

Abstract
The idea that tumors must “escape” from immune recognition contains the implicit assumption that tumors can be destroyed by immune responses either spontaneously or as the result of immunotherapeutic intervention. Simply put, there is no need for tumor escape without immunological pressure. Here, we review evidence supporting the immune escape hypothesis and critically explore the mechanisms that may allow such escape to occur. We discuss the idea that the central engine for generating immunoresistant tumor cell variants is the genomic instability and dysregulation that is characteristic of the transformed genome. “Natural selection” of heterogeneous tumor cells results in the survival and proliferation of variants that happen to possess genetic and epigenetic traits that facilitate their growth and immune evasion. Tumor escape variants are likely to emerge after treatment with increasingly effective immunotherapies.
Progress in molecular and cellular immunology during the past two decades has advanced our understanding of tumor-host interactions and opened extraordinary opportunities for the development of antitumor immunotherapies. The identification of tumor antigens has allowed us, for the first time, to tailor therapies to specific molecular targets expressed on tumor cells. In addition, advances in recombinant biotechnology have enabled us to design and develop more effective cancer vaccines for active immunization and cellular therapy for adoptive-transfer treatments. However, consistently effective immunotherapy has not yet been developed for any type of malignancy.
Although some maintain that the lack of broad success with current strategies is due to the “escape” of tumor cells from the immunotherapies directed against them, it is also possible—and even likely—that current immune-based treatments for malignancy are simply ineffective. For example, current T cell-based immunotherapies may not be eliciting enough of the right kinds of T cells in the right places at the right time to allow them to be consistently effective. To fully understand the potential role played by tumor escape in the failure of immunotherapeutic treatments in cancer patients, it is worth briefly discussing the basic principles of immunosurveillance and its potential role in shaping tumor phenotypes.
Assumptions of the tumor escape hypothesis
It is not often explicitly stated that the tumor escape hypothesis assumes that if left unchecked, the immune system would indeed spontaneously attack and destroy tumor. Simply put, immune escape implies immune attack. A Review by Schreiber and colleagues in this issue examines the evidence for immunosurveillance and attempts to resolve the long-standing controversy over its role in the early stages of tumor development. Schreiber’s group has demonstrated the existence of an interferon-γ (IFN-γ)-dependent extrinsic tumor suppressor system in mice1. However, it is not clear how large a role this plays in the immunosurveillance of tumors because only a minority of human tumor cell lines examined in this study exhibited a permanent and selective IFN-γ insensitivity1.
Two studies have examined the potential roles of natural killer (NK) and γδT cells bearing the stimulatory lectin-like receptor NKG2D in immunosurveillance. The first study explored antitumor immune responses elicited by tumors cells transduced with the NKG2D ligands, retinoic acid early transcript 1b (Rae-1b) or H-602 and found that NK and/or CD8+ T cells mediated potent antitumor immunity2. The study used NKG2D ligand-transduced cell lines that expressed high amounts of Rae-1b or H-60; NKG2D ligand expression in most spontaneous tumors may be much lower. Nevertheless, these findings demonstrated a possible mechanism of immunosurveillance that may involve NK cells. A second study uncovered a possible role for NKG2D+γδ T cells at the very early stages of tumor development3.
A note of caution is useful when interpreting these elegant mouse studies: there may be fundamental differences between the development of tumors injected into a mouse or induced by chemical carcinogens compared to naturally occurring spontaneous tumors in humans. For example, the act of injection itself and the presence of dead tumor cells as well as tissue-culture contaminants, such as fetal bovine serum in the inoculum, may induce local inflammatory reactions. In tumors induced by painting chemical carcinogens onto the skin of highly susceptible mice, Rae-1b and H-60 expression may be up-regulated, resulting in their rejection by γδ T cells. In addition, whereas carcinogenesis in humans may be a slow process that occurs over several years, mouse experiments use high doses of carcinogens, which cause transformation in just weeks or months. At these doses, carcinogens may cause massive and rapid mutagenesis, resulting in greatly increased number of neoantigens that may alert the immune system.
IFN-γ, perforin and tumor necrosis factor (TNF)-related apoptosis-inducing ligand (TRAIL) act as effector molecules in immune surveillance for the prevention of tumor development. Mice that are deficient in lymphocytes and/or in the IFN-γ signaling pathway have a much higher incidence of carcinogen-induced sarcomas, lymphoma and spontaneous epithelial tumors4, 5. Other studies have shown that in perforin-deficient mice, there is a higher rate of spontaneous lymphoma and lung adenocarcinoma4, 6. Upon transplantation into syngeneic wild-type mice, these lymphoma cells are promptly rejected by cytotoxic T lymphocyte (CTL)-mediated activity. Inhibition of TRAIL (by blocking antibody) promotes carcinogen-induced tumor development in mice; large numbers of these tumors are TRAIL-sensitive, which is something rarely seen in tumors developed in wild-type mice7.
Thus, evidence exists, in some models and under some conditions, that immunosurveillance can play an active role in suppressing the growth of very early tumors. In this paradigm, when tumors do successfully grow, they are thought of as having “escaped” from this immunosurveillance; these early surveillance mechanisms are viewed as “shaping” the tumor’s immunological phenotype. Another model that explains why established tumors grow and do not undergo immune-mediated rejection is that tumors function as immunologically normal tissue. In this paradigm, tumor cells appear immunologically as healthy growing cells that do not send out danger signals to activate the immune system because they express neither microbial immune-recognition patterns nor release distress signals to alarm the innate immune cells8. Tumor histology and phenotype may determine whether early tumors ultimately grow as a result of stealth and nonrecognition or as the result of escape and immunological sculpting.
Later in the natural history of a tumor, during the progressive growth phase, tumors may become more immune-activating for a variety of reasons (Fig. 1). Tumors can damage or disrupt surrounding tissue or trigger a stress response when they outstrip their oxygen and nutrient supplies. These processes can cause pH imbalance that results from metabolic disturbance, generation of reactive oxygen species (ROS), up-regulation of stress protective factors—such as heat shock proteins—and death by necrosis or apoptosis. Also, as tumors grow progressively, dysregulated genetic and epigenetic events lead to the expression of large numbers of neoantigens. One study estimated that the average malignancy contained more than 10,000 mutations9. All these factors may act as alarm signals to recruit and activate local innate immune cells such as dendritic cells (DCs), macrophages, neutrophils and NK cells, which, in turn, activate T cells to mount an adaptive immune response against tumors.
Figure 1. Activation versus suppression during tumor progression.

Immune activation during early tumor progression may be triggered by the expression of neoantigens. In addition, the progressive growth of tumors may be associated with the invasion and destruction of surrounding normal tissues. Other factors that may cause immune activation include generation of heat shock proteins (Hsps), which result from cellular stress, and ROS such as OH− and H2O2. Conversely, immune activation and function may be hampered by a lack of appropriate costimulation, the presence of immunosuppressive cytokines (for example, VEGF, IL-10 and TGF-β), and the impact of immunoregulatory cells such as CD4+CD25+ T cells and NKT cells. As tumors grow, these events occur in the presence of massive apoptosis and necrosis of tumor cells, infiltrating immune cells and surrounding stromal cells. The fate of tumors may be dictated by the net effect of immune activation and immune inhibition.
