucleotides not needed for copying of the proper target sequence are included as dideoxy analogues to reduce non-relevant priming from breaks in chromosomal DNA and cross-hybridizing probes to a minimum. Probes Telo2 and 8 were thus incubated with ddGTP rather than dGTP, and probes Telo3, 5 and 7 with ddCTP rather than dCTP. A complete list of relevant probes for PRINS studies of telomeres is given in Table 1.
Table 1.
Base Sequence and Name of Oligonucleotide Probes for Telomere Studies by PRINS.
| Telo1: (TTAGGG)7 |
| Telo2: (CCCTAA)7 |
| Telo3: (GGTTGG)4G |
| Telo4: (CCAACC)4C |
| Telo5: (AGGGTTT)5 |
| Telo6: (CCCTAAA)5 |
| Telo7: (GGTGAG)4GGTG |
| Telo8: TCAC (CCTCAC)4 |
All odd-numbered probes hybridize to the C-rich strand of the telomere, and all even-numbered to the G-rich. The probes come in pairs staining opposite strands of a particular telomeric repeat. Telo1 and 2 thus stain the standard telomeric repeat whereas Telo3 and 4 stain one type of variant repeat, Telo5 and 6 another variant, and Telo7 and 8 a third variant. For further details, please consult Ref. [16].
The FISH probe for 11qter was from AL Technologies, and it was used as specified by that company, except that the hybridization was preceded by a PRINS reaction (PRINS-painting [19]). The FISH probe for the 11q terminus was selected for the purpose because it was originally made from DNA material obtained by microdissection of 11qter. It must, therefore, be expected to cover sequences to the most terminal part of 11q.
Image Analysis
Relative size measurements after dideoxy-PRINS were achieved as previously reported [13]. In brief, metaphases were after the dideoxy-PRINS reaction selected in the counterstain (DAPI) on the basis of good chromosome spreading. Images were recorded with a CCD camera, and analyzed by the CGH program from VYSIS (QUIPS). In this program, the telomere signals were assigned as “tester”, whereas the DAPI stain was assigned as “reference”. The metaphases thus recorded were karyotyped, and data from Philadelphia-positive or Philadelphia-negative cells were assigned to the relevant category, according to the presence or absence of a Philadelphia chromosome. Finally, for comparison of telomere sizes at the individual chromosome ends, ratio profiles were created for each of the two categories of cells.
Results and Discussion
Anatomy of the Individual Telomeres
To get an overview of the telomere size at all chromosome ends in the Philadelphia-negative and the Philadelphia-positive subset of cells, a comparative relative telomere sizing was performed after staining of the telomeres with dideoxy-PRINS with Telo2. The resulting telomere profiles demonstrated three main differences between pooled Philadelphia-positive, and pooled Philadelphia-negative cells (Figure 1).
Figure 1.

Telomeric ratio profiles of Philadelphia-positive (upper part) and Philadelphia-negative cells (lower part). At multiple points along the axis of each chromosome, the QUIPS program calculates the ratio between the intensity of the dideoxy-PRINS reaction and the intensity of the DAPI staining. As the dideoxy-PRINS reaction stains the telomeres, whereas DAPI stains the entire chromosomes, the ratio is highest at the chromosome ends, as evidenced by the profile peaks. The higher a peak is at a certain chromosome end, the higher the ratio at that site. The blue curves represent average profiles over more than 10 pooled metaphases, whereas the red curves give the 95% confidence intervals. The Philadelphia-positive cells have the lowest telomere peaks, as well as the narrowest 95% confidence intervals. The increased peak height at the 11q telomere in the Philadelphia-positive cells is also evident.
First of all, the telomeric profile peaks were, in general, lower in the Philadelphia-positive subset of cells, demonstrating shorter telomeres in these cells [13].