Whatever the roles of immunosurveillance, immune attack and immune escape in humans, it is clear that once tumors are clinically detectable, spontaneous regression is exceedingly rare for the vast majority of histologies. Once solid tumors become established, vascularized and clinically detectable, it is simply not clear—given the available evidence—whether they need to “escape” from immune recognition; this is because it is unclear whether most tumors elicit immune responses that can cause tumor destruction8. Indeed, some observations contradict the idea that the immune system spontaneously mounts lethal attacks against tumor cells. If cycles of immune pressure and tumor escape were operative during tumor development, one might expect to observe (either by examination or by imaging modalities) progressive tumor growth that was interspersed with one or more periods of contraction. Ongoing immunological “shaping” or “sculpting” of tumors might be expected to result in the destruction of sensitive cells followed by proliferation of immunoresistant cells that would, in turn, form the bulk of a new tumor emerging from the bed of the old tumor (Fig. 2). However, solid tumors generally do not have growth curves with evidence of significant drops or depressions: tumors simply grow, and then grow larger.
Figure 2. Natural selection of tumor variants in the generation of “tumor escape” phenotypes.
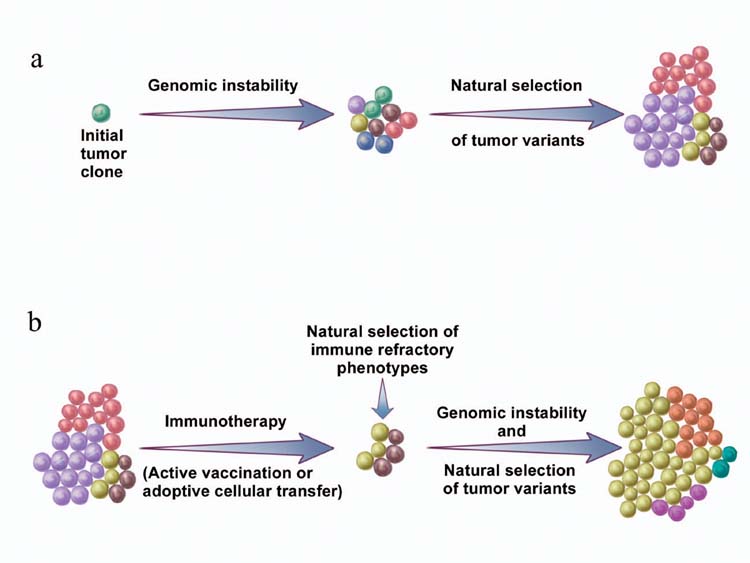
(a) Genomic instability gives rise to genetic diversity in tumors. Natural selection of tumor variants occurs by differential propagation of tumor subclones in their microenvironment. (b) The same concept also applies to tumor growth after effective immunotherapy.
On the other hand, it is also possible that selection is taking place at a cellular level, especially during early tumor development, as discussed above. Ongoing selection of tumor variants on a microscopic or cellular level would not necessarily result in gross or macroscopic changes, such as those easily seen histologically or with commonly used imaging modalities. Similarly, a lack of inflammation at the site of solid tumors may be interpreted by some to cast further doubt on the hypotheses of spontaneous immune attack and immune escape. Although immune cells can be observed in or around tumors, spontaneous local inflammation in an uninfected tumor is generally not seen clinically or histologically. Again, however, it is possible that this inflammation is occurring chronically and on a microscopic level. Thus, there is little direct evidence in mice or humans to support the concept of tumor “shaping” or “sculpting” by the unmanipulated immune system once tumors are vascularized and established. Nevertheless, these hypothesized processes may well turn out to be important upon further investigation.
The natural selection of tumor cell variants
The setting of partially successful antitumor immunotherapy is one scenario that might be expected to result in tumor escape. One of the most important factors that favors the survival and propagation of evolving organisms is genetic diversity. It is interesting to view the survival of cancer cells in a Darwinian light, where the survival of individual cancer cells is based on heritable variations and differential survival associated with certain genotypes. Natural selection is a passive process by which an organism has a survival advantage simply by possessing certain beneficial genes. Genomic instability, an inherent property shared by all tumors, gives rise to genetic diversity, as manifested by the great degree of heterogeneity seen in human tumors.
The law of natural selection is at work when cancer cells that possess genetic and epigenetic traits, which are beneficial to their survival and/or proliferation, have a growth advantage over other cells. The outcome of this passive process is determined by multiple factors in the tumor environment, such as growth factors, nutrient supply and immune pressure. Genomic instability simply creates a vast repertoire of tumor cells that will be selected according to environmental factors. Therefore, terms such as “tumor evasion” or “tumor escape” are misnomers because evasion and escape imply active acquisition of certain characteristics or phenotypes, rather than the differential propagation of tumor subclones (Fig. 2a). The same concept also applies to tumor growth in the face of an effective immunotherapy (Fig. 2b).
Loss or down-regulation of HLA class I antigens
It is certainly well documented that tumor cells can indeed lose HLA class I molecules through a panoply of mechanisms. Descriptions of HLA loss causing immune escape have a great deal of intuitive appeal, but are nevertheless correlative and indirect. There is little controlled evidence in humans or animals that loss of major histocompatibility complex (MHC) class I molecules actually leads to tumor escape or immunoresistance in the unmanipulated host. Indeed, loss of H-2 expression in transporter associated with antigen processing 1 (TAP-1)- or low-molecular weight protein 2 (LMP-2)-deficient mice does not increase the onset or incidence of a variety of spontaneous tumors10. On the other hand, some recurring tumors lose cell-surface MHC class I both in mice with pre-existing immunity induced by immunization with B7-1-transfected tumor cells11 and in humans after partial responses to T cell-based immunotherapy12. Decreased or absent HLA class I expression is associated with invasive and metastatic lesions13. Total loss of HLA class I expression is not uncommon in many tumors, including melanoma, colorectal carcinoma and prostate adenocarcinoma14. In breast carcinoma, the frequency of total HLA class I loss is >50%15. Thus, it is possible that in antigen-loss, HLA-loss and MHC class I processing-defective variants, the loss or down-regulation of HLA class I antigens occurs as a result of immunological “sculpting” of early tumor lesions (see the Review by Schreiber and colleagues in this issue) or escape from immune attack later in tumor development.
Some mechanisms that can result in the loss or down-regulation of HLA class I expression are shown (Fig. 3). The mechanisms that underlie total loss of HLA class I include mutations in one copy of the β2-microglobulin gene in association with loss of heterozygosity (LOH) involving the second allele16. The loss of β2-microglobulin has been observed in patients experiencing objective partial responses after T cell-based immunotherapy12. We recently observed loss of β2-microglobulin in tumor cells from two patients: one patient had an apparently complete response to immunotherapy for nearly a decade; the other had experienced an extraordinary response after adoptive transfer of tumor-specific T cells, then had a resurgence of tumor (unpublished observations), although a causal relationship in such clinical scenarios is difficult to demonstrate unequivocally. Other causes of total HLA class I down-regulation include defects in MHC genes and in the antigen processing and transport pathway. Down-regulation of the proteosome multicatalytic complex subunits LMP-2 and LMP-7 and of peptide transporters TAP-1 and TAP-2 have been reported in tumor histologies that include small cell lung carcinoma17, non-small cell lung cancer (NSCLC)18, prostate carcinoma19 and renal cell carcinoma20. In these situations, HLA class I can often be up-regulated by treatment with IFN-γ.
Figure 3. Molecular mechanisms responsible for HLA class I deficiency.

Tumor antigens are processed in the proteosome and generate peptides that are transported by TAP to the endoplasmic reticulum (ER); there they bind to certain HLA class I heavy chain in association with β2-microglobulin. The peptide-HLA complexes are then transported through the Golgi to the cell surface. Several different defects are associated with tumor “escape” from immune recognition: (a) defects in components of the antigen-processing machinery (such as LMP-2 and LMP-7) in the proteosome result in HLA class I down-regulation; (b) defects in peptide transporters TAP-1 or TAP-2 cause HLA class I down-regulation; and (c) LOH on chromosome 6 causes HLA class I haplotype loss. Defects in the transcriptional regulation of the HLA class I gene result in HLA class I locus loss. Point mutations or gene deletions involving the HLA class I heavy chain result in HLA class I allelic loss. (d) β2-microglobulin (β2M) mutation or deletion results in total loss of HLA class I.
Tumors can also express selective loss of HLA class I haplotype, locus or allele. HLA haplotype loss can be due to LOH on chromosome 621. Several mechanisms are involved in locus down-regulation, which is more frequent with HLA-B than HLA-A antigens. In melanomas, c-Myc oncogene overexpression correlates with selective HLA-B locus down-regulation22. Loss of transcription factor binding to locus-specific regulatory elements can induce HLA-B locus down-regulation in colon carcinoma cells23. In melanoma, the gene products of the HLA-C locus are often expressed poorly or not at all24. The defects underlying HLA class I allele-specific loss include mutations in the genes encoding HLA class I heavy chain25.
Descriptions of partial or complete losses of HLA class I as mechanisms of immune escape often fail to consider an increased susceptibility to NK cell lysis, which is a direct consequence of such a loss26. Why do HLA class I loss tumor cell variants continue to grow and are not destroyed by NK cells? NK cells express activating receptors, such as NKG2D, which bind to stress-induced ligands (MICA and MICB) that can be up-regulated in a variety of tumors. Activation of NK cells through this signaling pathway can overcome the inhibitory effect of HLA class I-binding receptors (KIRs)27, 28. Thus, although HLA class I-negative tumors should be susceptible to NK killing, the loss or down-regulation of MICA or MICB expression by actively growing tumors represents a potential escape strategy, but one that has not yet been demonstrated in human tumors29.
Alternative explanations for why tumor cells that have lost HLA class I are not destroyed by NK cells may be derived from the activation-inhibition model (Fig. 1). NK cells are rapidly activated in the presence of stimulatory factors such as interleukin 12 (IL-12), IL-2, IL-15 or type 1 IFNs in response to inflammatory conditions associated with microbial infection. In “sterile” environments, as seen with tumors or transplantation, such stimulatory factors may not be readily available, and the cross-talk between DCs and resting NK cells that induces NK cell activation may not occur30. In addition, a lack of expression of costimulatory molecules—such as B7-1 (also known as CD80), B7-2 (also known as CD86), CD40 and CD70—by tumors may also hinder optimal NK cell activation via CD28 and CD27 costimulation pathways31, 32, 33. It is possible that in some situations, tumors may produce immunomodulatory cytokines, such as transforming growth factor-β (TGF-β) or macrophage migration inhibitory factor (MIF)34, which can directly inhibit NK cell activation and function.
Loss of tumor antigens and immunodominance
Loss of surface antigen expression can occur independently of the dysregulation of HLA class I expression. It is well documented that tumor antigen expression is heterogeneous, even within the same tumor. Decreased expression of melanoma-melanocyte differentiation antigens (MDAs) such as gp100, melanoma antigen recognized by T cells 1 (MART1) and tyrosinase is associated with disease progression35. In one study, cells were MART1+ in 100% of stage I lesions but only 75% of stage IV lesions36. Decreased antigen expression has also been found in residual tumors after peptide vaccination37, 38. With a gp100 peptide (209-2M) vaccine, there was a decrease in gp100 expression in tumors after ztreatment (47% versus 32%), whereas expression of MART1 was unchanged (54% versus 54%)39. In addition, the amount of tumor antigen expressed may also be important for recognition40. Again, these are correlative studies and fully controlled human studies can be difficult or impossible to come by. It seems likely, however, that as T cell-based tumor immunotherapy becomes stronger, escape mechanisms such as antigen loss are likely to become more prominent.
The exact mechanisms that control the down-regulation of tumor antigens are not known in most cases; however, the propagation of such antigen loss variants may be facilitated by epitope immunodominance41. The phenomenon of immunodominance may be thought of as the preferential immunodetection of one or a few epitopes among many expressed on a given target. The theory of immunodominance, as it relates to tumor escape, predicts that one of the ways that antigen-loss variants within a tumor are shielded from immune pressure is that the parental tumor cells that carry the immunodominant epitope serve as a red flag for immune attack, thereby diverting attention from the tumor variants. Once the parental cells are eliminated, a new hierarchy is established among the variant subpopulations, and formerly immunorecessive epitopes become dominant41. A tumor variant that has lost the restricting HLA class I allele while retaining the immunodominant antigen could cross-present this antigen to CD8+ CTLs by DCs and maintain an immunodominant response to a “phantom” target at the expense of more appropriate and effective responses to other antigens41.
Defective death receptor signaling
Two death receptor ligands that play a role in immune surveillance against tumor development are Fas ligand (FasL) and TRAIL7, 42, 43, 44. Defective death receptor signaling is a mechanism that may contribute to the survival and proliferation of tumor cells. Death receptors have cytoplasmic sequences called “death domains” that are essential for the transmission of apoptotic signals via the caspase cascade. For example, upon engagement of the death receptor Fas by its ligand, Fas-associated death domain (FADD) engages caspase-8, which, in turn, autoactivates itself and cleaves downstream substrates such as caspase-3, caspase-6 and caspase-745. The caspase-8 inhibitor cellular FLICE-inhibitory protein (cFLIP) is expressed in various tumors. In these cases, cFLIP may render tumor cells resistant to death receptor-mediated apoptosis46. Increased expression of cFLIP by tumor cells may also contribute to immunoresistance to T cells in vivo47.
Down-regulation or loss of Fas expression in tumors may also contribute to their resistance to apoptosis. Missense mutations and loss of the gene encoding Fas have been identified in multiple myeloma48, non-Hodgkin’s lymphoma (NHL)49 and melanoma50. These mutations disrupt Fas signaling, resulting in a loss-of-function death receptor. In addition, mutations of the genes in the proximal pathways downstream of Fas signaling have been also found in NSCLC and lymphoma51, 52. These include inactivating mutations of FADD and caspase-10. Most were detected in metastatic lesions of NSCLC. Besides defective death receptor signaling, tumors can also block CTL-mediated cytotoxicity via the perforin pathway by overexpressing PI-9 (also known as SPI-6), a serine protease inhibitor that inactivates granzyme B53.