Secondly, the confidence intervals on the curves were narrower in the Philadelphia-positive cells (Figure 1). This finding may seem in contrast to the finding by Bryan et al. [7] of increased variation in telomere size when the telomere is maintained by alternative methods. However, as Bryan et al. employed the TRF assay for the telomere sizing, they were looking at the distribution of telomeres sizes in a pool of all the clones found in the particular case. Their observation is therefore comparable to what we would get if we pooled the data from the Philadelphia-positive and the Philadelphia-negative cells, rather than investigating the two sets of data individually. Thus, whereas the pooled data show a high level of variation (data not shown), telomere sizes in the dominant malignant clone do not. The lower variation in telomere size among the Philadelphia-positive cells, as compared with the Philadelphia-negative cells, could similarly reflect that whereas the former represents one clone of cells, the latter likely come from multiple clones of cells. Alternatively, it could reflect that short telomeres inhibit cell division [4]. As a consequence of this, cells with short telomeres should divide less willingly, if at all, thus loosing telomeric DNA slowly, if at all. Neighbouring cells with longer telomeres should, on the other hand, continue to divide unimpeded, thus continuously shortening their telomeres. In a population of cells where residual telomere size is limiting to the rate of cell division, the long telomeres would thus be expected to shorten faster than the short telomeres, resulting in a narrower distribution of telomere sizes. Interestingly, in a comparison between telomere sizes in lymphocytes from a (healthy) old donor and a related (healthy) young donor of lymphocytes, we observed a similar narrowing of the confidence intervals in the cells with the shorter telomeres (unpublished observation).
The third and final difference between the Philadelphia-positive and the Philadelphia-negative cells was that the long arm telomere on chromosome 11, unlike any of the other telomeres, was longer in the former [13]. From the telomeric profiles of each of the two homologous chromosomes 11 in the Philadelphia-positive cells, it can be seen that this was due to an increased staining of only one of the two homologues (Figure 2). The difference in telomere staining between homologues was directly visible in individual pairs of chromosomes (Figure 3). The comparative sizing approach applied here does not give estimated telomere sizes in kilobases, but tells whether a particular telomere is longer or shorter than another telomere. We, therefore, cannot give an approximate size of the elongated 11q telomere, except for what can be deduced from the visual appearance of the telomere signal. The intensity of the signal on this particular telomere was clearly beyond anything we have observed on normal telomeres, and considering the size range of normal telomeres, the size of the elongated 11q telomere should be at least 20 kb.
Figure 2.
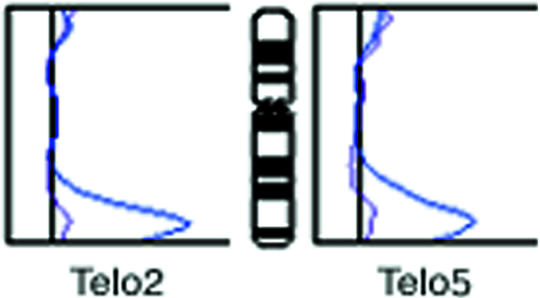
Telomeric ratio profiles on homologous chromosomes 11 after dideoxy-PRINS with Telo2 or Telo5. For each probe, two curves are shown, one representing the normal chromosome 11, and one representing the chromosome 11 with the 11q telomere expansion. Each of the two curves represent pooled data from more than 10 chromosomes.
Figure 3.
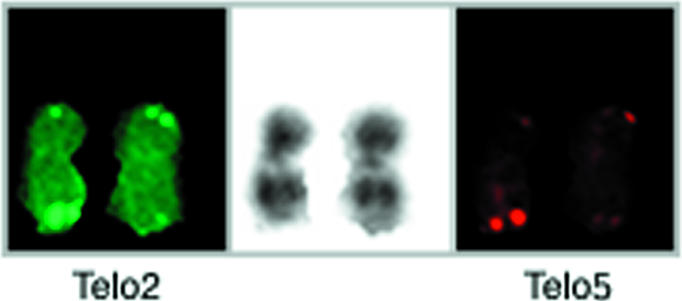
Telomeric staining on homologous chromosomes 11 by dual color PRINS with Telo2 and Telo5. Telo2-primed DNA synthesis was visualized through incorporation of fluorescein-dUTP (Boehringer Mannheim), whereas Telo5-primed DNA synthesis was visualized through incorporation of digoxigenin-dUTP (Boehringer Mannheim), and subsequent staining with rhodamine labeled anti-digoxigenin antibody (Boehringer Mannheim). The Telo2 signals are shown to the left, the Telo5 signals to the right, and the DAPI staining at the center.