In the case of TRAIL-mediated apoptosis, loss of expression of all TRAIL receptors by one of the following causes: chromosomal loss; loss of caspase-8 by chromosomal loss or mutation; no signaling from death-inducing signaling complex (DISC) due to FADD mutation, X-linked inhibitor of apoptosis protein (XIAP) inhibition of caspase-3 and low second mitochondria-derived activator of caspase or direct IAP-binding protein with low pI SMAC (also known as Diablo) release; and low expression of death receptors by post-transcriptional regulation are associated with tumor resistance to TRAIL-mediated apoptosis54. Thus in tumor cells, there may be defects at multiple sites in the death receptor pathways that can favor tumor escape.
Lack of costimulation
As discussed above, most tumors seem to grow in a noninflammatory microenvironment that is not conducive to immune activation. Histologically, tumors generally coexist innocuously with normal tissues, apparently without giving or inducing immune-activating signals, especially during the early stage of growth. Recognition of tumor antigens by DCs under these conditions will not lead to DC activation and maturation. In addition, lack of expression of costimulatory molecules by tumor cells may lead to T cell anergy55 and suboptimal activation of NK cells. In an experimental setting, insertion of genes encoding B7-1, B7-2 or both into tumors generally increases the immunogenicity of those tumors but does not necessarily lead to regression56.
Immunosuppressive cytokines
Activation or inhibition of T cells also depends on the presence or absence of cytokines in their immediate microenvironment. Tumor cells produce a variety of cytokines and chemokines that can negatively effect maturation and function of immune cells. Vascular endothelial growth factor (VEGF) is a cytokine that is secreted by most tumors57. In vitro studies show VEGF inhibits DC differentiation and maturation through suppression of the transcription factor NF-κB in hematopoietic stem cells58. Immunohistochemical staining of gastric carcinoma tissues revealed an inverse correlation between the density of DCs and VEGF expression. This finding was also associated with poor prognosis59. In patients with lung, head and neck, and breast cancers, there was a decrease in the function and number of mature DCs, which was associated with increased plasma concentrations of VEGF60. Besides VEGF, increased concentrations of another cytokine, IL-10, are frequently detected in the serum of patients with cancer. IL-10 can exert an inhibitory effect on DC differentiation from stem cell precursors61. In addition, maturation and the functional status of DCs are also compromised by IL-10. This cytokine also inhibits antigen presentation, IL-12 production and induction of T helper type 1 responses in vivo62, 63. IL-10 also enhances spontaneous DC apoptosis64 as well as susceptibility to autologous NK cell lysis65. IL-10 may protect tumor cells from CTLs by down-regulation of HLA classes I and II and ICAM-1 (intercellular adhesion molecule 1)66. The loss of HLA class I expression could also be due to IL-10-mediated down-regulation of TAP1 and TAP2 proteins in tumor cells67, 68.
The proinflammatory factor prostaglandin E2 (PGE2) is another cytokine that is expressed by tumors as a result of enhanced expression of the enzyme cyclooxygenase 2, which is the rate-limiting enzyme for PGE2 synthesis, in multiple human tumors69, 70, 71. PGE2 increases the production of IL-10 by lymphocytes and macrophages and inhibits IL-12 production by macrophages72. High concentrations of TGF-β are also frequently found in cancer patients and are associated with disease progression73 and poor responses to immunotherapy74. In addition to potentially being produced by some tumor cell lines, TGF-β may also be released by cells dying apoptotically75. TGF-β inhibits the activation, proliferation and activity of lymphocytes in vivo76. One point to consider is that tumors may not necessarily produce these cytokines as escape mechanisms: the hypothesized immunosuppressive functions may be mere side-effects of the angiogenic and growth factor functions of these cytokines.
Apoptosis of activated T cells
One of the more controversial mechanisms of tumor escape is the expression of death receptor ligands by tumor cells. A variety of cancer cells express functional FasL, which induces apoptosis of Fas+-susceptible target cells. These include lung carcinoma77, melanoma78, colon carcinoma79 and hepatocellular carcinoma80. The most recent study on this controversial topic asserts that FasL is coexpressed with melanosomal and lysosomal markers in multivesicular bodies in human melanoma cells81. Some of the data concerning FasL expression on tumor cells have been questioned due to a lack of appropriate negative controls and technical concerns, such as the use of nonspecific antibodies, non-intron-spanning polymerase chain reaction primers without proper controls and functional assays that use the Jurkat cell line, which can themselves be induced to express FasL82, 83. In our own work exploring FasL expression on human melanoma cells, we found no evidence of significant expression either at the mRNA or protein level. In addition, there was no killing of Fas+-susceptible targets by melanoma cells in controlled functional assays84. Another level of complexity in the role of FasL may be that it is actually proinflammatory in some circumstances. In fact, all well controlled in vivo experiments with membrane-bound FasL-transfected tumor cells show accelerated rejection accompanied by neutrophil infiltration85, 86, 87. However, the activity of FasL may be modulated if it is processed to a secreted soluble form or if it is elaborated in the presence or absence of certain cytokines within the tumor microenvironment. For example, TGF-β can regulate the proinflammatory effects of FasL88. The most crucial role of Fas-FasL interactions in the tumor setting may be the induction of activation-induced cell death (AICD) of antitumor T cells. Upon activation by tumor antigen recognition, T cells express high amounts of FasL, which induces apoptosis of these T cells (“suicide”) and between T cells (“fratricide”)84, 89 (Fig. 4).
Figure 4. Alternative models for the induction of FasL-mediated T cell death after encounter with tumor cells.
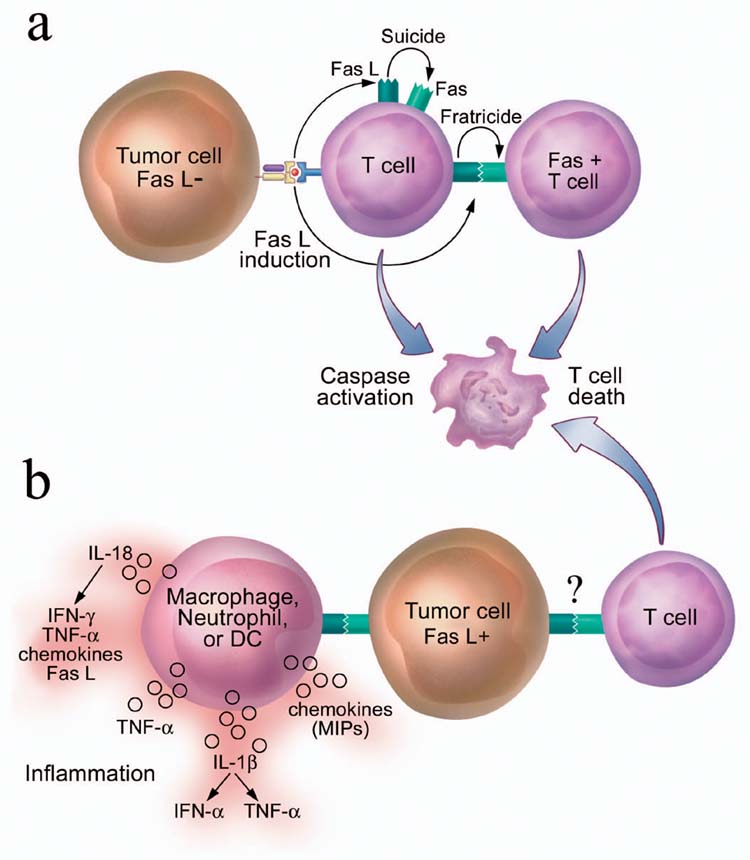
(a) AICD of T cells after recognition of tumor cells. Tumor recognition leads to activation of T cells and up-regulation of Fas and FasL on T cell surface, which results in the T cell killing of itself (“suicide”) and of other T cells (“fratricide”). (b) Proposed tumor “counterattack” model. Tumor cells express functional FasL and kill infiltrating Fas-expressing T cells via Fas-FasL binding, which leads to tumor escape. However, ligation of Fas expressed on innate immune cells such as neutrophils, macrophages and immature DCs by FasL expressed on tumor cells may also lead to release of multiple proinflammatory cytokines and chemokines, setting the stage for tumor rejection.