The reason for this exclusive expansion of a single 11q telomere can only be speculated, but it seems reasonable to consider it as a cellular response to a critically short or lost telomere. Not all telomeres within an individual cell are of exactly the same length, and it could be that this particular 11q telomere originally was the shortest of the telomeres in this case. Alternatively, this 11q terminus could have suffered a sudden substantial loss of telomeric DNA. In a study of the dynamics of the telomere on chromosome 13q in an immortal human cell line, Murnane et al. [20] have found that such rapid losses of telomere repeats do indeed occur, though the predominant pattern is a steady reduction in telomere size of 52 bases per cell division. We reason that since only one telomere is expanded, and since this expansion is found only in a fraction of cells, it is rather unlikely to be a product of telomerase activity. Instead, it must represent telomere elongation by alternative mechanism. In this respect, it would be interesting to test for telomerase activity. Unfortunately, stem cells in the bone marrow are telomerase-positive, and the available telomerase assays all work on material extracted from a substantial number of pooled cells. Therefore, even if the cells having the 11q telomere expansion are truly telomerase-negative, the assay might still be positive due to “contaminating” stem cells. Thus, in the absence of an in situ assay for telomerase activity, such activity was not tested for on the limited cell material. At least in vitro, alternative mechanism does not occur spontaneously at any measurable frequency except after viral or chemical perturbations. In the present case, it could be speculated that the chemotherapy given to the patient provoked the alternative event.
To investigate the sequence anatomy of the elongated telomere, staining was also attempted with primers specific for repeat variants [14,16]. It appeared that the elongated 11q telomere was strongly stained by dideoxy-PRINS with the variant repeat probe, “Telo5” (AGGGTTT)5 (Figure 3). Measured on single cells, there was no obvious correlation between the increase in Telo5 signal and the increase in Telo2 signal. However, averaged over a number of cells, the increase in variant telomeric repeat signals was of the same magnitude with both probes (Figure 2), as could be suspected for a repeat domain expanded by multiplication from the preexisting repeat domain at the same locus. An obvious caveat in this is the possibility of cross-reaction between Telo5 and the main repeat. However, considering that the Telo5 sequence contains a seven-base repeat unit, whereas the main repeat contains a six-base repeat, the match over the length of the Telo5 probe is modest (16 of 35 positions). Also, the intensity of staining with Telo5 does not vary in parallel with the intensity of the staining with Telo2 on telomeres, in general [16]. Unfortunately, none of the other variant repeat probes employed was able to produce further information from the few (low quality) cells left at this point of the investigation. As it was, Telo7 gave too high a background staining to allow safe conclusions on the result, whereas neither Telo3 nor Telo8 generated visible signals on 11qter.
This only partly successful pseudo-sequencing reaction illustrates a major limitation to the use of most of the techniques employed here, namely that they work on metaphase cells, which are often available in insufficient amounts to enable a complete analysis. The general applicability of such techniques would therefore increase substantially if computer programs were derived that could extract the relevant data also from interphase nuclei.
The 11q Telomere Identity
To investigate whether translocation played a role in the telomere elongation process, the identity of the chromosome terminus harbouring the expanded telomere was tested. A translocation of a foreign chromosome end to 11q would, on its own, not be sufficient to explain our observations, as the elongated telomere was much longer than any other telomere in these cells, but could still be a contributing mechanism. Also, translocations with break-points in 11q23 are not unusual in leukemia [21]. The tip of chromosome 11q was therefore hybridized with a chromosome-specific FISH probe for 11qter (distal to 11q23). As can be seen from Figure 4, both homologues of chromosome 11 were stained similarly at their long arm terminus with this probe.
Figure 4.
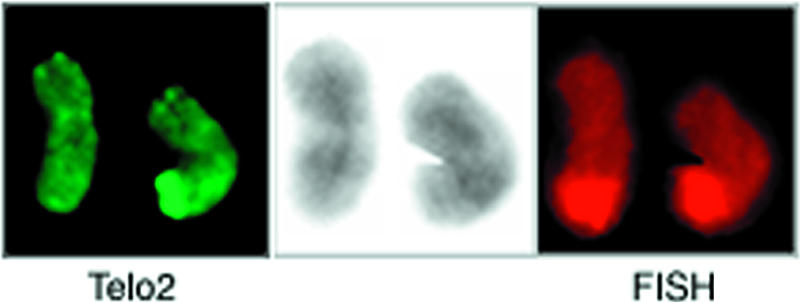
Staining of the 11q terminus by PRINS painting in a pair of chromosomes 11, one of which was positive for the 11q telomere expansion. The telomeric repeats were stained with Telo2 (left), whereas the tip of 11q was painted by FISH (right). The DAPI staining is shown at the center. The two 11q termini appeared equally stained by the FISH probe, and no site outside 11q showed apparent staining.