Other TNF family ligands, such as TRAIL, are expressed by tumors, but there is no convincing evidence that they play a major role in the induction of tumor-specific T cell apoptosis in human cancers90. Tumor-associated B7-H1 ligand also induces T cell apoptosis91. However, this study relied heavily on artificial models of B7-H1-transfected tumor cells that expressed high amounts of B7-H1. It remains unclear whether the relatively small increases in specific T cell apoptosis over background caused by endogenously expressed B7-H1 will be enough to enable human tumors to evade immune recognition.
The role of suppressor T cells
Immunoregulatory CD4+ CD25+ T cells (also known as Treg, TH3 and TR1 cells) control key aspects of immunological tolerance to self-antigens92, 93. Removal of these cells, which constitute 5–10% of CD4+ T cells in humans and rodents, induces autoimmune disease in the ovaries, thyroid gland, salivary glands and the mucosal linings of the stomach and small intestine and accelerate diabetes in nonobsese diabetic mice. Depletion of CD4+CD25+ cells plus injection of an antibody capable of blocking CTLA-4 (cytotoxic T lymphocyte antigen 4), enhanced reactivity to a known tumor-associated antigen (TAA)94. However, based on the available evidence from experimental mouse tumor models, it seems that simply blocking or even eliminating T regulatory cell function will not be enough to treat established tumors95.
Identification of the glucocorticoid-induced TNF receptor family-related gene (GITR, also known as TNFRSF18) expressed on T regulatory cells might afford new therapeutic opportunities, as stimulation of GITR with an activating antibody reverses CD4+CD25+ T cell-mediated suppression96, 97. Another possible therapeutic intervention could be blockade of signaling through the molecular pair of TNF-related activation-induced cytokine (TRANCE) and receptor-activator of NF-κ B (RANK), as these signals are involved in the generation and activation of CD4+CD25+ T regulatory cells98. In addition to CD4+CD25+ T cells, NKT cells can exert immunoinhibitory effects on tumor immunity. CD4+ NKT cells inhibit effective CTL-mediated tumor rejection by IL-13 via the IL-4R-STAT6 (interleukin 4 receptor-signal transducers and activators of transcription 6) pathway99. Another study has identified CD4+DX5+ NKT suppressor cells that regulate the growth of ultraviolet-induced skin cancers and mediate antigen-specific immune suppression100; thus, targeting NKT cells could be another therapeutic option. The challenge for immunotherapists now is to discover how to use an understanding of T regulatory function to enhance other immunotherapeutic interventions.
CONCLUSION
Despite recent progress in tumor immunobiology and technical advances in the field of tumor immunotherapy, current immunotherapeutic strategies used in the treatment of patients with cancer have not been successful in most cases. It is not clear whether tumor escape accounts for these failures or whether lack of observed tumor regression is due to inadequacies of the immunotherapies themselves.
The balance of immune activation and immune inhibition must favor the former if tumors are to come under immune pressure. However, even in some instances where effective antitumor responses can be achieved with immunotherapy, tumors often recur. It seems likely that as the tumor immunologist’s armamentarium becomes more powerful and immunotherapies become more effective, the selection of tumor variants with immunoresistant phenotypes will be observed with greater frequency. Tumor escape may ultimately thwart some dramatic responses to immunotherapy. Indeed, some tumors that recur after successful immunotherapy often possess immunoresistant phenotypes.
The challenge for the tumor immunologists now is to understand the mechanisms by which tumors become refractory to immune modulation. The optimal immunotherapy strategy may be to achieve three things concurrently: provision of appropriate immune activating signals, elimination of inhibitory factors and avoidance of the emergence of immunoresistant phenotypes. The latter might be achieved with a combination of modalities sustained for a long enough period of time to complete tumor destruction.
References
- 1.Kaplan DH, et al. Demonstration of an interferon γ-dependent tumor surveillance system in immunocompetent mice. Proc Natl Acad Sci USA. 1998;95:7556–7561. doi: 10.1073/pnas.95.13.7556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Diefenbach A, Jensen ER, Jamieson AM, Raulet DH. Rae1 and H60 ligands of the NKG2D receptor stimulate tumour immunity. Nature. 2001;413:165–171. doi: 10.1038/35093109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Girardi M, et al. Regulation of cutaneous malignancy by γδT cells. Science. 2001;294:605–609. [PubMed] [Google Scholar]
- 4.Street SE, Trapani JA, MacGregor D, Smyth MJ. Suppression of lymphoma and epithelial malignancies effected by interferon γ. J Exp Med. 2002;196:129–134. doi: 10.1084/jem.20020063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shankaran V, et al. IFN γ and lymphocytes prevent primary tumour development and shape tumour immunogenicity. Nature. 2001;410:1107–1111. doi: 10.1038/35074122. [DOI] [PubMed] [Google Scholar]
- 6.Smyth MJ, et al. Perforin-mediated cytotoxicity is critical for surveillance of spontaneous lymphoma. J Exp Med. 2000;192:755–760. doi: 10.1084/jem.192.5.755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Takeda K, et al. Critical role for tumor necrosis factor-related apoptosis-inducing ligand in immune surveillance against tumor development. J Exp Med. 2002;195:161–169. doi: 10.1084/jem.20011171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Restifo NP, et al. Assumptions of the tumor ‘escape’ hypothesis. Semin Cancer Biol. 2002;12:81–86. doi: 10.1006/scbi.2001.0399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stoler DL, et al. The onset and extent of genomic instability in sporadic colorectal tumor progression. Proc Natl Acad Sci USA. 1999;96:15121–15126. doi: 10.1073/pnas.96.26.15121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Johnsen AK, et al. Systemic deficits in transporter for antigen presentation (TAP)-1 or proteasome subunit LMP2 have little or no effect on tumor incidence. Int J Cancer. 2001;91:366–372. doi: 10.1002/1097-0215(200002)9999:9999<::aid-ijc1056>3.0.co;2-k. [DOI] [PubMed] [Google Scholar]
- 11.Zheng P, Sarma S, Guo Y, Liu Y. Two mechanisms for tumor evasion of preexisting cytotoxic T-cell responses: lessons from recurrent tumors. Cancer Res. 1999;59:3461–3467. [PubMed] [Google Scholar]
- 12.Restifo NP, et al. Loss of functional β2-microglobulin in metastatic melanomas from five patients receiving immunotherapy. J Natl Cancer Inst. 1996;88:100–108. doi: 10.1093/jnci/88.2.100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Garrido F, et al. Implications for immunosurveillance of altered HLA class I phenotypes in human tumours. Immunol Today. 1997;18:89–95. doi: 10.1016/s0167-5699(96)10075-x. [DOI] [PubMed] [Google Scholar]
- 14.Algarra I, Collado A, Garrido F. Altered MHC class I antigens in tumors. Int J Clin Lab Res. 1997;27:95–102. doi: 10.1007/BF02912442. [DOI] [PubMed] [Google Scholar]
- 15.Cabrera T, et al. High frequency of altered HLA class I phenotypes in invasive breast carcinomas. Hum Immunol. 1996;50:127–134. doi: 10.1016/0198-8859(96)00145-0. [DOI] [PubMed] [Google Scholar]
- 16.Hicklin DJ, et al. β2-microglobulin mutations, HLA class I antigen loss, and tumor progression in melanoma. J Clin Invest. 1998;101:2720–2729. doi: 10.1172/JCI498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Restifo NP, et al. Identification of human cancers deficient in antigen processing. J Exp Med. 1993;177:265–272. doi: 10.1084/jem.177.2.265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Korkolopoulou P, Kaklamanis L, Pezzella F, Harris AL, Gatter KC. Loss of antigen-presenting molecules (MHC class I and TAP-1) in lung cancer. Br J Cancer. 1996;73:148–153. doi: 10.1038/bjc.1996.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sanda MG, et al. Molecular characterization of defective antigen processing in human prostate cancer. J Natl Cancer Inst. 1995;87:280–285. doi: 10.1093/jnci/87.4.280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Seliger B, et al. Expression and function of the peptide transporters in escape variants of human renal cell carcinomas. Exp Hematol. 1997;25:608–614. [PubMed] [Google Scholar]
- 21.Ramal LM, et al. Molecular strategies to define HLA haplotype loss in microdissected tumor cells. Hum Immunol. 2000;61:1001–1012. doi: 10.1016/s0198-8859(00)00171-3. [DOI] [PubMed] [Google Scholar]
- 22.Versteeg R, et al. Suppression of class I human histocompatibility leukocyte antigen by c-myc is locus specific. J Exp Med. 1989;170:621–635. doi: 10.1084/jem.170.3.621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Soong TW, Hui KM. Locus-specific transcriptional control of HLA genes. J Immunol. 1992;149:2008–2020. [PubMed] [Google Scholar]
- 24.Marincola FM, et al. Locus-specific analysis of human leukocyte antigen class I expression in melanoma cell lines. J Immunother Emphasis Tumor Immunol. 1994;16:13–23. doi: 10.1097/00002371-199407000-00002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Koopman LA, van Der S, Giphart MJ, Fleuren GJ. Human leukocyte antigen class I gene mutations in cervical cancer. J Natl Cancer Inst. 1999;91:1669–1677. doi: 10.1093/jnci/91.19.1669. [DOI] [PubMed] [Google Scholar]
- 26.Porgador A, Mandelboim O, Restifo NP, Strominger JL. Natural killer cell lines kill autologous β2-microglobulin-deficient melanoma cells: implications for cancer immunotherapy. Proc Natl Acad Sci USA. 1997;94:13140–13145. doi: 10.1073/pnas.94.24.13140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bauer S, et al. Activation of NK cells and T cells by NKG2D, a receptor for stress-inducible MICA. Science. 1999;285:727–729. doi: 10.1126/science.285.5428.727. [DOI] [PubMed] [Google Scholar]
- 28.Groh V, et al. Broad tumor-associated expression and recognition by tumor-derived γδT cells of MICA and MICB. Proc Natl Acad Sci USA. 1999;96:6879–6884. doi: 10.1073/pnas.96.12.6879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Garrido F, Algarra I. MHC antigens and tumor escape from immune surveillance. Adv Cancer Res. 2001;83:117–158. doi: 10.1016/s0065-230x(01)83005-0. [DOI] [PubMed] [Google Scholar]
- 30.Gerosa F, et al. Reciprocal activating interaction between natural killer cells and dendritic cells. J Exp Med. 2002;195:327–333. doi: 10.1084/jem.20010938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Galea-Lauri J, et al. Expression of a variant of CD28 on a subpopulation of human NK cells: implications for B7-mediated stimulation of NK cells. J Immunol. 1999;163:62–70. [PubMed] [Google Scholar]
- 32.Carbone E, et al. A new mechanism of NK cell cytotoxicity activation: the CD40-CD40 ligand interaction. J Exp Med. 1997;185:2053–2060. doi: 10.1084/jem.185.12.2053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Takeda K, et al. CD27-mediated activation of murine NK cells. J Immunol. 2000;164:1741–1745. doi: 10.4049/jimmunol.164.4.1741. [DOI] [PubMed] [Google Scholar]
- 34.Apte RS, Mayhew E, Niederkorn JY. Local inhibition of natural killer cell activity promotes the progressive growth of intraocular tumors. Invest Ophthalmol Vis Sci. 1997;38:1277–1282. [PubMed] [Google Scholar]
- 35.de Vries TJ, et al. Heterogeneous expression of immunotherapy candidate proteins gp100, MART-1, and tyrosinase in human melanoma cell lines and in human melanocytic lesions. Cancer Res. 1997;57:3223–3229. [PubMed] [Google Scholar]
- 36.Hofbauer GF, Kamarashev J, Geertsen R, Boni R, Dummer R. Melan A/MART-1 immunoreactivity in formalin-fixed paraffin-embedded primary and metastatic melanoma: frequency and distribution. Melanoma Res. 1998;8:337–343. doi: 10.1097/00008390-199808000-00007. [DOI] [PubMed] [Google Scholar]
- 37.Jager E, et al. Inverse relationship of melanocyte differentiation antigen expression in melanoma tissues and CD8+ cytotoxic-T-cell responses: evidence for immunoselection of antigen-loss variants in vivo. Int J Cancer. 1996;66:470–476. doi: 10.1002/(SICI)1097-0215(19960516)66:4<470::AID-IJC10>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 38.Lee KH, et al. Functional dissociation between local and systemic immune response during anti-melanoma peptide vaccination. J Immunol. 1998;161:4183–4194. [PubMed] [Google Scholar]
- 39.Riker A, et al. Immune selection after antigen-specific immunotherapy of melanoma. Surgery. 1999;126:112–120. [PubMed] [Google Scholar]
- 40.Cormier JN, et al. Natural variation of the expression of HLA and endogenous antigen modulates CTL recognition in an in vitro melanoma model. Int J Cancer. 1999;80:781–790. doi: 10.1002/(sici)1097-0215(19990301)80:5<781::aid-ijc24>3.0.co;2-a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schreiber H, Wu TH, Nachman J, Kast WM. Immunodominance and tumor escape. Semin Cancer Biol. 2002;12:25–31. doi: 10.1006/scbi.2001.0401. [DOI] [PubMed] [Google Scholar]