Relative Timing of the 11q Telomere Elongation, the Philadelphia Translocation and the Overall Telomere Shortening
As the telomere elongation and the Philadelphia translocation could well be separate events, we tried to establish the relative timing of the two events by looking for individual cells that had one of these features, but not the other. No cells were found with only the Philadelphia translocation. However, 3 out of 20 Philadelphia-negative cells in metaphase actually had the long 11q telomere. From this, it seems that the telomere elongation happened first, whereas the Philadelphia translocation was a later event. There is, thus, no evidence that the telomere expansion contributed to the transformation of the neoplastic cells.
Telomeric ratio profiles from the three Philadelphia-negative cells with an 11q telomere expansion were compared to the profiles from all other Philadelphia-negative cells, as well as to the profiles from the Philadelphia-positive cells. From this, it appeared (data not shown) that the profiles from these three cells matched the profiles of the other Philadelphia-negative cells (except for the peak at 11q), rather than the profiles of the Philadelphia-positive cells. The general telomere shortening was thus associated with the Philadelphia chromosome, rather than with the 11q expansion. Considering that the Philadelphia-positive cells with the 11q telomere expansion outnumbered what must have been the parental cells, the Philadelphia-negative cells with that expansion, 17 to 3, a significant clonal expansion of the former must have occurred, possible accounting for their shorter telomeres. In conclusion, the order of events seems to have been: 11q telomere expansion, Philadelphia translocation, overall telomere shortening.
Acknowledgements
The authors wish to thank Bent Pedersen for critically reviewing the manuscript prior to its submission.
Footnotes
This work was supported by The Danish Cancer Society and by Boehringer Mannheim grant no. 301/99/0045 from the Grant Agency of the Czech Republic (GACR).
References
- 1.Moyzis RK, Jones MD, Meyne J, Ratliff RL, Wu JR. A highly conserved repetitive DNA sequence, (TTAGGG)n, present at the telomeres of human chromosomes. Proc Natl Acad Sci USA. 1988;85:6622–6626. doi: 10.1073/pnas.85.18.6622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Greider CW, Blackburn EH. A telomeric sequence in the RNA of tetrahymena telomerase required for telomere repeat synthesis. Nature. 1989;337:331–337. doi: 10.1038/337331a0. [DOI] [PubMed] [Google Scholar]
- 3.Hastie ND, Dempster M, Dunlop MG, Thompson AM, Green DK, Allshire RG. Telomere reduction in human colorectal carcinoma and with ageing. Nature. 1990;346:866–868. doi: 10.1038/346866a0. [DOI] [PubMed] [Google Scholar]
- 4.Harley CB. Telomeres and aging. In: Blackburn EH, Greider CW, editors. Telomeres. Cold Spring Harbor Laboratory Press; 1995. pp. 247–264. [Google Scholar]
- 5.de Lange T. Tumor telomeres. In: Blackburn EH, Greider CW, editors. Telomeres. Cold Spring Harbor Laboratory Press; 1995. pp. 264–293. [Google Scholar]
- 6.Counter CM, Avilion AA, LeFeuvre CE, Stewart NG, Greider CW, Harley CB, Bacchetti S. Telomere shortening associated with chromosome instability is arrested in immortal cells which express telomerase activity. EMBO J. 1992;11:1921–1929. doi: 10.1002/j.1460-2075.1992.tb05245.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bryan TM, Englezou A, Dalla-Pozza L, Dunham MA, Reddel RR. Evidence for an alternative mechanism for maintaining telomere length in human tumors and tumor-derived cell lines. Nat Med. 1997;3:1271–1274. doi: 10.1038/nm1197-1271. [DOI] [PubMed] [Google Scholar]