- 42.Straus SE, et al. The development of lymphomas in families with autoimmune lymphoproliferative syndrome with germline Fas mutations and defective lymphocyte apoptosis. Blood. 2001;98:194–200. doi: 10.1182/blood.v98.1.194. [DOI] [PubMed] [Google Scholar]
- 43.Davidson WF, Giese T, Fredrickson TN. Spontaneous development of plasmacytoid tumors in mice with defective Fas-Fas ligand interactions. J Exp Med. 1998;187:1825–1838. doi: 10.1084/jem.187.11.1825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Takeda K, et al. Involvement of tumor necrosis factor-related apoptosis-inducing ligand in surveillance of tumor metastasis by liver natural killer cells. Nature Med. 2001;7:94–100. doi: 10.1038/83416. [DOI] [PubMed] [Google Scholar]
- 45.Salvesen GS, Dixit VM. Caspase activation: the induced-proximity model. Proc Natl Acad Sci USA. 1999;96:10964–10967. doi: 10.1073/pnas.96.20.10964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Irmler M, et al. Inhibition of death receptor signals by cellular FLIP. Nature. 1997;388:190–195. doi: 10.1038/40657. [DOI] [PubMed] [Google Scholar]
- 47.Medema JP, de Jong J, van Hall T, Melief CJ, Offringa R. Immune escape of tumors in vivo by expression of cellular FLICE-inhibitory protein. J Exp Med. 1999;190:1033–1038. doi: 10.1084/jem.190.7.1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Landowski TH, Qu N, Buyuksal I, Painter JS, Dalton WS. Mutations in the Fas antigen in patients with multiple myeloma. Blood. 1997;90:4266–4270. [PubMed] [Google Scholar]
- 49.Gronbaek K, et al. Somatic Fas mutations in non-Hodgkin’s lymphoma: association with extranodal disease and autoimmunity. Blood. 1998;92:3018–3024. [PubMed] [Google Scholar]
- 50.Shin MS, et al. Alterations of Fas (Apo-1/CD95) gene in cutaneous malignant melanoma. Am J Pathol. 1999;154:1785–1791. doi: 10.1016/S0002-9440(10)65434-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Shin MS, et al. Alterations of Fas-pathway genes associated with nodal metastasis in non-small cell lung cancer. Oncogene. 2002;21:4129–4136. doi: 10.1038/sj.onc.1205527. [DOI] [PubMed] [Google Scholar]
- 52.Shin MS, et al. Inactivating mutations of CASP10 gene in non-Hodgkin lymphomas. Blood. 2002;99:4094–4099. doi: 10.1182/blood.v99.11.4094. [DOI] [PubMed] [Google Scholar]
- 53.Medema JP, et al. Blockade of the granzyme B/perforin pathway through overexpression of the serine protease inhibitor PI-9/SPI-6 constitutes a mechanism for immune escape by tumors. Proc Natl Acad Sci USA. 2001;98:11515–11520. doi: 10.1073/pnas.201398198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hersey P, Zhang XD. How melanoma cells evade trail-induced apoptosis. Nature Rev Cancer. 2001;1:142–150. doi: 10.1038/35101078. [DOI] [PubMed] [Google Scholar]
- 55.Schwartz RH. A cell culture model for T lymphocyte clonal anergy. Science. 1990;248:1349–1356. doi: 10.1126/science.2113314. [DOI] [PubMed] [Google Scholar]
- 56.Chen L, et al. Tumor immunogenicity determines the effect of B7 costimulation on T cell-mediated tumor immunity. J Exp Med. 1994;179:523–532. doi: 10.1084/jem.179.2.523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Toi M, et al. Clinical significance of the determination of angiogenic factors. Eur J Cancer. 1996;32:2513–2519. doi: 10.1016/s0959-8049(96)00397-8. [DOI] [PubMed] [Google Scholar]
- 58.Oyama T, et al. Vascular endothelial growth factor affects dendritic cell maturation through the inhibition of nuclear factor-κ B activation in hemopoietic progenitor cells. J Immunol. 1998;160:1224–1232. [PubMed] [Google Scholar]
- 59.Saito H, Tsujitani S, Ikeguchi M, Maeta M, Kaibara N. Relationship between the expression of vascular endothelial growth factor and the density of dendritic cells in gastric adenocarcinoma tissue. Br J Cancer. 1998;78:1573–1577. doi: 10.1038/bjc.1998.725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Almand B, et al. Clinical significance of defective dendritic cell differentiation in cancer. Clin Cancer Res. 2000;6:1755–1766. [PubMed] [Google Scholar]
- 61.Girolomoni G, Ricciardi-Castagnoli P. Dendritic cells hold promise for immunotherapy. Immunol Today. 1997;18:102–104. doi: 10.1016/s0167-5699(97)01030-x. [DOI] [PubMed] [Google Scholar]
- 62.De Smedt T, et al. Effect of interleukin-10 on dendritic cell maturation and function. Eur J Immunol. 1997;27:1229–1235. doi: 10.1002/eji.1830270526. [DOI] [PubMed] [Google Scholar]
- 63.Sharma S, et al. T cell-derived IL-10 promotes lung cancer growth by suppressing both T cell and APC function. J Immunol. 1999;163:5020–5028. [PubMed] [Google Scholar]
- 64.Ludewig B, et al. Spontaneous apoptosis of dendritic cells is efficiently inhibited by TRAP (CD40-ligand) and TNF-α, but strongly enhanced by interleukin-10. Eur J Immunol. 1995;25:1943–1950. doi: 10.1002/eji.1830250722. [DOI] [PubMed] [Google Scholar]
- 65.Carbone E, et al. Recognition of autologous dendritic cells by human NK cells. Eur J Immunol. 1999;29:4022–4029. doi: 10.1002/(SICI)1521-4141(199912)29:12<4022::AID-IMMU4022>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- 66.Yue FY, et al. Interleukin-10 is a growth factor for human melanoma cells and down-regulates HLA class-I, HLA class-II and ICAM-1 molecules. Int J Cancer. 1997;71:630–637. doi: 10.1002/(sici)1097-0215(19970516)71:4<630::aid-ijc20>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
- 67.Salazar-Onfray F, et al. Down-regulation of the expression and function of the transporter associated with antigen processing in murine tumor cell lines expressing IL-10. J Immunol. 1997;159:3195–3202. [PubMed] [Google Scholar]
- 68.Zeidler R, et al. Downregulation of TAP1 in B lymphocytes by cellular and Epstein-Barr virus-encoded interleukin-10. Blood. 1997;90:2390–2397. [PubMed] [Google Scholar]
- 69.Ristimaki A, Honkanen N, Jankala H, Sipponen P, Harkonen M. Expression of cyclooxygenase-2 in human gastric carcinoma. Cancer Res. 1997;57:1276–1280. [PubMed] [Google Scholar]
- 70.Sano H, et al. Expression of cyclooxygenase-1 and -2 in human colorectal cancer. Cancer Res. 1995;55:3785–3789. [PubMed] [Google Scholar]
- 71.Wolff H, et al. Expression of cyclooxygenase-2 in human lung carcinoma. Cancer Res. 1998;58:4997–5001. [PubMed] [Google Scholar]
- 72.Huang M, et al. Non-small cell lung cancer cyclooxygenase-2-dependent regulation of cytokine balance in lymphocytes and macrophages: up-regulation of interleukin 10 and down-regulation of interleukin 12 production. Cancer Res. 1998;58:1208–1216. [PubMed] [Google Scholar]
- 73.Gorsch SM, Memoli VA, Stukel TA, Gold LI, Arrick BA. Immunohistochemical staining for transforming growth factor β1 associates with disease progression in human breast cancer. Cancer Res. 1992;52:6949–6952. [PubMed] [Google Scholar]
- 74.Doran T, Stuhlmiller H, Kim JA, Martin EWJ, Triozzi PL. Oncogene and cytokine expression of human colorectal tumors responding to immunotherapy. J Immunother. 1997;20:372–376. doi: 10.1097/00002371-199709000-00006. [DOI] [PubMed] [Google Scholar]
- 75.Chen W, Frank ME, Jin W, Wahl SM. TGF-β released by apoptotic T cells contributes to an immunosuppressive milieu. Immunity. 2001;14:715–725. doi: 10.1016/s1074-7613(01)00147-9. [DOI] [PubMed] [Google Scholar]
- 76.Fontana A, et al. Transforming growth factor-β inhibits the generation of cytotoxic T cells in virus-infected mice. J Immunol. 1989;143:3230–3234. [PubMed] [Google Scholar]
- 77.Niehans GA, et al. Human lung carcinomas express Fas ligand. Cancer Res. 1997;57:1007–1012. [PubMed] [Google Scholar]
- 78.Hahne M, et al. Melanoma cell expression of Fas(Apo-1/CD95) ligand: implications for tumor immune escape. Science. 1996;274:1363–1366. doi: 10.1126/science.274.5291.1363. [DOI] [PubMed] [Google Scholar]
- 79.O’Connell J, O’Sullivan GC, Collins JK, Shanahan F. The Fas counterattack: Fas-mediated T cell killing by colon cancer cells expressing Fas ligand. J Exp Med. 1996;184:1075–1082. doi: 10.1084/jem.184.3.1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Strand S, et al. Lymphocyte apoptosis induced by CD95 (APO-1/Fas) ligand-expressing tumor cells-a mechanism of immune evasion? Nature Med. 1996;2:1361–1366. doi: 10.1038/nm1296-1361. [DOI] [PubMed] [Google Scholar]
- 81.Andreola G, et al. Induction of lymphocyte apoptosis by tumor cell secretion of FasL-bearing microvesicles. J Exp Med. 2002;195:1303–1316. doi: 10.1084/jem.20011624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Restifo NP. Not so Fas: Re-evaluating the mechanisms of immune privilege and tumor escape. Nature Med. 2000;6:493–495. doi: 10.1038/74955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Restifo NP. Countering the ‘counterattack’ hypothesis. Nature Med. 2001;7:259. doi: 10.1038/85357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Chappell DB, Zaks TZ, Rosenberg SA, Restifo NP. Human melanoma cells do not express Fas (Apo-1/CD95) ligand. Cancer Res. 1999;59:59–62. [PMC free article] [PubMed] [Google Scholar]
- 85.Arai H, Gordon D, Nabel EG, Nabel GJ. Gene transfer of Fas ligand induces tumor regression in vivo. Proc Natl Acad Sci USA. 1997;94:13862–13867. doi: 10.1073/pnas.94.25.13862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Kang SM, Lin Z, Ascher NL, Stock PG. Fas ligand expression on islets as well as multiple cell lines results in accelerated neutrophilic rejection. Transplant Proc. 1998;30:538. doi: 10.1016/s0041-1345(97)01396-1. [DOI] [PubMed] [Google Scholar]
- 87.Drozdzik M, Qian C, Lasarte JJ, Bilbao R, Prieto J. Antitumor effect of allogenic fibroblasts engineered to express Fas ligand (FasL) Gene Ther. 1998;5:1622–1630. doi: 10.1038/sj.gt.3300763. [DOI] [PubMed] [Google Scholar]
- 88.Chen JJ, Sun Y, Nabel GJ. Regulation of the proinflammatory effects of Fas ligand (CD95L) Science. 1998;282:1714–1717. doi: 10.1126/science.282.5394.1714. [DOI] [PubMed] [Google Scholar]
- 89.Zaks TZ, Chappell DB, Rosenberg SA, Restifo NP. Fas-mediated suicide of tumor-reactive T cells following activation by specific tumor: selective rescue by caspase inhibition. J Immunol. 1999;162:3273–3279. [PMC free article] [PubMed] [Google Scholar]
- 90.Cappello P, Novelli F, Forni G, Giovarelli M. Death receptor ligands in tumors. J Immunother. 2002;25:1–15. doi: 10.1097/00002371-200201000-00001. [DOI] [PubMed] [Google Scholar]
- 91.Dong H, et al. Tumor-associated B7-H1 promotes T-cell apoptosis: A potential mechanism of immune evasion. Nature Med. 2002;8:793–800. doi: 10.1038/nm730. [DOI] [PubMed] [Google Scholar]
- 92.McHugh RS, Shevach EM. Cutting edge: depletion of CD4+CD25+ regulatory T cells is necessary, but not sufficient, for induction of organ-specific autoimmune disease. J Immunol. 2002;168:5979–5983. doi: 10.4049/jimmunol.168.12.5979. [DOI] [PubMed] [Google Scholar]
- 93.Sakaguchi S, et al. Immunologic tolerance maintained by CD25+ CD4+ regulatory T cells: their common role in controlling autoimmunity, tumor immunity, and transplantation tolerance. Immunol Rev. 2001;182:18–32. doi: 10.1034/j.1600-065x.2001.1820102.x. [DOI] [PubMed] [Google Scholar]
- 94.Sutmuller RP, et al. Synergism of cytotoxic T lymphocyte-associated antigen 4 blockade and depletion of CD25+ regulatory T cells in antitumor therapy reveals alternative pathways for suppression of autoreactive cytotoxic T lymphocyte responses. J Exp Med. 2001;194:823–832. doi: 10.1084/jem.194.6.823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Antony PA, Restifo NP. Do CD4+ CD25+ immunoregulatory T cells hinder tumor immunotherapy? J Immunother. 2002;25:202–206. doi: 10.1097/00002371-200205000-00002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Shimizu J, Yamazaki S, Takahashi T, Ishida Y, Sakaguchi S. Stimulation of CD25+CD4+ regulatory T cells through GITR breaks immunological self-tolerance. Nature Immunol. 2002;3:135–142. doi: 10.1038/ni759. [DOI] [PubMed] [Google Scholar]
- 97.McHugh RS, et al. CD4+CD25+ immunoregulatory T cells: gene expression analysis reveals a functional role for the glucocorticoid-induced TNF receptor. Immunity. 2002;16:311–323. doi: 10.1016/s1074-7613(02)00280-7. [DOI] [PubMed] [Google Scholar]
- 98.Green EA, Choi Y, Flavell RA. Pancreatic lymph node-derived CD4+CD25+ Treg cells: highly potent regulators of diabetes that require TRANCE-RANK signals. Immunity. 2002;16:183–191. doi: 10.1016/s1074-7613(02)00279-0. [DOI] [PubMed] [Google Scholar]
- 99.Terabe M, et al. NKT cell-mediated repression of tumor immunosurveillance by IL-13 and the IL-4R-STAT6 pathway. Nature Immunol. 2000;1:515–520. doi: 10.1038/82771. [DOI] [PubMed] [Google Scholar]
- 100.Moodycliffe AM, Nghiem D, Clydesdale G, Ullrich SE. Immune suppression and skin cancer development: regulation by NKT cells. Nature Immunol. 2000;1:521–525. doi: 10.1038/82782. [DOI] [PubMed] [Google Scholar]