- 8.Allshire RC, Dempster M, Hastie ND. Human telomeres contain at least three types of G-rich repeats distributed non-randomly. Nucleic Acids Res. 1989;17:4611–4627. doi: 10.1093/nar/17.12.4611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Broccoli D, Chong L, Oelmann S, Fernald AA, Marziliano N, van Steensel B, Kipling D, Le Beau MM, de Lange T. Comparison of the human and mouse genes encoding the telomeric protein, TRF1: chromosomal location, expression and conserved protein domains. Hum Mol Genet. 1997;6:69–76. doi: 10.1093/hmg/6.1.69. [DOI] [PubMed] [Google Scholar]
- 10.van Steensel B, deLange T. Control of telomere length by the human telomeric protein TRF1. Nature. 1997;385:740–743. doi: 10.1038/385740a0. [DOI] [PubMed] [Google Scholar]
- 11.van Steensel B, deLange T. TRF2 protects human telomeres from end-to-end fusions. Cell. 1998;92:401–413. doi: 10.1016/s0092-8674(00)80932-0. [DOI] [PubMed] [Google Scholar]
- 12.Lansdorp PM, Verwoerd NP, van de Rijke FM, Dragowska V, Little MT, Dirks RW, Raap AK, Tanke HJ. Heterogeneity in telomere length of human chromosomes. Hum Mol Genet. 1996;5:685–691. doi: 10.1093/hmg/5.5.685. [DOI] [PubMed] [Google Scholar]
- 13.Krejcí K, Koch J. Improved detection and comparative sizing of human chromosomal telomeres in situ. Chromosoma. 1998;107:198–203. doi: 10.1007/s004120050297. [DOI] [PubMed] [Google Scholar]
- 14.Koch J. PRINS: PRimed IN Situ labeling and hybridization in one step. In: Kessler C, editor. Nonradioactive Labelling and Detection of Biomolecules. 2nd edn. Springer-Verlag; 1999. in press. [Google Scholar]
- 15.Koch J, Kølvraa S, Gregersen N, Bolund L. Oligonucleotide-priming methods for the chromosome-specific labeling of alpha satellite DNA in situ. Chromosoma. 1989;98:259–265. doi: 10.1007/BF00327311. [DOI] [PubMed] [Google Scholar]
- 16.Krejcí K, Koch J. An in situ study of variant telomeric repeats in human chromosomes. Genomics. 1999;58:202–206. doi: 10.1006/geno.1999.5809. [DOI] [PubMed] [Google Scholar]
- 17.Therkelsen AJ, Nielsen A, Koch J, Hindkjær J, Kølvraa S. Staining of human telomeres with primed in situ labeling (PRINS) Cytogenet Cell Genet. 1995;68:115–118. doi: 10.1159/000133903. [DOI] [PubMed] [Google Scholar]
- 18.Koch J, Hindkjær J, Kølvraa S, Bolund L. Construction of a panel of chromosome specific oligonucleotide probes (PRINS primers) useful for the identification of individual human chromosomes in situ. Cytogenet Cell Genet. 1995;71:142–147. doi: 10.1159/000134094. [DOI] [PubMed] [Google Scholar]
- 19.Hindkjær J, Brandt CA, Koch J, Lund TB, Kølvraa S, Bolund L. Simultaneous detection of centromere specific probes and chromosome painting libraries by a combination of PRimed IN Situ labeling and chromosome painting (PRINS-painting) Chromosome Res. 1995;3:41–44. doi: 10.1007/BF00711160. [DOI] [PubMed] [Google Scholar]
- 20.Murnane JP, Sabatier L, Marder BA, Morgan WF. Telomere dynamics in an immortal human cell line. EMBO J. 1994;13:4953–4962. doi: 10.1002/j.1460-2075.1994.tb06822.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rowley JD, Diaz MO, Espinosa R, III, Patel YD, van Melle E, Ziemen S, Taillon-Miller P, Lichter P, Evans GA, Kersey JH, Ward DC, Domer PH, Le Beau MM. Mapping chromosome band 11q23 in human acute leukemia with biotinylated probes: identification of 11q23 translocation breakpoints with a yeast artificial chromosome. Proc Natl Acad Sci USA. 1990;87:9358–9362. doi: 10.1073/pnas.87.23.9358. [DOI] [PMC free article] [PubMed] [Google Scholar]